
Abstract
Currently, three stocks of sei whales (Balaenoptera borealis) are defined in the North Atlantic; the Nova Scotian, Iceland-Denmark Strait and Eastern North Atlantic stocks, which are mainly based upon historical catch and sighting data. We analyzed mitochondrial control region DNA (mtDNA) sequences and genotypes from 7 to 11 microsatellite loci in 87 samples from three sites in the North Atlantic; Iceland, the Gulf of Maine and the Azores, and compared against the North Pacific using 489 previously published samples. No statistically significant deviations from homogeneity were detected among the North Atlantic samples at mtDNA or microsatellite loci. The genealogy estimated from the mtDNA sequences revealed a clear division of the haplotypes into a North Atlantic and a North Pacific clade, with the exception of one haplotype detected in a single sample from the Azores, which was included in the North Pacific clade. Significant genetic divergence between the North Atlantic and North Pacific Oceans was detected (mtDNA ΦST = 0.72, microsatellite Weir and Cockerham’s ϴ = 0.20; p < 0.001). The coalescent-based estimate of the population divergence time between the North Atlantic and North Pacific populations from the sequence variation among the mtDNA sequences was at 163,000 years ago. However, the inference was limited by an absence of samples from the Southern Hemisphere and uncertainty regarding mutation rates and generation times. The estimates of inter-oceanic migration rates were low (Nm at 0.007 into the North Pacific and at 0.248 in the opposite direction). Although estimates of genetic divergence among the current North Atlantic stocks were low and consistent with the extensive range of movement observed in satellite tagged sei whales, the high uncertainty of the genetic divergence estimates precludes rejection of multiple stocks in the North Atlantic.
Similar content being viewed by others

Introduction
The pelagic sei whale (Balaenoptera borealis) has a cosmopolitan distribution and undertakes seasonal migrations between high-latitude summer foraging grounds and low-latitude winter breeding grounds (Mizroch et al. 1984). Sei whales were commercially hunted from the 1950s to 1980s after populations of the larger baleen whales were depleted by whaling (Mizroch et al. 1984; Prieto et al. 2012). The current population trends are unknown and the International Union for the Conservation of Nature (IUCN) estimated the current global abundance of sei whales at approximately 20% of pre-whaling levelsFootnote 1. Although the International Whaling Commission (IWC) placed a moratorium on commercial whaling in 1986, sei whales are still occasionally targeted under special permits for scientific whaling and aboriginal subsistence huntingFootnote 2.
In 1977, the IWC divided the global sei whale population into distinct ‘stocks’ for management purposes. The stock divisions were based upon the distribution of catches and sightings as well as mark-recapture data, which was the nature of the data available at the time (Donovan 1991). The Southern Hemisphere was divided into six stocks, following IWC management practice for other baleen whale species. Initially, three distinct stocks were proposed in the North Pacific, but these were subsequently combined into a single stock, due to absence of conclusive evidence for a three-stock hypothesis (Donovan 1991).
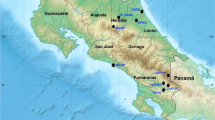
In the North Atlantic, sei whales were caught and sighted in eight main areas. However, the IWC did not presume these areas to represent different stocks and instead divided the North Atlantic sei whales into three stocks: the Nova Scotian, Iceland-Denmark Strait and Eastern North Atlantic stocks (Fig. 1). The possible presence of a fourth stock off Labrador north of the Nova Scotian stock boundary (Donovan 1991) was acknowledged, but this stock was never designated. After the cessation of commercial sei whaling, the overall research effort aimed specifically at sei whales was reduced and most efforts were directed towards the larger mysticetes (Prieto et al. 2012). As a result, the original stock boundaries for sei whales in the North Atlantic have remained unchanged, even though it is unclear whether they reflect an underlying ‘biological’ population structure (Donovan 1991).

Map with sampling locations in the North Atlantic and the current IWC stock boundaries
As is the case for other cosmopolitan mysticete species, such as fin (Balaenoptera physalus) and humpback whales (Megaptera novaeangliae), each major ocean basin (i.e. the Southern, the Pacific and the Atlantic Ocean) likely represents a distinct stock or subspecies (e.g. Archer et al. 2013; Jackson et al. 2014), and perhaps even different species in the case of right whales (Eubalaena spp.; Rosenbaum et al. 2000). The sei whale’s annual migration cycle between low and high latitudes is similar to the annual migration pattern assumed for many mysticetes resulting in an anti-tropical temporal separation between populations in different hemispheres (Mizroch et al. 1984). In addition, the populations in the Atlantic and the Pacific are geographically separated by continental land masses.
Genetic analysis of sei whale materials began with an allozyme study by Wada and Numachi (1991) who compared the allozyme variation at 45 loci in sei whales sampled in the Southern Ocean and the North Pacific. The authors reported statistically significant differences in allele frequencies between the two hemispheres. A more recent study compared mitochondrial DNA (mtDNA) control region sequence variation in samples collected from sei whales in the two aforementioned ocean basins and the North Atlantic (Baker et al. 2004). The study revealed that North Atlantic sei whales were genetically distinct from their North Pacific and Southern Hemisphere conspecifics. In contrast to earlier findings by Wada and Numachi (1991), Baker et al. (2004) failed to detect a clear differentiation between the Southern Ocean and the North Pacific.
The population genetic structure of sei whales within each ocean basin remains poorly understood as well. No data or analyses of the sei whale population genetic structure within the Southern Ocean have been presented so far. In the cases of the North Pacific and North Atlantic populations, few analyses and data have been presented. Kanda et al. (2006) failed to detect any spatial or temporal heterogeneity in the genetic variation at both microsatellite loci and later mtDNA sequences (Kanda et al. 2009) in 790 North Pacific sei whales. Similarly, Daníelsdottír et al. (1991) did not detect any temporal heterogeneity at 40 allozyme loci genotyped in 101 sei whales caught off Iceland between 1985 and 1988. Population genetic structure across the North Atlantic basin has yet to be assessed.
Recent satellite tagging studies (Olsen et al. 2009; Prieto et al. 2014) have shed some light on possible sei whale migratory routes in the North Atlantic. Olsen et al. (2009) deployed a satellite radio transmitter on a sei whale off the Azores, which was tracked to the Labrador Sea, revealing that some sei whales traverse the entire North Atlantic during the spring migration. Prieto et al. (2014) later deployed satellite radio transmitters on seven sei whales off the Azores during their spring migration, which were all tracked to summer foraging grounds in the Labrador Sea. Signals from two transmitters were lost when the tagged whales were moving toward the Iceland-Denmark Strait. The trajectory of these two tagged whales suggests that sei whales can move among different high-latitude summer foraging grounds, but whether different sei whale breeding populations also utilize the same foraging grounds, remains unknown. The exact location of the low-latitude winter breeding grounds is unknown, although there may be a located ground off northwestern Africa (Ingebrigtsen 1929; Prieto et al. 2012, 2014).
