
Abstract
Luminal breast cancer is the most frequently encountered type of human breast cancer and accounts for half of all breast cancer deaths due to metastatic disease. We have developed new in vivo models of disseminated human luminal breast cancer that closely mimic the human disease. From initial lesions in the tibia, locoregional metastases develop predictably along the iliac and retroperitoneal lymph node chains. Tumors cells retain their epithelioid phenotype throughout the process of dissemination. In addition, systemically injected metastatic MCF-7 cells consistently give rise to metastases in the skeleton, floor of mouth, adrenal glands, as well as in the lungs, liver, brain and mammary fat pad. We show that growth of luminal breast cancer metastases is highly dependent on estrogen in a dose-dependent manner and that estrogen withdrawal induces rapid growth arrest of metastatic disease. On the other hand, even though micrometastases at secondary sites remain viable in the absence of estrogen, they are dormant and do not progress to macrometastases. Thus, homing to and seeding of secondary sites do not require estrogen. Moreover, in sharp contrast to basal-like breast cancer metastasis in which transforming growth factor-β signaling plays a key role, luminal breast cancer metastasis is independent of this cytokine. These findings have important implications for the development of targeted anti-metastatic therapy for luminal breast cancer.
Similar content being viewed by others


Introduction
Metastasis is the predominant cause of breast cancer deaths [1]. Yet we know relatively little about this process compared to the primary events that drive breast cancer development such as loss of proliferative control and resistance to cell death [2]. The process of metastasis can be broken down into three distinct stages. The first stage of locoregional metastasis encompasses local invasion of cancer cells through basement membrane, followed by intravasation into nearby lymphatic capillaries, and transit of cancer cells through the lymphatic system into the blood stream. The second stage of systemic dissemination involves arrest of cancer cells in specific capillary beds (“homing”) followed by their exit into the parenchyma of secondary tissues (extravasation), and the establishment of micrometastases. The third and final stage involves the progression of micrometastatic lesions to macroscopic tumors [3].
Studies of human basal-like breast cancer have shown that specific sets of genes need to be activated for cells to home to and establish macrometastases in different secondary organ sites [4–6]. Dissemination of basal-like breast cancer appears to recapitulate the epithelial-to-mesenchymal transitions (EMT) utilized during development by precursor cells to migrate to their final destination in the embryo [7]. In fact, EMT is considered to be an absolute requirement for hematogenous metastasis of human basal-like breast cancer cells [3]. Moreover, transforming growth factor-β (TGF-β) has been identified as a key driver of EMT and has been validated as a therapeutic target of tumor cell dissemination in preclinical studies of human basal-like breast cancer [8–10]. Based on these findings, TGF-β signaling inhibitors are being evaluated in clinical trials as a new therapeutic modality for metastatic basal-like breast cancer [11].
Nonetheless, basal-like breast cancers account for only 15 % of human breast cancers, while the so called luminal subtype accounts for two-thirds of all breast cancers [1]. Moreover, approximately 70 % of all metastatic breast cancers are of the luminal type [1]. Human luminal breast cancer cell lines typically express estrogen receptors (ERα) and are dependent on estrogen for growth, are highly epithelioid and cohesive, have a low capability to invade the basement membrane surrogate, Matrigel®, and form tight spherical colonies in 3-D culture ([12], Grazioli and Reiss, unpublished observations). Moreover, they do not express mesenchymal markers typically associated with basal-like tumors [13, 14] and metastatic lesions typically retain the highly epithelioid phenotype of primary tumors [15–17]. These observations suggest the possibility that metastasis of luminal breast cancer does not require transition to a mesenchymal phenotype, and may be mediated by so-called cohesive (collective) migration [18].
In order to identify the drivers of metastasis of ERα-positive human breast cancer, we used the MCF-7 cell line to develop novel experimental models of ERα positive luminal breast cancer metastasis that closely and reproducibly mimic the human disease. We demonstrate that metastatic tumor growth is highly dependent on the ambient level of estrogen, while homing to and seeding of secondary sites does not appear to be. Moreover, luminal breast cancer cells retain their cohesive epithelial phenotype throughout the entire regional and systemic metastatic cascade. In contrast to basal-like breast cancer cells, metastatic luminal breast cancer cells are intrinsically incapable of responding to TGF-β because the TGFBR2 gene is transcriptionally silent. Moreover, tumor cell dissemination is entirely independent of TGF-β. These findings establish a new paradigm for metastatic breast cancer and have important implications for the treatment of the luminal subtype.
