

A new direction of the reaction of 6-amino-substituted 4-hydrazino-1,3,5-triazin-2(1H)-ones with formic acid was found leading to the formation of N-substituted (4H-1,2,4-triazol-3-yl)guanidines together with the expected 5-amino-substituted 1,2,4-triazolo[1,5-a][1,3,5]-triazin-7-ones as a result of opening of the triazine ring and subsequent decarboxylation of an intermediate of the Dimroth rearrangement. The discovered reaction is of interest as a novel method for the synthesis of N-substituted (4H-1,2,4-triazol-3-yl)guanidines.
Similar content being viewed by others

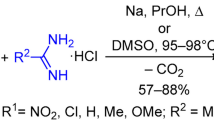

For some time, the reaction of hydrazinoazines with formic acid has been successfully used for the synthesis of fused 1,2,4-triazoloazines.1 The reaction proceeds via intermediate N-formylhydrazines, which in some cases can be preparatively isolated. In most cases, the closure of 1,2,4-triazole ring by the action of formic acid is accompanied by the Dimroth rearrangement (Scheme 1).2 At the same time, it is possible to achieve various directions for the cyclization if alternative reaction routes are possible.1a

ᅟ
Noteworthy, the mechanism of the Dimroth rearrangement involves opening and subsequent closure not of the 1,2,4-triazole ring but of the azine ring that is annulated to it.3 Thus, the reaction of hydrazinoazines with formic acid can serve as a convenient method for accessing a wide range of annulated 1,2,4-triazole heterocycles.
Previously, we obtained 5-amino-substituted 1,2,4-triazolo[1,5-a][1,3,5]triazin-7-ones 2a–e in a reaction of 6-aminosubstituted 4-hydrazino-1,3,5-triazin-2(1Н)-ones 1a–e with orthoformate.4 In search of alternative methods for the synthesis of 1,2,4-triazolo-1,3,5-triazines, we investigated the reaction of 6-amino-substituted 4-hydrazino-1,3,5-triazin-2(1Н)-ones with formic acid.
The reaction was conducted by heating hydrazines 1a–e under reflux in an excess of 100% formic acid. After removal of formic acid and washing the residue with water, 5-amino-substituted 1,2,4-triazolo[1,5-a][1,3,5]triazin-7-ones 2a–e were obtained in 50–69% yields (Table 1).
IR spectra as well as 1H and 13C NMR spectra of the obtained products 2a–e were identical with the spectra of 5-amino-substituted 1,2,4-triazolo[1,5-a][1,3,5]triazines synthesized previously by reacting hydrazine derivatives 1a–e with orthoformate (76-94% yields).4 Under the conditions of the reaction with formic acid, 7-amino-substituted 1,2,4-triazolo[1,5-a][1,3,5]triazin-5-ones 5a–e did not form, i.e., the 1,2,4-triazole ring closure proceeded exclusively at the nitrogen atom N-1 of the triazine ring (Scheme 2). In addition, no intermediate formylhydrazino-1,3,5-triazines 4a–e were detected in the reaction products.

ᅟ
The relatively low yields of the reaction products 2a–e (in some cases about 50%) can be attributed to the formation of water-soluble compounds that are removed during work-up of the reaction mixture with water. Such compounds may be formate salts of the starting hydrazino derivatives 1a–e, intermediate formylhydrazines 4a–e, or the final reaction products 2a–e.
Indeed, bringing the wash water pH to 9 with aqueous ammonia resulted in precipitation of crystalline products. However, analysis of IR, 1H and 13C NMR, and 1H-13C HMBC data of the products isolated from the aqueous phase, as well as the analysis of the X-ray structural data of compound 3a obtained after basification of the watersoluble substance formed in the synthesis of revealed that the second reaction product is N-substituted (4Н-1,2,4-triazol-3-yl)guanidines 3a–e (Table 1).
In the 1H NMR spectra of N-substituted (4Н-1,2,4-triazol-3-yl)guanidines 3a–e, signals of the amino group protons at 7.06–7.81 ppm are observed as a broad singlet, signals of the protons of the 1,2,4-triazole ring appear at 7.50–7.53 ppm (CH) and 12.32–12.42 ppm (NH). In the 13C NMR spectra of compounds 3a–e, three downfield signals are evident: two signals of 1,2,4-triazole ring carbons at 147.6–147.8 ppm (C-5) and 159.7–159.9 ppm (C-3), as well as the signal of guanidine carbon at 155.1–157.1 ppm.
According to X-ray structural analysis data, the molecule of compound 3a is planar within 0.1 Å (with the exception of the hydrogen atoms of the methyl groups). The bond lengths and valence angles indicate a strongly conjugated molecular structure. The lengths of all C(sp2)–N bonds are within 1.35(2) Å while bonds C(sp 3)–N have a length of 1.45(5) Å, typical for conjugated π-systems. In particular, the differences in lengths of the bonds between formally single and double bonds in the imino group C(4)=N(4)–C(3)Het are less than 0.03 Å, and are in good agreement with both the bond length of the heterocyclic moiety and the distance C(4)–N(5)H2 in the amino group. NH2 group is located in the cis position relative to the heterocyclic ring. This configuration is further stabilized by the intramolecular hydrogen bond NH···N with N(5)···N(3) distance of 2.674(2) Å (Fig. 1).