Given the documented long seasonal migrations of sei whales in the North Atlantic (Olsen et al. 2009; Prieto et al. 2014) and wide summer ranges (see above), it is plausible that the genetic heterogeneity among North Atlantic sei whale summer grounds is low as in the case of the North Pacific sei whale (Kanda et al. 2006, 2009). Here we present the results of the first assessment of the population genetic structure of sei whales in three different locations in the North Atlantic, representing two of the three putative stocks; off Iceland, in the Gulf of Maine and in the Azores. Under the three-stock hypothesis, we expected Iceland and the Azores to be genetically similar, and different from the Gulf of Maine. However, Iceland and the Gulf of Maine most likely represent summer foraging grounds and the Azores a migratory corridor where whales from different winter breeding grounds potentially mix (Olsen et al. 2009; Prieto et al. 2014). We estimated the effective population size, divergence time and migration rates of sei whales in two different ocean basins: the North Atlantic and North Pacific Oceans. To this end, the genetic data on North Atlantic sei whales from the present study were combined with previously published genetic data collected from North Pacific sei whales (Kanda et al. 2006, 2009).
Materials and methods
Sample collection
The genetic data from the North Atlantic were obtained from tissue samples collected from sei whales caught during special-permit whaling operations off Iceland (1986–1988; n = 43), and as skin biopsies obtained from free-ranging sei whales using a crossbow and biopsy tips (Palsbøll et al. 1991) in the Gulf of Maine (1999, 2002–2004; n = 18) and the Azores (2005, 2008–2010; n = 26) (Fig. 1). Biopsy collection was conducted under national permits and according to national regulations. The laboratory methods described below pertain to the North Atlantic samples.
Data from previous studies
Genetic data from the North Pacific (collected 2002–2007) were obtained from previously published studies (n = 489; Kanda et al. 2006, 2009; Tamura et al. 2009). A single additional Antarctic sei whale mtDNA control region sequence was obtained from GenBank™ (accession number NC_006929.1; Sasaki et al. 2005).
DNA extraction and sexing
Total-cell DNA was extracted using the Qiagen DNeasy™ Blood and Tissue Kit (Qiagen Inc.) according to the manufacturer’s instructions. The extracted DNA was re-suspended in 1× TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). Samples were sexed using the ZFY/ZFX multiplexing system as described by Bérubé and Palsbøll (1996a, b).
Genotyping microsatellite loci
Eleven microsatellite loci were genotyped using the polymerase chain reaction (PCR; Mullis and Faloona 1987). The specific loci genotyped were: EV094 and EV037 (Valsecchi and Amos 1996), GATA028, GATA053, GATA098 (Palsbøll et al. 1997), GT011 (Bérubé et al. 1998), GT023 and GT211 (Bérubé et al. 2000) as well as AC082, CA232 and GT541 (Bérubé et al. 2005).
PCR amplifications of the above microsatellite loci were performed in 10 µL reaction volumes containing 1× Taq buffer (Fermentas Inc.), 3.2 mM dNTPs, 0.4 units Taq DNA polymerase (Fermentas Inc.) and 1 ng extracted DNA. The concentration of each PCR primer pair differed among loci. The concentrations of the forward and reverse primers were: 0.25 µM for locus EV094, GATA028, GATA053 and GATA098, and 0.50 µM for locus AC082, CA232, EV037, GT011, GT023, GT211 and GT541. The PCR amplifications were conducted using a MJ Research PTC-100™ (MJ Research Inc.) in the case of locus GATA028 and GT023, a MJ Research Dyad™ thermocycler (MJ Research Inc.) for locus AC082, CA232, EV037, EV094, GATA053, GATA098, GT011 and GT541, and a Stratagene Robocycler™ (Stratagene Inc.) for locus GT211. PCR cycling profiles were as described in the original publications of each locus.
The experimental conditions employed for the data generation of the North Pacific samples were described by Kanda et al. (2006, 2009). The microsatellite genotypes from the two datasets (the North Pacific and North Atlantic) were calibrated by re-genotyping the above microsatellite loci in 55 North Pacific samples.
Sequencing the mtDNA control region
The first 487 base pairs of the 3′ end of the mtDNA control region were amplified and the nucleotides sequenced. The fragment corresponds to positions 15,476–15,963 in the published sei whale mitochondrial genome (Árnason et al. 1993; Sasaki et al. 2005). The PCR primers used for the amplification were MT4F (Árnason et al. 1993) and Mn312-R (Palsbøll et al. 1995), as well as BP16071R (Drouot et al. 2004).
For the North Atlantic samples, PCR amplification was performed in a final volume at 15µL containing: 1 µM of each PCR primer, 1× Taq DNA polymerase buffer (Fermentas Inc.), 3.2 mM dNTPs, 0.09 units Taq DNA polymerase (Fermentas Inc.), and 1 ng of extracted DNA. The PCR amplifications were conducted using an MJ Research PTC-100™ thermocycler (MJ Research Inc.) and occurred in 25 reaction cycles, each consisting of a denaturing step of 30 s at 94 °C, a 30 s annealing step at 54 °C and a 120 s extension step at 72 °C. These 25 cycles were preceded by a single 120 s denaturing step at 94 °C.
Unincorporated ddNTPs and PCR primers were removed using the Shrimp Alkaline Phosphate/Exo-I protocol described by Werle et al. (1994). Cycle-sequencing of the PCR products obtained by the above described amplifications was performed using the BigDye Terminator™ ver. 3.1 Cycle Sequencing Kit (Applied Biosystems Inc.) following the manufacturer’s instructions, in both directions using the same primers as used for the initial PCR amplification. The cycle-sequencing products were purified by ethanol/sodium acetate precipitation (Sambrook and Russell 2001). The order of labeled sequencing fragments was resolved by capillary electrophoresis on an ABI 3730 DNA Genetic Analyzer™ (Applied Biosystems Inc.).
Analysis of microsatellite genotypes
Quality control and levels of polymorphism
Microsatellite alleles were visually checked and sized using GENEMAPPER™ (ver. 4.0, Applied Biosystems Inc.). All 87 samples were re-typed once at all 11 loci to estimate a genotyping ‘inconsistency rate’ per genotype. We estimated the number of alleles (A), the expected (HE), the observed heterozygosity (HO), and the probability of identity (I; Paetkau et al. 1995). I was subsequently employed to detect duplicate samples from the same individuals. HE and HO were estimated using ARLEQUIN (ver. 3.5.2.2, Excoffier and Lischer 2010) and I was estimated using GENALEX (ver. 6.5, Peakall and Smouse 2006, 2012). The 95% confidence interval for the mean HE and HO was estimated by bootstrapping over loci (10,000 replicates) using the R package POPGENKIT (Paquette 2012) in R (ver. 3.2.5, R Development Core Team 2016).
Controlling procedure for multiple comparisons
The false discovery rate correction developed by Benjamini and Hochberg (FDR; Benjamini and Hochberg 1995) was applied in all instances when multiple simultaneous tests were conducted, using a critical alpha-value at 0.05.
Assessing deviations from Hardy–Weinberg expectations and linkage disequilibrium
Deviations from the expected Hardy–Weinberg genotype proportions and linkage disequilibrium were assessed using Fisher’s exact test (Fisher 1935) implemented in GENEPOP (ver. 4.1.4, Raymond and Rousset 1995; Rousset 2008) using the default analysis parameters and a complete enumeration whenever possible.
Homogeneity tests and genetic divergence
The degree of genetic differentiation was estimated as ϴ (Weir and Cockerham 1984). The probability of ϴ being equal to or larger than the observed value of ϴ under the null hypothesis of a panmictic population was estimated from 10,000 permutations (without replacement) as implemented in ARLEQUIN (ver. 3.5.2.2, Excoffier and Lischer 2010). The 95% confidence intervals of the observed estimates were obtained from 10,000 bootstrap replicates as implemented in the package DIVERSITY (Keenan et al. 2013) in R (ver. 3.2.5, R Development Core Team 2016).