Materials and methods
Reagents
Human recombinant TGF-β1 (1 µg/mL; Austral Biologicals, San Ramon, CA) was dissolved in 4 mmol/L HCl and 1 mg/mL bovine serum albumin (Sigma, St. Louis, MO). 1D11 and the isotype-matched murine IgG1 monoclonal control antibody, 13C4, directed against Shigella toxin (Genzyme, Framingham, MA) were diluted in formulation buffer composed of 0.1 M glycine, 70 mM Na2HPO4, 0.0011 % Tween 20 for both in vitro and in vivo studies. The Alk4, -5 and -7 kinase inhibitor SD-093 (Scios, Inc., Sunnyvale, CA) and the Alk2, -3 and -6 kinase inhibitor Dorsomorphin (Sigma, St. Louis, MO) were dissolved in DMSO and stored at −70 ºC.
Cell culture
Parental MCF-7 cells were obtained from Dr. Lorna Rodriguez (The Cancer Institute of New Jersey, New Brunswick, NJ). Parental MCF-7-ERE-FLuc cells as well as fulvestrant-resistant MCF-7-ERE-FLuc-FR and tamoxifen-resistant MCF-7-ERE-FLuc-TR cells were obtained from Dr. Kenneth Nephew, Indiana University [19, 20]. MCF-7-ERE-FLuc cells were derived from a clonal derivative of MCF-7 cells that had been stably transfected with an ERα-responsive luciferase reporter (ERE-pS2-Luc) as previously described [20, 21]. MCF7-ERE-FLuc cells were maintained in MEM (Invitrogen, Carlsbad, CA) supplemented with 2 mmol/L l-glutamine, 0.1 mmol/L nonessential amino acids, 50 units/mL penicillin, 50 μg/mL streptomycin, 6 ng/mL insulin, and 10 % FBS. Estrogen-deficient medium consisted of phenol red-free MEM supplemented with 2 mmol/L l-glutamine, 0.1 mmol/L nonessential amino acids, 50 units/mL penicillin, 50 μg/mL streptomycin, 6 ng/mL insulin, and 10 % charcoal-stripped FBS. This medium was further supplemented with 10−7 mol/L 4-hydroxytamoxifen (for the tamoxifen-resistant subline “MCF7-TR”), or with 10−7 mol/L fulvestrant (for the fulvestrant-resistant subline “MCF7-FR”). All other cell lines were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10 % (v/v) fetal bovine serum (FBS) (Sigma, St Louis, MO). BT474, T47D, HCC1419, HCC1954, and HCC2218 cells were obtained from the ATCC. MDA-MB-231 cells and its bone-tropic subclone, SCP2, were obtained from Dr. Yibin Kang (Princeton University, Princeton, NJ).
In vivo selection and labeling of cell lines
Bone tropic cell variants of parental MCF-7 cells were generated by in vivo selection. Briefly, 1 × 105 ERα-positive parental MCF-7 or MCF-7-ERE-FLuc breast cancer cells were inoculated into tibiae of ovariectomized nude mice supplemented with pellets that delivered 17β-estradiol at 2 μg/day (Innovative Research of America, Sarasota, FL). To isolate tumor cells from bone lesions, the affected hind limbs were separated from the body at the joints. Both ends of the tibia were cut open after skin and muscle were removed using a scalpel. A 1 ml syringe with a 26G needle was filled with PBS and inserted into one end of the tibia. Mouse bone marrow cells as well as tumor cells were forced out from the other end by applying pressure to the syringe. Cells were collected by centrifugation and washed once with PBS before being plated in 5 cm tissue culture plates using regular MCF-7 culture medium. Mouse bone marrow cells did not attach to the plate and could be washed off with PBS after the tumor cells became attached. After one to two weeks of culture, a pure population of human cancer cells was obtained. Secondary (MCF-7-5624, MCF-7-6012-ERE-FLuc) and tertiary (MCF-7-5624A, -B, and -C) intratibia inoculates gave rise to bone lesions much more rapidly than the initial inoculates, indicating that we were selecting increasingly bone tropic cell populations. For subsequent metastasis assays, MCF-7-5624A cells were labeled with pGreenFire1-CMV (pTRH1 CMV dscGFP T2A Fluc, System Biosciences, Mountain View, CA) using lentiviral infection following the manufacturers instructions and isolated using fluorescence activated cell sorting.
