
Abstract
Background
Reversible N6-methyladenosine (m6A) modifications in messenger RNAs can be categorized under the field of “RNA epigenetics.” However, the potential role of m6A-related genes in gastric cancer (GC) prognosis has not been systematically researched.
Aims
This study was aimed at providing insights into the prognostic role of m6A-related gene expression, at both mRNA and protein levels.
Methods
Kaplan–Meier (KM) plotter database and The Cancer Genome Atlas (TCGA) database were used to explore the prognostic significance of individual m6A-related genes in overall survival (OS) and progression-free survival at the mRNA level. For independent validation, the protein level of genes significantly associated with prognosis in both databases was further detected in 450 paired GC and corresponding adjacent non-tumor tissues using tissue microarray (TMA)-based immunohistochemistry (IHC). The relationship between the FTO and ALKBH1 expression and the clinicopathological characteristics was explored.
Results
Among nine m6A-related genes, aberrantly high mRNA expression of FTO and ALKBH1 was associated with poor OS in the KM and TCGA cohorts. However, the TMA-IHC indicated that protein expression of FTO and ALKBH1 was markedly downregulated in GC tissues. A lower protein level of ALKBH1 was closely correlated with larger tumor sizes (≥ 5 cm) and more advanced TNM stages, while lower FTO protein expression was associated with shorter OS in GC patients.
Conclusions
Aberrant expression of demethylase genes, FTO and ALKBH1, has a distinct prognostic value in GC patients, indicating that FTO and ALKBH1 may play vital roles in GC progression and metastasis.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is one of the most common malignancies worldwide [1] and the second biggest health burden in China [2]. The basis of treatment is surgical removal of the tumor mass. The 5-year survival rate for R0 surgical resection ranges from 30 to 50% for patients with stage II disease and from 10 to 25% for patients with stage III disease [3]. However, these patients have a high likelihood of local or systemic relapse. At present, there are no solid biomarkers for GC, except the tissue expression level of human epidermal growth factor receptor 2 (HER2) and regular clinicopathological parameters, such as TNM stage, which predict prognosis and response to therapy [4]. Thus, there is an urgent need to locate and identify robust prognostic biomarkers for GC.
In recent years, another form of gene regulation at the RNA level, RNA epitranscriptomics, has attracted the attention and interest of the research community. N6-methyladenosine (m6A), the most abundant internal modification in eukaryotic messenger RNAs (mRNAs) and long non-coding RNAs (lncRNAs), is known to play critical roles in various bioprocesses such as tissue development, stem cell self-renewal and differentiation, heat-shock or DNA damage response, and maternal-to-zygotic transition [5]. The m6A patterns involve a series of proteins identified as “writers,” “erasers,” and “readers” of m6A. Methyltransferase-like 3 and methyltransferase-like 14 (METTL3 and METTL14) and their cofactor, Wilms’ tumor 1-associated protein (WTAP), compose the m6A methyltransferase complex (MTC), which catalyzes m6A modification [6]. Fat mass and obesity-associated protein (FTO), alkB homolog 1 (ALKBH1), and ALKBH5 have been shown to act as m6A demethylases, oxidizing the N-methyl group of m6A site to a hydroxymethyl group [7, 8]. Similar to DNA methylation [9], information contained in m6A codes needs to be deciphered by “readers” such as YTHDF1, YTHDF2, and YTHDF3, directly or indirectly [10,11,12]. All of the above m6A-related genes were reported to regulate vital biological processes associated with a range of malignancies, such as hepatocellular carcinoma [13,14,15], glioblastoma [16, 17], acute myeloid leukemia [18], breast cancer [19, 20], lung cancer [21], and GC [22]. However, the association between m6A-related genes and GC has not been studied in detail. The present study evaluated the prognostic role of mRNA expression of nine m6A-related genes using the Kaplan–Meier (KM) plotter database and The Cancer Genome Atlas (TCGA) database. Next, genes stated as being significantly associated with prognosis in both databases were further detected in 450 paired GC and corresponding adjacent non-tumor tissues using tissue microarray (TMA)-based immunohistochemistry (IHC) for independent validation (Fig. 1).