Bayesian clustering
The software STRUCTURE (ver. 2.3.4, Pritchard et al. 2000; Falush et al. 2007) was employed to assess possible cryptic population genetic structure. We followed the recommendation by Wang (2017). In each assessment, we employed the admixture and the ‘F’ model, the sample location as a prior, and 100,000 burn-in Markov chains, followed by 200,000 Markov chains. Fifteen replicates were conducted per value of K, ranging from one to five. Lambda was inferred per ‘population’. The remaining estimation parameters were the software default values. The output was summarized using the program CLUMPAK (Kopelman et al. 2015). The most probable value of K was determined from the posterior mean likelihood values (Pritchard et al. 2000).
Analysis of mtDNA control region sequences
Levels of polymorphism
The chromatograms of the mtDNA control region sequences were visually checked using CHROMAS™ (ver. 2.13, Technelysium Inc.) and sequences were aligned using CLUSTALW (ver. 1, Thompson et al. 1994) with default parameter settings as implemented in MEGA (ver. 6.06, Tamura et al. 2013). DNASP (ver. 5.10, Librado and Rozas 2009) was employed to estimate the haplotype (HD) and nucleotide diversity (π; Nei 1987). Coalescent simulations (Hudson 1990, implemented in DNASP) were employed to estimate the 95% confidence interval for both HD and π from 10,000 replicates.
Estimation of mtDNA haplotype sequence genealogy
Nucleotide positions subject to alignment gaps were deleted from the entire dataset. The genetic distances among the haplotypes were estimated and visualized using MEGA (ver. 6.06, Tamura et al. 2013). Genetic distances were estimated using Kimura’s 2-parameter model of nucleotide substitution (Kimura 1980) using a transition–transversion ratio (R) estimated from the data. R was estimated at 15 using the maximum-likelihood method in MEGA. The mtDNA genealogy was estimated using the maximum-likelihood method from the genetic distances estimated as described above. The consensus genealogy and support for each node was inferred from 10,000 bootstrap (over nucleotide positions) replicates (Felsenstein 1985). The genealogy was rooted with the homologous mtDNA control region sequences from a North Atlantic fin whale, Balaenoptera physalus, (GenBank™ accession number NC_001321.1; Árnason et al. 1991) and a North Pacific Bryde’s whale, B. brydei (GenBank™ accession number NC_006928.1; Sasaki et al. 2005). Furthermore, a neighbor-joining genealogy (Saitou and Nei 1987; Tamura et al. 2004) was estimated in MEGA using the same settings as for the maximum-likelihood genealogy and default settings for tree inference. Haplotype networks of both genealogies (without the Antarctic haplotype and the two outgroups) were estimated using the software HAPLOTYPE VIEWER (Ewing 2010).
Homogeneity tests and genetic divergence
The degree of differentiation was estimated as ΦST (Excoffier et al. 1992) using ARLEQUIN (ver. 3.5.2.2, Excoffier and Lischer 2010) applying Kimura’s 2-parameter model (Kimura 1980). The probability of ΦST being equal to or larger than the observed value of ΦST under the null hypothesis of a panmictic population was estimated from 10,000 permutations (without replacement) as implemented in ARLEQUIN. The 95% confidence intervals of the observed estimates were obtained from 10,000 bootstrap replicates as implemented in the package DIVERSITY (Keenan et al. 2013) in R (ver. 3.2.5, R Development Core Team 2016).
Estimation of effective population size, divergence time and migration rates
Effective population size, population divergence time and migration rates were estimated employing the coalescent approach implemented in the software IMA2P (ver. 1.0, Sethuraman and Hey 2016) which applies the Isolation with Migration model to genetic data. Compared to other demographic inference methods such as the methods implemented in the software BEAST (Drummond and Rambaut 2007) or MIGRATE-N (Beerli and Felsenstein 2001), which allow the estimation of either effective population size and divergence time or effective population size and migration rate, IMA2P allows the estimation of all three parameters (i.e., effective population size, divergence time and migration rate).
We applied the HKY model of sequence evolution (Hasegawa et al. 1985) and an annual, per-locus mutation rate at 2.58 × 10−5 (based upon a per-site mutation rate at 5.30 × 10−8 from Alter and Palumbi 2009) and a prior range from 4.87 × 10−6 to 4.87 × 10−5. The generation time was 26.19 years; i.e. the average of 29.08 years (Pacifici et al. 2013) and 23.30 years (Taylor et al. 2007). The inheritance scalar was set at 0.25.
The priors were defined from the posterior distribution from preliminary estimations (see Table S1 and Fig. S1) varying priors of θ (4Neµ, where Ne denotes the effective population size and µ the generational mutation rate), m (Nm/µ, where Nm denotes the number of migrants per generation) and divergence time (t = Tdivµ, where Tdiv denotes the time since splitting in generations) parameters. The final prior parameter values were set at θ = 250, m = 1.5 and t = 10 for the upper bound and zero for the lower bound for all parameters. The final Markov Chain Monte Carlo (MCMC) sampling comprised 1.0 × 108 steps, with samples drawn from the posterior every 100 steps and a preliminary burn-in at 1.0 × 106 steps. The Metropolis-Coupled Markov Chain Monte Carlo (MC3) was applied to improve the mixing. Stationarity was considered achieved when no perceivable trends were observed in the plot trend and an effective sample size (ESS) > 500 was obtained for all values. In addition, six independent runs, i.e. with different random number seeds, were examined for consistency in the final parameter estimates. The final parameter estimates of Ne, t and 2mNe were the average value of the six replicates (Table S2).
Tests of mutation-drift equilibrium and mismatch distributions
Estimates of Tajima’s D (Tajima 1989) and Fu and Li’s F* (Fu and Li 1993) and their statistical significance were computed using DNASP (ver. 5.10, Librado and Rozas 2009) to assess possible deviations from neutral evolution. Coalescent simulations (Hudson 1990; implemented in DNASP) were employed to estimate 95% confidence intervals for D and F* from 10,000 replicates. Furthermore, frequency distributions of the observed pairwise nucleotide site differences (‘mismatch distributions’) per sampling location were computed using DNASP and compared to the expected distribution for a population of exponentially expanding size (Watterson 1975; Slatkin and Hudson 1991; Rogers and Harpending 1992). The degree of statistical deviation from the expected distribution was evaluated with the χ2 test (Lindgren 1975).
Data access
All North Atlantic mtDNA haplotypes have been deposited in GenBank™ under accession numbers MH035689–MH035695. Interested readers are encouraged to contact the corresponding author(s) for microsatellite genotypes and access to raw data.
Results
Data analysis of microsatellite genotypes
Duplicate samples and missing data
The probability of identity (I) was estimated at 5.0 × 10−9 for the North Atlantic samples (a total of 11 loci, Tables 1 and 2) and at 1.1 × 10−5 for the North Pacific samples (a total of 7 loci, Table 1). Consequently, the expected number of pairs of unrelated individuals matching at all loci was at 4.2 × 10−7 in the North Atlantic and at 5.3 × 10−3 in the North Pacific. No matching pairs of multi-locus genotypes were observed among the North Pacific samples. A total of three pairs of matching multi-locus genotypes were detected among the North Atlantic samples; two sample pairs from the Gulf of Maine and one pair from the Azores. Also considering the samples’ corresponding sex and mtDNA haplotype, these were inferred as duplicate samples from the same individuals. Accordingly, only data from one sample of each identical pair were retained in the final dataset.