Molecular structure of compound 3a with atoms represented as thermal vibration ellipsoids with 50% probability.
Two routes for the formation of (4Н-1,2,4-triazol-3-yl)-guanidines 3 are conceivable. The first one is waterinduced transformation of 5-amino-substituted 1,2,4-triazolo[1,5-a][1,3,5]triazin-7-ones 2 (Scheme 3). Addition of water at the carbonyl group of compounds 2 leads to two geminal diols 6, which are converted to carbamic acids 7. Acids 7 are unstable under the reaction conditions and undergo decarboxylation to form guanidines 3.

ᅟ
A number of similar reactions of opening of the azine ring by water followed by decarboxylation of the resulting carbamic acid to condensed 1,2,4-triazoloazines have been described,5 for example in imidazo[1,5-a][1,3,5]triazinones.6 To test the first hypothesis, hydrolysis of 5-propylamino- 1,2,4-triazolo[1,5-a][1,3,5]triazin-7(6H)-one (2b) was attempted in boiling 80% aqueous formic acid. However, even after 48 h of heating under reflux the starting compound 2b underwent no change and was quantitatively recovered from the reaction mixture.
A second possible route of formation of guanidines 3 is decarboxylation of the carbamic acids 7 which form as an intermediates in the Dimroth rearrangement (Scheme 4).