Wound healing assay
Tumor cells were grown to confluency in 6-well dishes in RPMI 1640 with 10 % FBS. The wells were washed once with PBS. A straight wound was made across each lawn of cells using a sterile yellow pipette tip aided by a ruler. Sloughed off cells were washed off with PBS. Next, RPMI supplemented with 5 % (v/v) charcoal-stripped FBS (Sigma, St. Louis, MO) was added to each well. In order to block cell proliferation, cytosine-β-d-arabinofuranoside-hydrochloride (Ara-C, Sigma) was added to a final concentration of 10 μM. One hour following the addition of Ara-C, cultures were treated with 1 nM 17β-estradiol (Sigma) or DMSO only. The wound was digitally imaged using phase contrast microscopy immediately after it was made (0 h) and 24 h later. Photographs were taken of three fields (200×) per wound and the area quantified using ImageJ (v.1.45, National Institutes of Health, USA).
Western blot analysis
To determine the effects of TGF-β on Smad phosphorylation, cells were incubated overnight in serum free medium and treated with 1 μM SD-093, 10 μM Dorsomorphin or vehicle for 15 min, followed by the addition of 100 pM TGF-β1 for 1 h. Cells were then lysed in situ using buffer composed of 150 mM NaCl, 10 mM Tris–HCl (pH 8.0), 1 mM EGTA, 1 % (v/v) Triton-X-100 in the presence of protease inhibitors and phosphatase inhibitors (Complete Mini Protease Inhibitor Cocktail Tablets with EDTA, and PhosSTOP, Roche Diagnostics Corporation, Indianapolis, IN), for 30 min at 4 °C. Cell lysates were collected and clarified by centrifugation at 12,000 rpm for 10 min at 4 °C. The clarified lysates were then subjected to SDS-PAGE and transferred to nitrocellulose membranes using a Panther™ Semidry Electroblotter (Owl Separation Systems, Portsmouth, NH). Activated Smad2 (pSmad2), Smad3 (pSmad3) and Smad1/5/8 (pSmad1/5/8), were detected using rabbit monoclonal anti-human pSmad2 or polyclonal anti-human pSmad3 antibodies (Cell Signaling, Danvers, MA) at 1:1,000 dilution. Total Smad2, Smad3 and Smad1 were detected using mouse monoclonal anti-human Smad2 (Cell Signaling, Danvers, MA), rabbit monoclonal anti-human Smad3 (Zymed Laboratories, South San Francisco, CA) and rabbit monoclonal anti-human Smad1 (Cell Signaling, Danvers, MA) antibodies at 1:1,000 dilution. Blots were developed using a 1:5,000 dilution of horseradish peroxidase-tagged goat anti-rabbit (Calbiochem, San Diego, CA) or anti-mouse (Vector Labs, Burlingame, CA) IgG antibody and the bands visualized using ECL™ (Amersham, Piscataway, NJ) reagent. Blots were scanned using an Epson Perfection V700 Photo scanner and integrated optical densities of individual bands on scanned images were determined using Image J software.
RNA extraction and quantitative RT-PCR
Transcript levels of individual genes were assayed by quantitative real time (qRT)-PCR, using the QuantiTect™ Probe RT-PCR Kit (QIAGEN, Valencia, CA). For the PCR, 50 μl reactions were set up with 100 ng of RNA, 0.4 μM primer, 0.2 μM dual labeled probe, 0.5 μl of QuantiTect™ Reverse Transcriptase Mix and QuantiTect™ Probe RT-PCR Master Mix. Real time PCR was performed using a Mx4000® Multiplex Quantitative PCR System (Stratagene, La Jolla, CA) with each sample assayed in triplicate. Three mRNA species were quantified, including TGFBR2 and ESR1 and the reference gene, GAPDH. Standard curves for all three genes were generated using serial dilution of RNA isolated from control cells. The relative mRNA amounts for each of the genes in the individual RNA samples were calculated from the standard curves. The primers used were Assay on Demand Gene Expression probes from Applied Biosystems. ESR1: HS01046818_m1; TGFBR2: Hs00559661_m1 and GAPDH: Hs02758991_g1.