The calibration with the North Atlantic dataset (i.e. size-calling of North Pacific alleles after amplification with North Atlantic primers) failed for four North Pacific samples, which were thus discarded. No ambiguous genotypes were found after re-typing the North Atlantic samples for all loci, yielding an inconsistency rate of < 5.2 × 10−4 per genotype.
The final microsatellite dataset was comprised of 569 unique multi-locus genotypes; n = 485 for the North Pacific, n = 43 for Iceland, n = 16 for the Gulf of Maine and n = 25 for the Azores. In total, eight genotypes were missing from the final dataset (i.e. 0.2%). Of the four additional microsatellite loci genotyped only in the North Atlantic samples, 6% of genotypes were missing.
Diversity estimates
Tables 1 and 2 list the diversity estimates observed for the microsatellite loci. The number of alleles ranged from 3 (GATA053) to 13 (GT023). The mean number of alleles was 8.4. Private alleles were detected in both ocean basins, as well as in each of the three North Atlantic sampling locations. When considering all 59 different alleles observed in the total dataset for 7 loci, 21 (35.6%) of these were private to the North Pacific and 10 (16.9%) were private to the North Atlantic. When considering all 68 different alleles observed in the North Atlantic dataset for 11 loci, 1 allele (1.5%) was private to the Gulf of Maine, 5 alleles (7.4%) were private to Iceland and 2 (2.9%) were private to the Azores.
Mean HE for all seven microsatellite loci was similar in each ocean basin (Table 1); HE was estimated at 0.60 in the North Atlantic (ranging from 0.19 to 0.76) and at 0.58 in the North Pacific (ranging from 0.31 to 0.81). The mean HO was also estimated at 0.60 in the North Atlantic (ranging from 0.17 to 0.88) and at 0.56 in the North Pacific (ranging from 0.30 to 0.77). The estimates of the mean HE and HO at each North Atlantic sampling location were in the same range as the estimates obtained from the pooled samples in both ocean basins. The mean HE and HO for all 11 loci estimated from the North Atlantic samples were also similar (Table 2).
Deviations from Hardy–Weinberg expectations and linkage disequilibrium
In the total sample (i.e. the combined North Atlantic and North Pacific dataset), significant deviations from the Hardy–Weinberg genotype frequencies were detected at five (EV094, GATA053, GATA098, GT011 and GT023) of the seven loci after FDR correction (p-values < 0.0036). No significant deviations from the expected Hardy–Weinberg genotype frequencies were detected in either the North Atlantic or the North Pacific datasets after applying the FDR correction. Several instances of statistically significant linkage disequilibrium were detected among the seven loci after applying the FDR procedure (p-values < 0.0047) in the combined North Atlantic and North Pacific dataset. In contrast, no statistically significant degree of linkage disequilibrium was detected among samples from each ocean basin after applying FDR correction.
Homogeneity tests and genetic divergence
Pairwise estimates of ϴ ranged from 0.003 (Iceland-Azores comparison) to 0.20 (North Atlantic–North Pacific comparison, Table 3). Homogeneity was rejected for all loci separately and combined (p-values < 0.0001) between the North Atlantic and North Pacific Ocean basins. In contrast, no significant deviations from homogeneity were detected within the North Atlantic Ocean.
Bayesian clustering
The most probable value of K in the combined dataset (i.e. both North Pacific and North Atlantic) was estimated at two from the posterior mean likelihood values (P (K = 2|D) = ~ 1.0, Table S3). All samples from the same ocean basin were allocated to the same cluster (Fig. 2) at admixture probabilities of 100%. K = 1 was the most probable estimate for the combined North Atlantic dataset (P (K = 1|D) = ~ 1.0, Table S3).

Structure plots for the total dataset (top) and for the North Atlantic dataset (bottom), showing estimated probabilities of assignment to each of two populations (K) for all individuals’ microsatellite genotypes. Each column represents one individual’s genotype
Data analysis of the mtDNA control region nucleotide sequences
Levels of polymorphism
The final dataset of mtDNA control region DNA sequences was comprised of the first 487 nucleotides at the 3′ end of the mtDNA control region in 572 samples (n = 488 for the North Pacific due to one failed mtDNA sequence and n = 84 for the North Atlantic; each sample representing a unique multi-locus microsatellite genotype). In total, 41 segregating sites which defined 65 different mtDNA sequence haplotypes were identified (Fig. 3), with none shared between ocean basins. Among the 41 segregating sites, three were segregating for three nucleotides, resulting in a total of 44 observed substitutions; one inferred insertion-deletion event, 38 transitions and five transversions. There were seven mtDNA haplotypes detected among the North Atlantic samples and 58 among the North Pacific samples. The mean haplotype and nucleotide diversity for each sampling location separately and for all samples together are listed in Table 4.

Frequency of mtDNA control region haplotypes per sampling location
MtDNA genealogy
The final alignment of sei, Bryde’s and fin whale mtDNA control region sequences yielded a consensus sequence of 491 nucleotides (including alignment gaps). The maximum-likelihood genealogy (Fig. 4) estimated from the aligned sequences was comprised of two clades with sei whale mtDNA sequences supported by a bootstrap value at 90%. One clade contained six mtDNA haplotypes detected among the North Atlantic samples. The other clade contained all the mtDNA haplotypes detected in the North Pacific, the only Antarctic mtDNA haplotype as well as one North Atlantic mtDNA haplotype. The neighbour-joining genealogy showed a similar topology and similar bootstrap values (Fig. S2). The haplotype networks (see Fig. 4 and Fig. S2) were similar with a sister position of North Atlantic haplotype Hap_6.

Maximum-likelihood haplotype network (left) and genealogy (right) of mtDNA control region haplotypes. Each node in the haplotype network represents a haplotype and node sizes are proportional to haplotype frequencies. Each line segment between nodes represents one nucleotide difference. For convenience, only the North Atlantic haplotype designations and the designations of the four most frequently occurring North Pacific haplotypes are shown. The haplotype tree is drawn to scale, with branch lengths in the evolutionary distance unit of number of base substitutions per site. Only bootstrap values above 60% are shown
Homogeneity tests and estimates of genetic divergence
Homogeneity was rejected (ΦST = 0.72, p < 0.001) between the North Atlantic and North Pacific Oceans (Table 3). However, no significant deviations from homogeneity were detected among the three North Atlantic sampling locations.
Estimation of effective population sizes, divergence time and migration rates
The parameter \(\theta\), which can be viewed as a proxy for long-term historic effective population sizes, was estimated at 6.2 (95% credible interval: 2.2–14) and 53 (95% credible interval: 39–73) for the North Atlantic and North Pacific samples, respectively; a difference of almost one order of magnitude (Table 5). The divergence time between the North Atlantic and North Pacific populations was estimated at ~ 163 thousand years ago (kya, 95% credible interval: 57–386 kya; Table 5). The number of effective migrants 2mNe from the North Pacific population into the North Atlantic population was estimated at 0.248 (95% credible interval: 0–1.97, Table 5) and at 0.007 from the North Atlantic population into the North Pacific population (95% credible interval: 0–1.47, Table 5).
Tests of neutrality and mismatch distributions
The observed estimates of Tajima’s D and Fu and Li’s F* for the separate and pooled sampling locations were all negative (Table 4), suggestive of population expansion. However, F* was only statistically significant for the Azores’ sample (p < 0.05) and for the pooled North Atlantic sample (p < 0.02) and D was only significant for the Azores’ sample (p < 0.05). The observed mismatch distributions (Fig. 5) corresponded to the expected frequency distributions of pairwise nucleotide site differences in an exponentially growing population.