A diagrammatic summary of the research strategy in this study
Methods
Predicting Prognosis of GC by KM Plotter
KM plotter database includes 1065 GC samples with follow-up data. The median overall survival (OS) was 28.9 months, and median progression-free survival (PFS) was 18.3 months [23]. Unprocessed CEL files were MAS5 normalized in the R statistical environment (http://www.r-project.org) using the Affy Bioconductor library, and only arrays passing quality criteria were utilized. Gene expression and clinical data were integrated using PostgreSQL. The KM curve indicated the association between the candidate gene and survival, in which the samples are grouped by the median expression of each gene. KM survival plots with hazard ratio (HR), 95% confidence interval (CI), and log-rank p were obtained online.
Predicting Prognosis of GC by TCGA Database
The mRNA sequencing level 2 data (preprocessed and normalized) and clinical patient data of 415 GC cases were downloaded from TCGA database (http://download.cbioportal.org/stad_tcga_pub.tar.gz?raw=ture). The cutoffs dividing high and low expression groups were decided via receiver operating characteristic curve (ROC) analysis by SPSS Statistics 23.0 (IBM Corp., Armonk, NY, USA).
Tissue Samples
For tissue microarray (TMA) detection, 450 cases of gastric tumor and matched non-tumor tissues were collected in the department of pathology of Sun Yat-Sen University Cancer Center. To be specific, two different pieces of GC tissues and one piece of non-tumor tissue, taken at a distance of > 3 cm from the tumor margin, were collected from each individual, paraffin-embedded blocks, and arranged in recipient ones. These patients had not received any preoperative anticancer therapy. The clinicopathological features of patients are provided (Table 1). Tumor differentiation grades and clinical stages were reclassified according to the 8th American Joint Committee on Cancer (AJCC) TNM classification. The OS was calculated as the duration from the date of surgery to the date of death or last follow-up. The use of human tissue samples and clinical data was approved by the Ethics Committee of Sun Yat-Sen University Cancer Center, all patients provided signed informed consent, and the research was carried out in accordance with the Helsinki Declaration.
Immunohistochemistry (IHC) Staining and Evaluation
IHC staining was performed according to a previously described protocol [24]. Tissue sections were deparaffinized with dimethylbenzene and rehydrated via a graded alcohol series. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide for 15 min. Slides were boiled in tris(hydroxymethyl) aminomethane–EDTA buffer (pH 8.0) in a microwave for 30 min to retrieve antigen. Nonspecific antigens were blocked with 10% normal goat serum for 20 min. Next, the slides were incubated with monoclonal rabbit anti-FTO antibody (ab124892, 1:100 dilution, Abcam, Cambridge, UK) and anti-ALKBH1 antibody (ab126596, 1:50 dilution, Abcam) overnight at 4 °C in a moist chamber. Slides were incubated with horseradish peroxidase (DAKO ChemMate™ EnVision™ Detection Kit, Copenhagen, Denmark) for 30 min at 37 °C and subsequently incubated with 3,3′ diaminobenzidine (DAB) solution for visualization.
Two independent observers blinded to clinicopathological data conducted the immunoreactivity score (IRS) assessment for FTO and ALKBH1 expression. Scoring criteria for staining intensity were as follows: 0 (negative), 1 (weak), 2 (moderate), 3 (strong). The staining extent score was as follows: 0 (< 10%), 1 (11–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). The final expression score, ranging from 0 to 12, was calculated as intensity score × extent score. Total scores of 6 or higher were categorized as the high expression group, and the rest as the low expression group. The staining result for each case was the average IRS determined by two pathologists. The specimens would be rescored if the difference between the two pathologists was greater than 3.