In vivo metastasis assays
For locoregional metastasis assays, MCF-7-5624, MCF-7-5624A-GF or MCF-7-ERE-FLuc tumor cells were injected into the left tibia (1 × 105 cells) of viral antibody-free 4- to 5-week-old female ovariectomized athymic nude mice (Harlan Laboratory, Indianapolis, IN). For distant metastasis assays, MCF-7-5624A-GF or MCF-7-6012-ERE-FLuc tumor cells were injected into the left cardiac ventricle (5 × 105 cells). Estrogen supplementation was provided by implanting time-release pellets (Innovative Research of America, Sarasota, FL) that deliver 17β-estradiol at a rate of 2 μg/day. Body weight and bioluminescence were monitored weekly. For bioluminescence imaging (BLI), anesthetized mice were injected with 100 mg/kg d-Luciferin (Xenogen, Alameda, CA) in PBS intraperitoneally, and images were acquired using an IVIS Spectrum instrument (Calipers Life Sciences, Hopkinton, MA). Acquisition time was adjusted to avoid signal saturation. Analysis of the images was performed using Living Image Software Version 4.1 by measuring photon flux of a region of interest (ROI) drawn around a BLI signal of a single metastatic lesion or a rectangular ROI around an entire mouse. Results are reported as average BLI per treatment group. Post mortem, lungs, bones, liver, kidneys, adrenal glands, mammary fat pads, and major lymph node groups were visually inspected for the presence of tumor metastases.
Micro-CT scanning procedures and bone morphometry
Scanning was performed using a Siemens Inveon Multimodality PET/CT scanner equipped with 80 W X-ray source with 50 μm focal spot size and 30 μm maximal spatial resolution, and a 125 mm X-ray detector. An Inveon 64-bit workstation and CT Real-time Reconstruction System running Inveon Acquisition Workplace (version 1.4.5) were used for image acquisition and reconstruction (modified Feldkamp algorithm). Anesthesia was induced using 3 % isofluorane and maintained using 1–2 % isofluorane with an oxygen flow rate of 2 L/min. The acquisition parameters were as follows: 80 kVp peak, 500 μA, exposure 350 ms per frame with 200 ms in-between-frame settle time, 1° angle increments, and 210 views per scan. The field of view of X-ray source and detector were adjusted to encompass either an entire mouse or only a single leg in the frame. Voxel resolution: 93 μM.
CT images were converted to DICOM and analyzed using the 64-bit OsiriX v.3.9 (Pixmeo) and Image J software packages. To quantify relative bone mass, a mid-sagittal CT image of the tibia was localized using orthogonal multiplanar reconstruction (MPR), and the proximal third of the bone was selected as the region of interest. Following thresholding on bone, the Image J particle counter plug-in was used to quantify bone area. Total bone area was defined as the fraction of the ROI that had the density of cortical and trabecular bone [22].
Histology and immunohistochemistry
Bones of mice were excised, fixed in 10 % neutral-buffered formalin, decalcified in 0.5 M EDTA, and stored at 4 °C in 70 % ethanol until processing. Soft tissue and visceral lesions were removed from the affected mice, fixed in 10 % neutral-buffered formalin overnight and stored at 4 °C in 70 % ethanol until processing. Tissues were embedded in paraffin and 5 μm sections prepared. Tissue sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E), as well as with rabbit monoclonal antihuman ERα IgG (Clone SP1, Spring Bioscience, CA), rabbit monoclonal antihuman PR IgG (Clone SP2, Spring Bioscience, CA), rabbit monoclonal antihuman c-erbB-2/HER-2 IgG (Clone SP3, Spring Bioscience, CA), rabbit monoclonal antihuman E-Cadherin (Clone: EP700Y, Cell Marque, CA) and rabbit polyclonal pan-cytokeratin (Invitrogen, CA) antibodies. Control slides were stained using appropriate isotype control antibodies. Biotinylated secondary antibodies (1:150; Zymed, San Francisco, CA) were used for detection. In addition, bone sections were stained with orange G and phloxine to visualize new bone (1.2 % eosin in 90 % alcohol plus 1 % phloxine B and 2 % orange G), and with tartrate resistant acid phosphatase (TRAP) stain to visualize osteoclasts. For TRAP staining, slides were incubated with pre-warmed 10 % (v/v) naphthol–ether (0.044 M 7-bromo-3-hydroxy-2-naphthoic-o-anisidide phosphate in ethylene glycol monoethyl ether) in basic incubation medium (0.112 M sodium acetate, 0.05 M disodium tartrate dihydrate) at 37 °C for 30 min. Slides were then transferred directly into 2 % (v/v) color reaction medium (1:1 mixture of 0.058 M NaNO2 and 0.154 M pararosaniline chloride in 2 M HCl in basic incubation medium), and incubated for 5–30 min at room temperature. Once optimal staining was achieved, slides were rinsed in deionized water and counterstained using Harris’s acid hematoxylin. The number of TRAP positive cells per mm2 of tumor adjacent to bone were used as a measure of osteoclast activity [10]. All sections were viewed on an Olympus BX51 microscope outfitted with a Q-Imaging Retiga 1300 Cooled CCD digital camera with color wheel. Images were captured using Q-Imaging v.2.8 software.