Frequency distributions of the observed pairwise nucleotide site differences, or mismatch distributions, for the North Pacific, the North Atlantic and the separate North Atlantic sampling locations, compared to expected frequency distributions for a population of exponentially expanding size (red dotted line). For each distribution comparison, the χ2 value and number of degrees of freedom (d.f.) are given. None of the observed mismatch distributions deviated significantly from the expected distribution. The observed frequency distribution for the combined North Atlantic and North Pacific dataset is also given, but no expected distribution can be shown because assumptions (i.e. panmictic population) for their estimation do not hold
Discussion
At an initial glance our results were consistent with the notion of a single panmictic population of sei whales in at least the western and central North Atlantic (but see below) which appear to have undergone a historic population expansion. Our results also supported the inference drawn by Baker et al. (2004) that sei whales in the North Atlantic and North Pacific Ocean are genetically distinct. The previous results were based solely on mtDNA sequences, and this study augmented the conclusion with nuclear microsatellite genotypes.
Differentiation within the North Atlantic Ocean
Low levels of genetic differentiation
We failed to detect any significant genetic heterogeneity among the three distinct sampling locations in the North Atlantic (the Gulf of Maine, off Iceland and the Azores) at nuclear or mtDNA loci. These findings suggested an absence of genetic population structure within the western and central North Atlantic. Pairwise estimates of ΦST and ϴ at mtDNA and nuclear loci were low, most close to zero (Table 3). The program STRUCTURE also failed to identify significant levels of genetic structure within the North Atlantic (and North Pacific). In other words, our analyses did not yield any results supporting the current designation of two of the three sei whale management units in the North Atlantic by the IWC (Fig. 1). Samples from the third, Eastern North Atlantic stock would provide for a more complete assessment, but according to the IUCN, sei whales seem to have been depleted in that area with no signs of recoveryFootnote 3.
However, low levels of genetic differentiation do not necessarily imply a single stock of sei whales in the North Atlantic but could have other causes (Palsboll et al. 2010). Firstly, our samples originated from two summer feeding grounds; namely, the Gulf of Maine and Iceland, and from a migratory corridor; the Azores. The sei whales utilizing these areas may have formed mixed assemblages of sei whales from different breeding populations and therefore do not show population structure. North Atlantic minke whales (Balaenoptera acutorostrata) present a similar problem of cryptic population structure. For instance, where Daníelsdottír et al. (1992) and Andersen et al. (2003) were able to detect some differentiation between minke whales from West Greenland, the Central and Northeast Atlantic, Anderwald et al. (2011) were not. The optimal sampling scheme would include the identification and sampling of sei whale breeding grounds as well as additional migratory corridors and feeding areas.
Secondly, it is important to consider the uncertainty of the divergence estimates as well as the assumptions underlying equating divergence estimates with contemporary connectivity. The upper bounds of 95% confidence intervals estimated for the point estimates of FST among the North Atlantic sampling locations ranged from 0.08 to 0.14 and from 0.03 to 0.05 for mtDNA and microsatellite data, respectively. Applying Wright’s drift-migration equilibrium, the relation between FST and Nm, i.e. FST = 1/(4 Nm + 1), implies that these upper bounds would correspond to between 3 and 8 migrants per generation (females for mtDNA). The failure of the program STRUCTURE to detect more than a single cluster among the North Atlantic samples should similarly be interpreted with caution. Several in silico assessments (e.g. Latch et al. 2006; Waples and Gaggiotti 2006) of the program have shown that STRUCTURE fails to detect more than one cluster when the degree of population genetic divergence is below 0.05–0.025, which corresponds to 5–10 migrants per generation assuming drift-migration equilibrium. In other words, the 95% confidence intervals of our divergence estimates included levels of divergence that both support a single stock (i.e. FST ~ 0) and multiple stocks (i.e. 3–8 migrants per generation). Along the same vein, the failure of STRUCTURE to detect more than one cluster in the North Atlantic does not negate the presence of multiple stocks given the relatively low migration rates possible given the observed outcome. From a conservation point of view, genetic differentiation alone might therefore not be a sufficient criterion to delineate useful management stocks.
Possible historic population expansion
The degree of population genetic divergence estimated as FST does not necessarily reflect contemporary gene flow (i.e. migration) but is heavily influenced by population history. The negative values of the observed estimates of Tajima’s D and Fu and Li’s F* were indicative of a historic population expansion, which makes sense given the geological history of the North Atlantic Ocean. The Gulf of Maine and the seas off Iceland were inaccessible to baleen whales during the last glacial maximum (LGM, 26.5–19 kya; Clark et al. 2009). The ice caps and summer sea ice extent have since retreated making the current summer foraging areas, such as the Gulf of Maine and the waters off Iceland, accessible to sei whales. Our results suggested an expansion of the North Atlantic sei whale population(s) after the LGM during the retreat of the summer sea ice as previously reported in case of the North Atlantic fin whale (Balaenoptera physalus; Bérubé et al. 1998) and minke whale (B. acutorostrata; Pastene et al. 2007; Anderwald et al. 2011). Albeit all values being negative, estimates of Fu and Li’s F* were only significant for the pooled North Atlantic sample and for the Azores, and Tajima’s D only for the Azores (Table 4). However, when the single individual from the Azores with haplotype Hap_6 (see below) was excluded, all estimates of D and F* became statistically insignificant. The observed frequency distributions of pairwise nucleotide site differences (Fig. 5) fitted the expected distribution for an exponentially growing population. Thus, we found a trend toward population expansion, but statistically insignificant, likely due to low statistical power from low sample sizes, and limited sequence variation in the sei whale mtDNA control region.
Among the seven mtDNA haplotypes detected in the North Atlantic, six differed from each other by a single substitution, suggesting a recent coalescence of these lineages consistent with the presumed recent population expansion. The seventh mtDNA haplotype (Hap_6) was detected in a single sample taken in the Azores. The haplotype differed from the remaining six North Atlantic mtDNA haplotypes by twelve substitutions and was placed as a sister group to the North Pacific haplotypes in the genealogy estimated in our study (Fig. 4). However, the bootstrap support for this haplotype’s branch was low leaving its position rather uncertain. Increased outgroup sampling and additional markers (i.e. the partial or complete mitogenome) may provide a more strongly supported topology. Although anecdotal at this point, the seventh divergent mtDNA haplotype might represent a recent immigrant maternal lineage, e.g. from the South Atlantic, or represent a rare North Atlantic mtDNA lineage. More data and samples are required to discern among these two possibilities.
Population expansion reduces the rate of genetic drift and hence the rate of population genetic divergence compared to constant-sized populations (e.g. Rogers and Harpending 1992; Kimmel et al. 1998; Waxman 2012). This effect would be even stronger if the ‘new’ populations were founded from the same historical population (e.g. Avise et al. 1988). In other words, a recent population history and expansion of sei whales in the North Atlantic may have contributed to the low levels of spatial population genetic divergence observed in our study. Basic population genetic theory relates the degree of genetic divergence among populations to the number of migrations, more precisely the product of the effective population size (Ne) and the probability that an individual is an immigrant (m; Wright 1951; Slatkin and Barton 1989). In other words, the number of immigrants per generation (i.e. 2mNe) determines the degree of genetic divergence among populations, meaning that populations with large Ne’s will diverge at a slower rate compared to populations with smaller Ne’s. Consequently, expanding populations will diverge at decreasing rates compared to similar-sized non-expanding populations, all other factors being equal. One example is depicted in Fig. S3, which illustrates the pronounced difference in estimates of FST in constant and expanding populations in the time following a population divergence.