ᅟ
Addition of formic acid at the carbonyl group of compounds 8 and subsequent opening of the 1,3,5-triazine ring lead to the formation of the mixed anhydrides of formic acid and carbamic acids 11 which are presumably more stable under the conditions of the reaction in comparison with carbamic acids 7. After rotation of the linear fragment and elimination of formic acid, compounds 11' form the Dimroth rearrangement product 5-aminosubstituted 1,2,4-triazolo[1,5-a][1,3,5]triazin-7-ones 2. In the reaction of hydrazines 1 with formic acid, 2 moles of water are formed, which may compete with formic acid in the subsequent Dimroth rearrangement. The competitive addition of water at the carbonyl group of compounds 8 and subsequent opening of the 1,3,5-triazine ring lead to the formation of carbamic acids 7 which gives N-substituted guanidines 3 after decarboxylation. Addition of water is the dominant of the competing reactions, as the molar ratio of water to formic acid in the reaction mixture is about 1:20 while the ratio of the yields of compounds 2 and 3 ranges from 3:1 to 1:1 (Table 1, yields in 100% formic acid).
To test the second proposition, we conducted the reaction of compounds 1a–e in excess of formic acid with different concentration (from 90 to 60%), i.e., successively increasing the water content (Table 1). Addition of 40% of water to the reaction mixture does not prevent the formylation reaction and the subsequent formation of 1,2,4-triazole ring. However, it does completely inhibit the addition of formic acid to the double bond of the carbonyl group. As a result, water becomes the only reagent that initiates the Dimroth rearrangement, and the only reaction products are N-substituted (4Н-1,2,4-triazol-3-yl)guanidines 3a–e.
To conclude, two conditions must be met for the formation of N-substituted (4Н-1,2,4-triazol-3-yl)guanidines: first, a carbonyl group must be present in the resulting molecule, and second, water must be present in the reaction mixture. Thus, under anhydrous conditions even in the presence of carbonyl group in the molecule the Dimroth rearrangement proceeds smoothly and with a high yield.7 On the other hand, if a carbonyl group is absent in the 1,2,4-triazoloazine molecule, the Dimroth rearrangement successfully proceeds in an aqueous medium in the presence of both acids and bases.3
The discovered directional reaction of 4-aminosubstituted 6-hydrazino-1,3,5-triazin-2(1H)-ones with dilute formic acid enables facile synthesis of N-substituted (4H-1,2,4-triazol-3-yl)guanidines.
Experimental
IR spectra were registered on an Avatar 360ESP FT-IR spectrometer using the attenuated total reflectance (ATR) attachment. 1H and 13C NMR spectra were acquired on a JEOL JNM ECX-400 (400 and 100 MHz, respectively) spectrometer in DMSO-d6, with TMS as internal standard. Elemental analysis was performed on a Eurovector EA 3000 Elemental Analyzer. Melting points were determined on a Gallenkamp apparatus and are uncorrected. Monitoring of the reaction progress and assessment of the purity of synthesized compounds was done by TLC on Silufol UV-254 plates (visualization with UV light at 254 nm).
Synthesis of 5-amino-substituted 1,2,4-triazolo[1,5- a ]-[1,3,5]triazin-7-ones 2a–e and N -substituted (4 H -1,2,4-triazol-3-yl)guanidines 3a–e (General method). A suspension of 4-amino-substituted 6-hydrazino-1,3,5-triazin-2(1H)-one 1a–e (10 mmol) in НСООН of the desired concentration (14 ml) was stirred and heated under reflux for 40–48 h. The reaction mixture was evaporated under reduced pressure, Н2О (50 ml) was added to the residue and the resulting mixture stirred for 15 min. The crystalline precipitate of 5-amino-substituted 1,2,4-triazolo[1,5-a][1,3,5]-triazin-7-one 2а–е was filtered off, washed with water, and dried in air, then recrystallized from 70% aqueous EtOH. The aqueous wash was evaporated under reduced pressure. The residue washed with acetone (10 ml), dissolved in Н2О (10 ml), and made pH 8–9 by the addition of aqueous ammonia. The formed precipitate of N-substituted (4H-1,2,4-triazol-3-yl)guanidine 3а–е was filtered and dried in air. The spectral characteristics of the prepared compounds 2a–e were identical to those of products prepared earlier.4