Statistics
The Chi-squared test was used to analyze the association between candidate gene expression and clinicopathological characteristics of GC patients. Survival curves were evaluated using the Kaplan–Meier method, and differences between survival curves were tested by the log-rank test. Cox proportional hazards regression model was used to examine univariate and multivariate analyses. Only significantly different variables in univariate analysis were included in the multivariate analysis. Statistical significance was based on two-tailed tests at p < 0.05. SPSS 23.0 (IBM Corp.) and GraphPad Prism 6 (San Diego, CA, USA) software were used for statistical analyses and graphical representations.
Results
Prognostic Values of m6A-Related Genes in KM Plotter Cohort
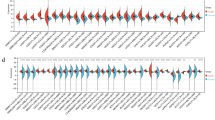
First, nine candidate genes were analyzed using the KM plotter database. The clinicopathological information of GC patients in this cohort was summarized previously (Table S1). The survival curves revealed that high mRNA expression of METTL3, METTL14, FTO, ALKBH1, and ALKBH5 and low mRNA expression of WTAP, YTHDF2, and YTHDF3 were associated with worse OS (Fig. 2) and worse PFS (Fig. 3), respectively. The mRNA expression of YTHDF1 seemed to have little effect on the prognosis of GC patients.

Prognostic influences of nine m6A-related genes on OS in the KM cohort. High mRNA expression levels of METTL3 (a), METTL14 (b), FTO (d), ALKBH1 (e), and ALKBH5 (f) and low mRNA expression levels of WTAP (c), YTHDF2 (h), and YTHDF3 (i) were associated with worse OS, while YTHDF1 (g) had no significant effect on OS in the KM cohort

Prognostic influences of nine m6A-related genes on PFS in the KM cohort. High mRNA expression levels of METTL3 (a), METTL14 (b), FTO (d), ALKBH1 (e), and ALKBH5 (f) and low mRNA expression levels of WTAP (c), YTHDF2 (h), and YTHDF3 (i) were associated with worse PFS, while YTHDF1 (g) had no significant influence on PFS in the KM cohort
Prognostic Values of m6A-Related Genes in TCGA Cohort
Next, we tested the associations between mRNA expression of the above genes and prognosis of GC patients in TCGA database to select even more reliable targets. The characteristics of GC patients in TCGA cohort are described in Table S2. The results suggested that higher mRNA levels of FTO and ALKBH1 lead to worse OS (Fig. 4a, b), while higher mRNA level of FTO, but not ALKBH1, was correlated with worse PFS (Fig. 4c, d), indicating that FTO and ALKBH1 were more likely to exert a significant influence on progression of GC compared to the other m6A-related genes. This also indicated that T stage, N stage, M stage, TNM stage, differentiation grade, FTO, and ALKBH1 expression had significant impact on OS in univariate analysis, while only FTO and ALKBH1 expression levels remained observably significant in multivariate analysis (Table S3). Patients diagnosed with stage III or IV had observably shorter OS than those with stage I or II (22.0 vs. 59.5 months, Fig. 4e). In addition, patients showing high expression levels of both FTO and ALKBH1 (OS: 19.9 vs. 72.2 months) and patients with high expression level of one gene (OS: 26.5 vs. 72.2 months) had observably shorter OS than patients with low expression levels of both genes (Fig. 4f).