Effects of whaling on population structure
It is possible that whaling of sei whales may have influenced the contemporary population genetic structure among North Atlantic sei whales. However, the possible effects could either increase or decrease post-whaling population genetic structure (Baker and Clapham 2004). For instance, differential rates of post-whaling recovery among populations could lead to source-sink dynamics and hence reduce pre-whaling population genetic divergence. In contrast, severe reductions of abundance in some populations might result in reduced levels of gene flow among populations and elevated rates of genetic drift which increase pre-whaling divergence. Among the baleen whales there are examples of both rapid post-whaling recolonization, i.e. source-sink dynamics (e.g. Best 1993; Clapham et al. 1999; George et al. 2004; Rugh et al. 2005), as well as slow or absent post-whaling recovery, i.e. increased isolation (e.g. Clapham et al. 1999; Wade et al. 2011; Pomilla et al. 2014), although none of these studies have demonstrated any discernible effects on change in pre-whaling population genetic divergence.
Timing and level of gene flow between the North Atlantic and North Pacific Oceans
We detected high and significant degrees of genetic divergence between the samples from the North Atlantic and North Pacific oceans (Table 3). Haplotype diversity was high for the North Pacific and intermediate for the North Atlantic (Table 4). The estimates of nucleotide diversity were low but within the range reported for other rorquals (e.g. Bérubé et al. 1998; Anderwald et al. 2011). The global haplotype genealogy revealed a clear separation of the North Atlantic and North Pacific haplotypes (Fig. 4). The single Antarctic mtDNA haplotype included in our analysis clustered together with the North Pacific mtDNA haplotypes, which was consistent with previous findings by Baker et al. (2004).
The inter-oceanic migration rate estimates pointed to migration rates of only ~ 1 migrant per four generations or less (Table 5). Divergence time estimates suggested that the North Atlantic and North Pacific sei whale populations separated ~ 163 kya during the penultimate Pleistocene glaciation; the Illinoian glaciation (140–350 kya; Lisiecki and Raymo 2005a, b). This is known to be one of the coldest glacial periods over the last million years (Colleoni et al. 2016). The extent of sea ice during colder conditions might have facilitated the population divergence between the North Atlantic and North Pacific sei whales, as has been suggested for other species and populations during the Pleistocene glaciations (e.g. Hewitt 2000, 2004).
The estimates of \(\theta\), a proxy for effective population size, indicated that the median effective population size of the North Pacific sei whale population was much larger (approximately nine times) compared to the North Atlantic population (Table 5). This was also reflected in the differences in haplotype and nucleotide diversities between the two oceans (Table 4). Looking at heterozygosity alone (Tables 1, 2) we saw no indication of a genetic bottleneck in the North Atlantic preceding the presumed population expansion after the LGM, which could have explained the differences in genetic diversity between the two oceans. However, providing detailed insight into the demographic history of both populations is reserved to future studies.
Although the estimates of \(\theta\) can be converted into estimates of effective female population sizes, we refrained from doing so given that the interpretation of such an estimate is far from straightforward (as reviewed by Palsbøll et al. 2013). Similarly, the inferred population divergence time should not be taken too literally. Direct gene flow between the North Atlantic and North Pacific after the rise of the Panama Isthmus (~ 3.5 million years ago; e.g. Coates et al. 1992) has only been possible through the Northwest Passage during a few brief periods with elevated temperatures. Our divergence time estimate was likely heavily influenced by past periods of gene flow between the hemispheres, as well as the mtDNA mutation rate and generation time employed in our estimation (Avise et al. 1988). The inclusion of samples from the Southern Hemisphere would likely result in very different estimates.
Concluding remarks
In conclusion, while our results did not seem to support the current division by the IWC of North Atlantic sei whales into three different stocks, the uncertainty in our estimates was sufficiently high that we could not rule out the presence of multiple stocks either. The available satellite tagging data suggests that sei whales travel across wide latitudinal and longitudinal ranges, which might explain the low levels of genetic divergence estimated in this study. In order to aid further efforts in the management and conservation of sei whales, we propose additional sampling across the species’ entire range, including breeding and feeding grounds and migratory corridors, as well as increased sample sizes. The low levels of variation in the North Atlantic sei whale suggest that increasing the number of loci may also enhance the precision of estimates of divergence and gene flow (e.g. single nucleotide polymorphism, or SNP, genotypes from genotyping-by-sequencing approaches).
Notes
Information available at http://www.iucnredlist.org/details/2475/0. Accessed 4 March, 2018.
Information available at https://iwc.int/total-catches. Accessed 4 March, 2018.
Information available at http://www.iucnredlist.org/details/2475/0. Accessed 4 March, 2018.
References
Alter SE, Palumbi SR (2009) Comparing evolutionary patterns and variability in the mitochondrial control region and cytochrome b in three species of baleen whales. J Mol Evol 68(1):97–111
Andersen LW, Born EW, Dietz R, Haug T, Øien N, Bendixen C (2003) Genetic population structure of minke whales Balaenoptera acutorostrata from Greenland, the North East Atlantic and the North Sea probably reflects different ecological regions. Mar Ecol Prog Ser 247:263–280
Anderwald P, Daníelsdottír AK, Haug T, Larsen F, Lesage V, Reid RJ, Víkingsson GA, Hoelzel AR (2011) Possible cryptic stock structure for minke whales in the North Atlantic: implications for conservation and management. Biol Conserv 144(10):2479–2489
Archer FI, Morin PA, Hancock-Hanser BL, Robertson KM, Leslie MS, Bérubé M, Panigada S, Taylor BL (2013) Mitogenomic phylogenetics of fin whales (Balaenoptera physalus spp.): genetic evidence for revision of subspecies. PLoS ONE 8(5):e63396. https://doi.org/10.1371/journal.pone.0063396
Árnason Ú, Gullberg A, Widegren B (1991) The complete nucleotide sequence of the mitochondrial DNA of the fin whale, Balaenoptera physalus. J Mol Evol 33(6):556–568
Árnason Ú, Gullberg A, Widegren B (1993) Cetacean mitochondrial DNA control region: sequences of all extant baleen whales and two sperm whale species. Mol Biol Evol 10(5):960–970
Avise JC, Ball RM, Arnold J (1988) Current versus historical population sizes in vertebrate species with high gene flow: a comparison based on mitochondrial DNA lineages and inbreeding theory for neutral mutations. Mol Biol Evol 5(4):331–344
Baker CS, Clapham PJ (2004) Modelling the past and future of whales and whaling. Trends Ecol Evol 19(7):365–371
Baker CS, Dalebout ML, Funahashi N, Ma YU, Steel D, Lavery S (2004) Market surveys of whales, dolphins and porpoises in Japan and Korea, 2003–2004, with reference to stock identity of sei whales. Paper SC/56/BC3 presented to the IWC Scientific Committee, June 2004 (unpublished). Available from the IWC Office