N , N -Dimethyl- N '-(4 H -1,2,4-triazol-3-yl)guanidine (3a). White crystals, mp 238–240°C (decomp.). IR spectrum, ν, cm−1: 3322, 3176, 3050, 2915, 2794, 2713, 1637, 1558, 1500, 1427, 1409, 1305, 1270, 1201, 1103, 1056, 973, 889, 844, 813, 796, 752, 734. 1H NMR spectrum, δ, ppm: 2.94 (6H, s, N(CH3)2); 7.53 (1H, s, H-5 triazole); 7.70 (2H, br. s, NH2); 12.35 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 36.8 (N(CH3)2); 147.7 (C-5 triazole); 157.1 (C guanidine); 159.7 (C-3 triazole). Found, %: C 39.12; H 6.71; N 54.17. C5H10N6. Calculated, %: C 38.95; H 6.54; N 54.51.
N -Propyl- N '-(4 H -1,2,4-triazol-3-yl)guanidine (3b). White crystals, mp 145–147°C (decomp.). IR spectrum, ν, cm−1: 3338, 3166, 3064, 2964, 2931, 2890, 1677, 1591, 1564, 1488, 1479, 1454, 1365, 1303, 1265, 1209, 1143, 1106, 1079, 1031, 962, 879, 821, 784, 729. 1H NMR spectrum, δ, ppm (J, Hz): 0.88 (3H, t, J = 7.4, (CH2)2CH 3); 1.48 (2H, sext, J = 7.4, CH2CH 2Me); 3.11 (2H, q, J = 6.8, NHCH 2); 6.45 (1H, br. s, NHCH2); 7.07 (2H, br. s, NH2); 7.50 (1H, s, H-5 triazole); 12.32 (1H, br. s, NH triazole). 13C NMR spectrum, δ, ppm: 11.4 ((CH2)2 CH3); 22.6 (CH2 CH2Me); 42.2 (NHCH2); 147.8 (C-5 triazole); 156.6 (C guanidine); 159.8 (C-3 triazole). Found, %: C 42.67; H 7.34; N 49.99. C6H12N6. Calculated, %: C 42.85; H 7.19; N 49.96.
N -Cyclohexyl- N '-(4 H -1,2,4-triazol-3-yl)guanidine (3c). White crystals, mp 268–270°C (decomp.). IR spectrum, ν, cm−1: 3286, 3265, 3160, 3103, 2929, 2852, 1633, 1556, 1519, 1450, 1348, 1307, 1261, 1201, 1147, 1081, 1043, 972, 889, 804, 746. 1H NMR spectrum, δ, ppm: 1.05–1.38 (5H, m), 1.46–1.59 (1H, m), 1.59–1.75 (2H, m) and 1.76– 1.94 (2H, m, cyclo-C6H11); 3.59 (1H, br. s, NHCH cyclo-C6H11); 6.38 (1H, br. s, NHC6H11); 7.06 (2H, br. s, NH2); 7.52 (1H, s, H-5 triazole); 12.39 (1H, br. s, NH triazole). 13C NMR spectrum, δ, ppm: 24.5 (CH2); 25.3 (CH2); 33.0 (CH2); 48.3 (NHCH cyclo-C6H11); 147.7 (C-5 triazole); 155.7 (C guanidine); 159.9 (C-3 triazole). Found, %: C 52.06; H 7.88; N 40.06. C9H16N6. Calculated, %: C 51.90; H 7.74; N 40.35.
N -(4 H -1,2,4-Triazol-3-yl)pyrrolidine-1-carboxamide (3d). White crystals, mp 166–168°C (decomp.). IR spectrum, ν, cm−1: 3328, 3178, 2973, 2879, 2796, 2709, 1633, 1546, 1502, 1461, 1403, 1355, 1299, 1267, 1218, 1099, 1078, 1006, 972, 879, 835, 756. 1H NMR spectrum, δ, ppm (J, Hz): 1.84 (4H, dt, J = 6.1, J = 3.2, 3,4-CH2 pyrrolidine); 3.35 (4H, t, J = 6.0, 2,5-CH2 pyrrolidine); 7.51 (1H, s, H-5 triazole); 7.59 (2H, br. s, NH2); 12.42 (1H, s, NH). 13C NMR spectrum, δ, ppm: 25.0 (3,4-CH2 pyrrolidine); 46.1 (2,5-CH2 pyrrolidine); 147.6 (C-5 triazole); 155.1 (C guanidine); 159.8 (C-3 triazole). Found, %: C 46.52; H 6.54; N 46.94. C7H12N6. Calculated, %: C 46.65; H 6.71; N 46.63.
N -(4 H -1,2,4-Triazol-3-yl)piperidine-1-carboxamide (3e). White crystals, mp 169–171°C (decomp.). IR spectrum, ν, cm−1: 3326, 3178, 3054, 2995, 2937, 2923, 2850, 1637, 1556, 1525, 1504, 1463, 1442, 1386, 1353, 1295, 1265, 1203, 1106, 1079, 1068, 1016, 973, 881, 858, 831, 756, 734. 1H NMR spectrum, δ, ppm (J, Hz): 1.46 (4H, quin, J = 5.8, J = 5.0, 4,5-CH2 piperidine); 1.57 (2H, dt, J = 10.4, J = 5.4, 3-CH2 piperidine); 3.41–3.52 (4H, m, 2,6-CH2 piperidine); 7.53 (1H, s, H-5 triazole); 7.81 (2H, br. s, NH2); 12.34 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 24.1 (CH2); 25.3 (CH2); 44.9 (2,6-CH2 piperidine); 147.7 (C-5 triazole); 156.0 (C guanidine); 159.7 (C-3 triazole). Found, %: C 49.35; H 7.42; N 43.23. C8H14N6. Calculated, %: C 49.47; H 7.26; N 43.27.
Hydrolysis of 5-dimethylamino-1,2,4-triazolo[1,5- a ]-[1,3,5]triazin-7-one (2a). A suspension of compound 2a (1.8 g, 10 mmol) in 80% aqueous HCOOH (14 ml) was stirred at reflux temperature for 48 h. The reaction mixture was evaporated under reduced pressure. Н2О (50 ml) was added to the residue and the resulting mixture stirred for 15 min. The crystalline precipitate was filtered and dried in air. As a result, starting compound 2a (1.73 g, 96%) was recovered.