Prognostic values of FTO and ALKBH1 on OS and PFS in the TCGA cohort. a, b High mRNA expression levels of FTO and ALKBH1 were correlated with poor OS. c, d High mRNA expression level of FTO but not ALKBH1 was correlated with poor PFS. e Patients diagnosed with stages III or IV had noticeably shorter OS than those with stages I or II. f Patients with high expression levels of both FTO and ALKBH1 (19.9 vs. 72.2 months) and patients with high expression level of one gene (26.5 vs. 72.2 months) had observably shorter OS than patients with low expression levels of both genes
Independent Validation of Prognostic Value of FTO and ALKBH1
Considering the findings of the in silico analysis, we validated the prognostic value of protein expression of demethylase genes FTO and ALKBH1 using TMA-based IHC to detect 450 paired GC and corresponding adjacent non-tumor tissues. Different staining intensities of FTO and ALKBH1 are displayed (Fig. 5a, b). Interestingly, as opposed to their mRNA levels, both FTO and ALKBH1 protein levels were significantly downregulated in signet ring cells compared to the adjacent normal gland cells (Fig. 5c, d). The proportion of strong and moderate staining was significantly lower in GC tissues than in adjacent non-tumor tissues for both FTO (p < 0.0001) and ALKBH1 (p < 0.0001) (Fig. 6a, b). Consistently, the IRSs of FTO and ALKBH1 were markedly lower in GC tissues than in adjacent non-tumor tissues (Fig. 6c, d), and lower protein expression of FTO, but not of ALKBH1, was closely associated with a shorter OS of GC patients (HR = 0.74, 95% CI: 0.56–0.98, and p = 0.0364) (Fig. 6e, f). In addition, the lower protein level of ALKBH1 was closely correlated with larger tumor sizes (≥ 5 cm, p < 0.001) and more advanced TNM stages (III/IV, p = 0.033) (Table 1). In the TMA-IHC cohort, T stage, N stage, M stage, TNM stage and the protein expression of FTO, but not ALKBH1, were correlated with OS in univariate analysis, while only the M stage, TNM stage, and FTO expression were significant in multivariate analysis, indicating that FTO protein expression was as valid as M stage and TNM stage in prognosis prediction in GC patients (Table 2).

FTO and ALKBH1 protein expression in GC tissue samples determined by TMA-IHC. a, b Representative images of different staining intensities of FTO and ALKBH1 protein. FTO mainly localizes in nucleus, and ALKBH1 mainly localizes in cytoplasm. c, d Representative images showing that both FTO and ALKBH1 protein expression levels were significantly downregulated in signet ring cells compared to adjacent normal gland cells in the same tissue sample

The IRSs of FTO and ALKBH1 and their influences on OS of GC patients. a, b The proportion of strong and moderate staining was significantly lower in GC tissues than that in adjacent non-tumor tissues for both FTO (p < 0.0001) and ALKBH1 (p < 0.0001). c, d The IRSs of FTO and ALKBH1 in GC tissues were both remarkably lower than those in corresponding non-tumor tissues. e, f The lower protein expression of FTO, but not that of ALKBH1, was closely associated with shorter OS in GC patients
Discussion
m6A, which is the most abundant modification in RNAs, is linked to human diseases, but its function in mammalian development is poorly understood. In addition, the role of m6A-related genes has been established in several malignancies, but the link between m6A expression and GC has not been elucidated [13,14,15,16,17,18,19,20,21,22]. Here, we evaluated the prognostic role of mRNA expression of nine m6A-related genes in GC.
Thousands of mRNA and lncRNAs show conserved m6A patterns, including transcripts encoding core pluripotency transcription factors. Thus, m6A can be taken as a mark of transcriptome flexibility required for stem cells to differentiate into specific lineages. FTO, also known as ALKBH9, belongs to the non-heme Fe II/α-KG-dependent dioxygenase AlkB family of proteins that also contains ALKBH1 to ALKBH8 [7]. It was reported that FTO was overexpressed at both protein and mRNA levels in GC tissues compared to corresponding adjacent non-tumor tissues, where high FTO expression was significantly associated with poor prognosis in GC patients [22]. By contrast, via analysis of RNAseq data from KM plotter and TCGA databases, we demonstrated that high mRNA expression of demethylase genes, FTO and ALKBH1, predicted a worse prognosis. However, TMA-IHC staining indicated that FTO and ALKBH1 were significantly downregulated at the protein level in GC tissue specimens, suggesting that high FTO protein expression is indicative of a better OS for GC patients.