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 98(8):4563–4568
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57(1):289–300
Bérubé M, Palsbøll P (1996a) Identification of sex in cetaceans by multiplexing with three ZFX and ZFY specific primers. Mol Ecol 5(2):283–287
Bérubé M, Palsbøll P (1996b) Erratum of identification of sex in cetaceans by multiplexing with three ZFX and ZFY specific primers. Mol Ecol 5(4):602
Bérubé M, Aguilar A, Dendanto D, Larsen F, Notarbartolo di Sciara G, Sears R, Sigurjónsson J, Urban-R J, Palsbøll PJ (1998) Population genetic structure of North Atlantic, Mediterranean Sea and Sea of Cortez fin whales, Balaenoptera physalus (Linneaus 1758): analysis of mitochondrial and nuclear loci. Mol Ecol 7(5):585–599
Bérubé M, Jørgensen H, McEwing R, Palsbøll PJ (2000) Polymorphic di-nucleotide microsatellite loci isolated from the humpback whale, Megaptera novaeangliae. Mol Ecol 9(12):2181–2183
Bérubé M, Rew MB, Skaug H, Jørgensen H, Robbins J, Best P, Sears R, Palsbøll PJ (2005) Polymorphic microsatellite loci isolated from humpback whale, Megaptera novaeangliae and fin whale, Balaenoptera physalus. Conserv Genet 6(4):631–636
Best PB (1993) Increase rates in severely depleted stocks of baleen whales. ICES J Mar Sci 50(2):169–186
Clapham PJ, Young SB, Brownell RL (1999) Baleen whales: conservation issues and the status of the most endangered populations. Mamm Rev 29(1):35–60
Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, Mitrovica JX, Hostetler SW, McCabe AM (2009) The last glacial maximum. Science 325(5941):710–714
Coates AG, Jackson JBC, Collins LS, Cronin TM, Dowsett HJ, Bybell LM, Jung P, Obando JA (1992) Closure of the Isthmus of Panama: the near-shore marine record of Costa Rica and western Panama. Geol Soc Am Bull 104(7):814–828
Colleoni F, Wekerle C, Näslund JO, Brandefelt J, Masina S (2016) Constraint on the penultimate glacial maximum Northern Hemisphere ice topography (≈ 140 kyrs BP). Quat Sci Rev 137:97–112
Daníelsdottír AK, Duke EJ, Joyce P (1991) Preliminary studies on genetic variation at enzyme loci in fin whales (Balaenoptera physalus) and sei whales (Balaenoptera borealis) from the North Atlantic. Rep Int Whal Commn 13:115–124
Daníelsdottír AK, Duke EJ, Árnarson A (1992) Genetic variation at enzyme loci in North Atlantic minke whales, Balaenoptera acutorostrata. Biochem Genet 30(3–4):189–202
Donovan GP (1991) A review of IWC stock boundaries. Rep Int Whal Commn 13:39–68
Drouot V, Bérubé M, Gannier A, Goold JC, Reid RJ, Palsbøll PJ (2004) A note on genetic isolation of Mediterranean sperm whales (Physeter macrocephalus) suggested by mitochondrial DNA. J Cetacean Res Manage 6(1):29–32
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. https://doi.org/10.1186/1471-2148-7-214
Ewing GB (2010) Haplotype viewer. Center for Integrative Bioinformatics Vienna, Vienna, Austria. http://www.cibiv.at/~greg/haploviewer
Excoffier L, Lischer HEL (2010) Arlequin suite ver. 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131(2):479–491
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7(4):574–578
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39(4):783–791
Fisher RA (1935) The logic of inductive inference. J R Stat Soc A 98(1):39–82
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133(3):693–709
George JCC, Zeh J, Suydam R, Clark C (2004) Abundance and population trend (1978–2001) of western Arctic bowhead whales surveyed near Barrow, Alaska. Mar Mamm Sci 20(4):755–773
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22(2):160–174
Hewitt GM (2000) The genetic legacy of the quaternary ice ages. Nature 405(6789):907–913
Hewitt GM (2004) Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc B 359(1442):183–195
Hudson RR (1990) Gene genealogies and the coalescent process. Oxf Surv Evol Biol 7:1–44
Ingebrigtsen A (1929) Whales captured in the North Atlantic and other seas. Rapp P-V Reun Cons Int Explor Mer 56:1 – 26
Jackson JA, Steel DJ, Beerli P, Congdon BC, Olavarría C, Leslie MS, Pomilla C, Rosenbaum H, Baker CS (2014) Global diversity and oceanic divergence of humpback whales (Megaptera novaeangliae). Proc R Soc B. https://doi.org/10.1098/rspb.2013.3222
Kanda N, Goto M, Pastene LA (2006) Genetic characteristics of western North Pacific sei whales, Balaenoptera borealis, as revealed by microsatellites. Mar Biotechnol 8:86–93
Kanda N, Goto M, Yoshida H, Pastene LA (2009) Stock structure of sei whales in the North Pacific as revealed by microsatellite and mitochondrial DNA analyses. Paper SC/J09/JR32 presented to the IWC Scientific Committee, January 2009 (unpublished). Available from the IWC Office
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) Diversity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4(8):782–788
Kimmel M, Chakraborty R, King JP, Bamshad M, Watkins WS, Jorde LB (1998) Signatures of population expansion in microsatellite repeat data. Genetics 148(4):1921–1930
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191
Latch EK, Dharmarajan G, Glaubitz JC, Rhodes OE (2006) Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv Genet 7(2):295–302
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452
Lindgren BW (1975) Basic ideas of statistics. Macmillan Publishers Ltd., New York
Lisiecki LE, Raymo ME (2005a) A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography 20(1):PA1003. https://doi.org/10.1029/2004PA001071
Lisiecki LE, Raymo ME (2005b) Correction to “A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records”. Paleoceanography 20(2):PA2007. https://doi.org/10.1029/2005PA001164
Mizroch SA, Rice DW, Breiwick JM (1984) The sei whale, Balaenoptera borealis. Mar Fish Rev 46(4):25–29
Mullis KB, Faloona FA (1987) Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol 155:335–350
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Olsen E, Budgell P, Head E, Kleivane L, Nøttestad L, Prieto R, Silva MA, Skov H, Víkingsson GA, Waring G, Øien N (2009) First satellite-tracked long-distance movement of a sei whale (Balaenoptera borealis) in the North Atlantic. Aquat Mamm 35(3):313–318
Pacifici M, Santini L, Di Marco M, Baisero D, Francucci L, Grottolo Marasini G, Visconti P, Rondinini C (2013) Generation length for mammals. Nat Conserv 5:87–94
Paetkau D, Calvert W, Stirling I, Strobeck C (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol 4(3):347–354
Palsbøll PJ, Larsen F, Hansen ES (1991) Sampling of skin biopsies from free-ranging large cetaceans in West Greenland: development of new biopsy tips and bolt designs. Rep Int Whal Commn 13:71–79
Palsbøll PJ, Clapham PJ, Mattila DK, Larsen F, Sears R, Siegismund HR, Sigurjónsson J, Vasquez O, Arctander P (1995) Distribution of mtDNA haplotypes in North Atlantic humpback whales: the influence of behaviour on population structure. Mar Ecol Prog Ser 116:1–10
Palsbøll PJ, Bérubé M, Larsen AH, Jørgensen H (1997) Primers for the amplification of tri-and tetramer microsatellite loci in baleen whales. Mol Ecol 6(9):893–895
Palsbøll PJ, Zachariah Peery M, Bérubé M (2010) Detecting populations in the ‘ambiguous’ zone: kinship-based estimation of population structure at low genetic divergence. Mol Ecol Resour 10(5):797–805
Palsbøll PJ, Zachariah Peery M, Olsen MT, Beissinger SR, Bérubé M (2013) Inferring recent historic abundance from current genetic diversity. Mol Ecol 22(1):22–40