Gene expression profiling
For gene expression profiling, medium was aspirated and cell cultures were washed with ice-cold PBS, followed by RNA extraction using the RNeasy RNA Extraction Kit (Qiagen, Valencia, CA) with the on-column DNase I digestion option. RNA was eluted into RNase free water and quantified. The concentration was adjusted to 1 μg/μl and quality assessed on an RNA chip using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Isolated total RNA was processed as recommended by Affymetrix, Inc. (Santa Clara, CA). In brief, cDNA was synthesized from the total RNA using the SuperScript® Double Stranded cDNA Synthesis kit (Invitrogen Corp., Carlsbad, CA) and T7 Oligo (dT) primers. Using the double stranded cDNA as template, biotin labeled cRNA was generated by in vitro transcription using the BioArray™ HighYield™ RNA Transcript Labeling Kit (T7) (Enzo Life Sciences, Inc., Farmingdale, NY). The cRNA was fragmented to 35–200 bases length using Affymetrix protocols and hybridized to the GeneChip® Human Gene 1.0 ST Array at 45 °C for 16 h in an Affymetrix GeneChip® Hybridization Oven 320. Each Gene Array® was then washed and stained with Streptavidin–Phycoerythrin conjugate (SAPE; Invitrogen Corp.) using an Affymetrix Fluidics Station 400 and scanned on a GeneArray laser scanner (Agilent Technologies).
Data analysis
Gene expression profiling experiments were run in triplicate on the Affymetrix Human Gene 1.0 ST Array platform, on which 28,869 well-annotated genes are represented by approximately 26 probes spread across the full length of each gene (1.4 million probes). Using GeneSpring GX 11.5.1 (Agilent Technologies, Inc., Santa Clara, CA, USA), raw exon expression signals were combined and summarized with ExonRMA16 (RMA) using all transcripts (28,829 transcript clusters from RefSeq and full-length GenBank mRNAs). The data were further quantile normalized with baseline transformation by the median of all samples. Further, the normalized expression signals were averaged between biological replicates. Gene expression data were first filtered by percentile cutoff, resulting in removal of genes with low signal—genes were removed if their expression signal intensity in any one of the three MCF-7-5624A-GF or three MCF-7 parental replicates was below the 10th percentile threshold cutoff of all expression values. Gene lists displaying differentially expressed behavior were generated by performing pairwise comparisons. The 336 genes which have been shown to change between MCF-7-5624A-GF and MCF-7 parental samples were identified by looking for a significant fold change of >2.0 and an unpaired t test p value with Benjamini Hochberg FDR correction of <0.05.
Statistical analysis
Results were reported as mean ± standard error of the mean. Two-sided independent Student’s t test without equal variance assumption or the Wilcoxon signed-rank test were performed to analyze individual time points of BLI data. Repeated measures analysis of variance (ANOVA) was used to assess statistical significance of whole BLI curves encompassing several time points between two treatment groups. The two-sided independent Student’s t test was also performed to analyze end-points of in vitro luciferase assays and histology data. One-way ANOVA and t tests were performed using InStat (GraphPad Software, Inc., version 3.1a). Two-way repeated measures ANOVA and survival analyses were carried out using JMP (v. 8.0; SAS Institute Inc.).