Notably, results of several studies are in total or partial agreement with our findings. Pi et al. believed that high level of FTO expression inhibited GC cell line MGC-803 proliferation, migration, and invasion [25]. Liu et al. demonstrated a noticeable increase in proliferation in ALKBH1 knockdown HeLa cells caused by elevated methylation of transfer RNA (tRNA). This dynamic process was used by human cells in response to glucose deprivation [26]. Zhou et al. suggested that a lack of ALKBH1 restricted its binding with the promoter of ATF4, an osteoblast-enriched transcriptional factor indispensable for osteogenic differentiation, and led to transcriptional silencing of ATF4 with increasing m6A levels [27]. Gao et al. reported that ALKBH2, another member of the dioxygenase AlkB family, was downregulated in GC tissues and cell lines as determined by real-time PCR and confirmed by IHC and Western blot. Overexpression of ALKBH2 significantly inhibited GC cell proliferation and induced G1 arrest of the cell cycle in GC cell lines [28]. The above-mentioned observations reveal the suppressive role of FTO and ALKBH1 in GC and are substantiated by findings of the current study. However, it has been established that a high level of transcription does not necessarily guarantee a high level of translation that produces proteins that orchestrate complex cellular processes. These discrepant findings between mRNA and protein levels may be caused, at least in part, by various post-transcriptional regulation mechanisms, such as 5′ capping of mRNA, polyadenylation, alternative splicing, and multiple covalent RNA modifications [29]. The specific mechanisms underlying these discrepancies may have to be elucidated via more sophisticated experiments in the future.
In the last few years, modification in mRNAs or non-coding RNAs has been reported to play a critical role in virtually all major normal bioprocesses. Modification of mRNAs is now being pushed to the forefront of biology due to the discovery of the “writers,” “erasers,” and “readers” that add, remove, or preferentially bind to the m6A site [30]. RNA m6A patterns are involved in various aspects of mRNA metabolism including mRNA export, translation, and decay and perform an important function in post-transcriptional regulation of gene expression and protein translation [12]. In the non-coding RNA field, m6A modification acts as a barrier to tRNA accommodation and translation elongation [31]. Reversible demethylation of tRNA mediated by ALKBH1 results in attenuated translation initiation and tRNA decreased use in protein synthesis [26]. Moreover, researchers expanded the concept of the RNA epitranscriptome to circular RNAs (circRNAs), providing evidence that m6A modifications in circRNAs are written and read by the same machinery used for mRNAs, but often at different locations [32].
We acknowledge several limitations in our study. Firstly, mRNA expression levels of candidate genes were not substantiated in GC and non-tumor tissues via experiments. Secondly, the effect of FTO and ALKBH1 expression on the biological function of GC cells remains to be elucidated. Thirdly, the reason for the contradictory trends between mRNA and protein expression levels of FTO and ALKBH1 in GC remains to be further investigated.
In conclusion, the aberrantly high mRNA expression of demethylase genes, FTO and ALKBH1, was markedly associated with poor OS in light of the in silico analysis, while FTO and ALKBH1 expression was significantly downregulated in GC tissues at the protein level, according to the TMA-IHC staining results. Additionally, lower ALKBH1 protein expression level was closely correlated with larger tumor size (≥ 5 cm) and more advanced TNM stages (III/IV). Lower FTO protein expression was markedly associated with a shorter OS in GC patients.
References
Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132.
Climent M, Hidalgo N, Vidal O, et al. Postoperative complications do not impact on recurrence and survival after curative resection of gastric cancer. Eur J Surg Oncol. 2016;42:132–139.
Cui Y, Li SB, Peng XC, et al. Trastuzumab inhibits growth of HER2-negative gastric cancer cells through gastrin-initialized CCKBR signaling. Dig Dis Sci. 2015;60:3631–3641.
Deng X, Su R, Weng H, et al. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507–517.