X-ray structural analysis of compound 3a. Crystals of compound 3a suitable for X-ray structural analysis were obtained by evaporating MeOH solution. X-ray structural analysis of a single crystal of compound 3a was performed on an Xcalibur S automatic diffractometer using λMoKα beam.
Collection of data, obtaining and refinement of the parameters of the unit cell was carried out using the program CrysAlis CCD,8 processing of diffraction data was done using the program CrysAlis RED.8 The structure was solved with the direct method using SHELXL9 program set, refining was done on F 2 by the least-squares technique in the full-matrix anisotropic (isotropic for H atoms) approximation. Representation of the molecular structure in crystal was rendered using the program Mercury.10 The full set of X-ray structural data was deposited at the Cambridge Crystallographic Data Center (deposit CCDC 1438751).
This work was supported by funding from the Ministry of Education and Science of the Russian Federation within the framework of the Project part of the State Assignment to Samara State Technical University (Project № 4.813.2014/K).
Supplementary information file containing synthetic procedures, 1H, 13C NMR, and 1H–13C HMBC spectra of compounds 3a–e, and crystallographic data for compound 3a is available online at http://link.springer.com/journal/10593.
References
(a) Gizatullina, É. M.; Kartsev, V. G. Chem. Heterocycl. Compd. 1993, 29, 1369. [Khim. Geterotsikl. Soedin. 1993, 1614.] (b) Marckwald, W.; Meyer, E. Chem. Ber. 1900, 33, 1885. (c) Babichev, F. S.; Kovtunenko, V. A. Chem. Heterocycl. Compd. 1977, 13, 117. [Khim. Geterotsikl. Soedin. 1977, 147.]
(a) Allen, C. F. H.; Beilfuss, H. R.; Burness, D. M.; Reynolds, G. A.; Tinker, J. F.; VanAllan, J. A. J. Org. Chem. 1959, 24, 787. (b) Allen, C. F. H.; Beilfuss, H. R.; Burness, D. M.; Reynolds, G. A.; Tinker, J. F.; VanAllan, J. A. J. Org. Chem. 1959, 24, 793. (c) Shirakawa, K. J. Pharm. Soc. Jpn. 1959, 79, 903. (d) Makisumi, Y.; Kano, H. Chem. Pharm. Bull. 1959, 7, 907. (e) Paudler, W. W.; Helmick, L. S. J. Heterocycl. Chem. 1966, 3, 269.
Ashry, E. S. H. E.; Kilany, Y. E.; Rashed, N.; Assafir, H. Adv. Heterocycl. Chem. 1999, 75, 79.
Bakharev, V. V.; Parfenov, V. E.; Ul'yankina, I. V.; Zavodskaya, A. V.; Selezneva, E. V.; Gidaspov, A. A.; Eltsov, O. S.; Slepukhin, P. A. Tetrahedron 2014, 70, 6825.
(a) Rusinov, V. L.; Ulomskii, E. N.; Aleksandrov, G. G.; Parshin, V. E.; Chupakhin, O. N. Chem. Heterocycl. Compd. 1991, 27, 561. [Khim. Geterotsikl. Soedin. 1991, 700.] (b) Baig, G. U.; Stevens, M. F. G. J. Chem. Soc., Perkin Trans. 1 1987, 665. (c) Kappe, T.; Roschger, P.; Färber, G. J. Heterocycl. Chem. 1993, 30, 1267. (d) Rusinov, V. L.; Ulomskii, E. N.; Kozhevnikov, D. N.; Chupakhin, O. N.; Aleksandrov, G. G. Russ. J. Org. Chem. 1996, 32, 738. [Zh. Org. Khim. 1996, 32, 770.] (e) Orihuela, S.; Sánchez, M. P.; Quirós, M.; Molina, J.; Faure, R. J. Mol. Struct. 1997, 415, 285. (f) Schroeter, G.; Finck, E. Chem. Ber. 1938, 71, 671.
Golankiewicz, B.; Januszczyk, P.; Zeidler, J.; Popenda, M. Nucleosides, Nucleotides Nucleic Acids 2004, 23, 127.
Balicki, R. Pol. J. Chem. 1983, 57, 1219.
Oxford Diffraction (2006). CrysAlis CCD (Version 1.171.29.9) and CrysAlis RED (Version 1.171.29.9). Oxford Diffraction Ltd.: Abingdon.
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112.
Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. J. Appl. Crystallogr. 2008, 41, 466.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2015, 51(11/12), 1014–1018
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary information file
(PDF 2304 kb)
Rights and permissions
About this article

Cite this article
Bakharev, V.V., Parfenov, V.E., Ul’yankina, I.V. et al. An unexpected direction of the reaction of hydrazino-1,3,5-triazines with formic acid. Synthesis of (4H-1,2,4-triazol-3-yl)guanidines. Chem Heterocycl Comp 51, 1014–1018 (2015). https://doi.org/10.1007/s10593-016-1812-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-016-1812-z