Paquette SR (2012) PopGenKit: useful functions for (batch) file conversion and data resampling in microsatellite datasets. R package version 1.0. http://CRAN.R-project.org/package=PopGenKit
Pastene LA, Goto M, Kanda N, Zerbini AN, Kerem D, Watanabe K, Bessho Y, Hasegawa M, Nielsen R, Larsen F, Palsbøll PJ (2007) Radiation and speciation of pelagic organisms during periods of global warming: the case of the common minke whale, Balaenoptera acutorostrata. Mol Ecol 16(7):1481–1495
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28(19):2537–2539
Pomilla C, Amaral AR, Collins T, Minton G, Findlay K, Leslie MS, Ponnampalam L, Baldwin R, Rosenbaum H (2014) The world’s most isolated and distinct whale population? Humpback whales of the Arabian Sea. PLoS ONE 9(12):e114162. https://doi.org/10.1371/journal.pone.0114162
Prieto R, Janiger D, Silva MA, Waring GT, Gonçalves JM (2012) The forgotten whale: a bibliometric analysis and literature review of the North Atlantic sei whale Balaenoptera borealis. Mamm Rev 42(3):235–272
Prieto R, Silva MA, Waring GT, Gonçalves JMA (2014) Sei whale movements and behaviour in the North Atlantic inferred from satellite telemetry. Endanger Species Res 26:103–113
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
R Development Core Team (2016) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. https://www.R-project.org
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86(3):248–249
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9(3):552–569
Rosenbaum HC, Brownell RL, Brown MW, Schaeff C, Portway V, White BN, Malik S, Pastene LA, Patenaude NJ, Baker CS, Goto M, Best PB, Clapham PJ, Hamilton P, Moore M, Payne R, Rowntree V, Tynan CT, Bannister JL, DeSalle R (2000) World-wide genetic differentiation of Eubalaena: questioning the number of right whale species. Mol Ecol 9(11):1793–1802
Rousset F (2008) GENEPOP ‘007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol 8(1):103–106
Rugh DJ, Hobbs RC, Lerczak JA, Breiwick JM (2005) Estimates of abundance of the eastern North Pacific gray whales (Eschrichtius robustus) 1997 to 2002. J Cetacean Res Manage 7:1–12
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Sambrook J, Russell DW (eds) (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sasaki T, Nikaido M, Hamilton H, Goto M, Kato H, Kanda N, Pastene LA, Cao Y, Fordyce RE, Hasegawa M, Okada N (2005) Mitochondrial phylogenetics and evolution of mysticete whales. Syst Biol 54(1):77–90
Sethuraman A, Hey J (2016) IMa2p—parallel MCMC and inference of ancient demography under the Isolation with migration (IM) model. Mol Ecol Resour 16(1):206–215
Slatkin M, Barton NH (1989) A comparison of three indirect methods for estimating average levels of gene flow. Evolution 43(7):1349–1368
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129(2):555–562
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA 101(30):11030–11035
Tamura T, Matsuoka K, Fujise Y (2009) Methodology and survey procedure under the JARPN II—offshore component—with special emphasis on whale sampling procedures. Paper SC/J09/JR4 presented to the IWC Scientific Committee, January 2009 (unpublished). Available from the IWC Office
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Taylor BL, Chivers SJ, Larese J, Perrin WF (2007) Generation length and percent mature estimates for IUCN assessments of cetaceans. Administrative report LJ-07-01. Southwest Fisheries Science Center, La Jolla, California
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22(22):4673–4680
Valsecchi E, Amos W (1996) Microsatellite markers for the study of cetacean populations. Mol Ecol 5(1):151–156
Wada S, Numachi K (1991) Allozyme analyses of genetic differentiation among the populations and species of the Balaenoptera. Rep Int Whal Commn 13:125–154
Wade PR, Kennedy A, LeDuc R, Barlow J, Carretta J, Shelden K, Perryman W, Pitman R, Robertson K, Rone B, Salinas JC, Zerbini A, Brownell RL Jr, Clapham PJ (2011) The world’s smallest whale population? Biol Lett 7(1):83–85
Wang J (2017) The computer program STRUCTURE for assigning individuals to populations: easy to use but easier to misuse. Mol Ecol Resour. https://doi.org/10.1111/1755-0998.12650
Waples RS, Gaggiotti O (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15(6):1419–1439
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7(2):256–276
Waxman D (2012) Population growth enhances the mean fixation time of neutral mutations and the persistence of neutral variation. Genetics 191(2):561–577
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370
Werle E, Schneider C, Renner M, Völker M, Fiehn W (1994) Convenient single-step, one tube purification of PCR products for direct sequencing. Nucl Acids Res 22(20):4354–4355
Wright S (1951) The genetical structure of populations. Ann Hum Eugen 15:323–354
Acknowledgements
We would like to thank Pauline Gauffier, Yvonne Verkuil and Vania Rivera for assistance with the laboratory and data analyses. We would also like to thank David Mattila and other field personnel involved in the collection of the samples. The Center for Information Technology of the University of Groningen is acknowledged for IT support and access to the Peregrine high performance-computing cluster. We thank the anonymous referees for their constructive comments on a draft of this paper. This study was in part funded by: the University of Groningen; Fundação para a Ciência e Tecnologia (FCT); Fundo Regional da Ciência, Tecnologia; the Marine and Environmental Sciences Centre (MARE); the Center for Coastal Studies; the Marine and Freshwater Research Institute; the Institute of Cetacean Research; the National Research Institute of Far Seas Fisheries. This work is an output of research projects TRACE-PTDC/MAR/74071/2006, and of the FCT-Exploratory project IF/00943/2013/CP1199/CT0001 (FEDER, the Competitiveness Factors Operational [COMPETE], QREN, POPH, European Social Fund, Portuguese Ministry for Science and Education European Social Fund). We acknowledge funds provided by FCT to MARE, through the strategic project UID/MAR/04292/2013. Co-authors MAS and RP are supported by FCT through contract (IF/00943/2013) and postdoctoral grant (SFRH_BPD_108007_2015), respectively. Fieldwork and sample collection in the Azores were approved by the relevant authorities (Regional Directorate for Sea Affairs, Autonomous Region of the Azores) under research permits 7/CN/2005, 76/2007/DRA, 20/2009/DRA and 16/2010/DRA. Fieldwork and sample collection in the Gulf of Maine were conducted under U.S. National Oceanic and Atmospheric Administration research permits 633–1483 and 633–1778 and licenses obtained from the Canadian Department of Fisheries and Oceans. The Icelandic samples were archived from sei whales taken in 1986–1988 as a part of a special permit issued by the government of Iceland in compliance with the rules of the International Whaling Commission. The North Pacific samples were archived from sei whales taken in 2000–2016 during the JARPNII program under a permit issued by the Government of Japan.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Huijser, L.A.E., Bérubé, M., Cabrera, A.A. et al. Population structure of North Atlantic and North Pacific sei whales (Balaenoptera borealis) inferred from mitochondrial control region DNA sequences and microsatellite genotypes. Conserv Genet 19, 1007–1024 (2018). https://doi.org/10.1007/s10592-018-1076-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-018-1076-5