Kobayashi M, Ohsugi M, Sasako T, et al. The RNA methyltransferase complex of WTAP, METTL3, and METTL14 regulates mitotic clonal expansion in adipogenesis. Mol Cell Biol. 2018;38:e00116-18.
Alemu EA, He C, Klungland A. ALKBHs-facilitated RNA modifications and de-modifications. DNA Repair Amst. 2016;44:87–91.
Fedeles BI, Singh V, Delaney JC, et al. The AlkB family of Fe(II)/alpha-ketoglutarate-dependent dioxygenases: repairing nucleic acid alkylation damage and beyond. J Biol Chem. 2015;290:20734–20742.
Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551–565.
Liao S, Sun H, Xu C. YTH domain: a family of N(6)-methyladenosine (m(6)A) readers. Genom Proteom Bioinform. 2018;16:99–107.
Wang X, He C. Reading RNA methylation codes through methyl-specific binding proteins. RNA Biol. 2014;11:669–672.
Dai D, Wang H, Zhu L, et al. N 6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124.
Zhao X, Chen Y, Mao Q, et al. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 2018;21:859–868.
Yang Z, Li J, Feng G, et al. MicroRNA-145 modulates N(6)-methyladenosine levels by targeting the 3′-untranslated mRNA region of the N(6)-methyladenosine binding YTH domain family 2 protein. J Biol Chem. 2017;292:3614–3623.
Ma JZ, Yang F, Zhou CC, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–543.
Cui Q, Shi H, Ye P, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634.
Zhang S, Zhao BS, Zhou A, et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606 e596.
Weng H, Huang H, Wu H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22:191–205 e199.
Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113:E2047–2056.
Zhang C, Zhi WI, Lu H, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527–64542.
Liu J, Ren D, Du Z, et al. m(6)A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem Biophys Res Commun. 2018;502:456–464.
Xu D, Shao W, Jiang Y, et al. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol Rep. 2017;38:2285–2292.
Szász AM, Lánczky A, Nagy Á, et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1065 patients. Oncotarget. 2016;7:49322–49333.
Li Y, Wu Z, Yuan J, et al. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer Lett. 2017;395:31–44.
Pi J, Zhang J, Wang X, et al. Down-regulation of FTO in human gastric cancer and its effect on cell line MGC-803 function. Basic Clin Med. 2017;37:907–911.
Liu F, Clark W, Luo G, et al. ALKBH1-mediated tRNA demethylation regulates translation. Cell. 2016;167:816–828 e816.
Zhou C, Liu Y, Li X, et al. DNA N(6)-methyladenine demethylase ALKBH1 enhances osteogenic differentiation of human MSCs. Bone Res. 2016;4:16033.
Gao W, Li L, Xu P, et al. Frequent down-regulation of hABH2 in gastric cancer and its involvement in growth of cancer cells. J Gastroenterol Hepatol. 2011;26:577–584.
Frye M, Blanco S. Post-transcriptional modifications in development and stem cells. Development. 2016;143:3871–3881.
Wang S, Sun C, Li J, et al. Roles of RNA methylation by means of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett. 2017;408:112–120.
Choi J, Ieong KW, Demirci H, et al. N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat Struct Mol Biol. 2016;23:110–115.
Zhou C, Molinie B, Daneshvar K, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 2017;20:2262–2276.
Acknowledgments
This study was funded by the Natural Science Foundation of Guangdong Province (2017A030310192) and Fundamental Research Funds for the Central Universities (17ykpy84).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The use of human tissue samples and clinical data was approved by the Ethics Committee of Sun Yat-Sen University Cancer Center (Guangzhou, China), all patients provided signed informed consent, and the research was carried out in accordance with the Helsinki Declaration. This article does not contain any studies with animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, Y., Zheng, D., Wang, F. et al. Expression of Demethylase Genes, FTO and ALKBH1, Is Associated with Prognosis of Gastric Cancer. Dig Dis Sci 64, 1503–1513 (2019). https://doi.org/10.1007/s10620-018-5452-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-018-5452-2