Results
The primary goal of this study was to address how luminal breast cancers disseminate and metastasize. Because luminal cancers predominantly metastasize to the skeleton [23], and the resulting lesions represent a major source of morbidity, we used in vivo selection to isolate bone-tropic metastatic cell lines. First, we injected MCF-7 luminal breast carcinoma cells directly into the tibiae of nude mice, and monitored tumor formation by microCT. Secondary isolates of MCF-7 (MCF-7-5624) readily gave rise to tumor growth in the bone marrow microenvironment after reinjection into the tibia. Moreover, these tumor lesions retained ERα expression, and were characterized by a highly epithelioid phenotype, as demonstrated by expression of cytokeratins and membrane associated E-cadherin (Supplemental Fig. 1). In addition, these lesions induced a strong osteoblastic response of the surrounding bone, as shown by orange G and phloxine positivity, a measure of new bone formation. On the other hand, there was little or no evidence of osteolytic activity (TRAP negativity). Comparison was made with the SCP2 bone-tropic subclone of the basal-like human breast cancer cell line, MDA-MB-231. When injected into the tibia, SCP2 cells also gave rise to bone lesions. However, the phenotype of these tumors was quite distinct from that of MCF-7-derived lesions in several key respects (Supplemental Fig. 1). As expected, SCP2-derived tumors failed to expressed ERα or progesterone receptors (PR) (not shown). Secondly, SCP2-induced lesions were associated with significant osteolysis, as evidenced by TRAP positivity. Finally, SCP2-derived tumors clearly displayed mesenchymal features not seen in MCF-7-5624 derived lesions. Specifically, pan-cytokeratin expression in SCP2-derived tumors was significantly weaker than in MCF-7-5624 and E-cadherin was absent from the cell membrane (Supplemental Fig. 1).
Strikingly, detailed histological analysis of the entire skeleton and other organs of mice that had been inoculated with MCF-7-5624 cells into one tibia revealed metastatic lesions at neighboring sites within the skeleton such as the fibula and femur. In addition, loco-regional lymphatic channels as well as retroperitoneal lymphatics and lymph nodes were filled with tumor (Supplemental Fig. 2). To determine the frequency and time course of these locoregional metastases, second generation MCF-7-5624A bone-tropic cells were infected with a lentiviral vector so that they would constitutively express firefly luciferase to allow localization and quantification of tumor burden in vivo. Animals were inoculated with MCF-7-5624A-GF or MCF-7-ERE-Fluc cells in one tibia and monitored by serial bioluminescence imaging (BLI) in vivo. Metastases first became detectable 12 weeks after tumor cell inoculation and were seen in 5 of 15 mice (Supplemental Fig. 2A). These lesions appeared in a predictable sequence, with the BLI signal first appearing in the iliac lymph nodes, followed by lumbar, and, eventually, high retroperitoneal lymph nodes. To further characterize this regional dissemination, the entire lymph node chain was dissected post mortem and examined histologically (Supplemental Fig. 2B, C). We were able to confirm the presence of extensive tumor deposits throughout retroperitoneal lymphatic vessels and lymph nodes. In addition, in one case, we also found clusters of tumor cells in the right cardiac ventricle as well as in both lungs (Supplemental Fig. 2, 3). Most strikingly, in-transit metastases in retroperitoneal lymphatic vessels had a highly cohesive appearance, suggesting that entire cohorts of cells were disseminating collectively. Moreover, the tumor cells appeared to remain confined to the lymphatic system, as no extravasation was observed. These observations suggested that tumor cells might be metastasizing from the initial lesions in the tibia by collective migration rather than as individual mesenchymal cells. To test whether we were dealing with cohesive clusters of cells or with random aggregates of single cells, we performed immunostaining for E-cadherin. As can be seen in Supplemental Fig. 3, in all of the metastatic lesions in the lymphatics, lymph nodes, heart and lungs, E-cadherin was strongly expressed at the cell membrane. Thus, we concluded that tumor cells disseminate regionally from initial lesions in the tibia as collective sheets or strands along lymphatic channels. Moreover, the process appears to be directional, as we never observed lymphatic metastases distal to the originating tibia lesion.
In order to determine whether MCF-7-5624- and MCF-7-ERE-Fluc-derived tibia lesions had retained their estrogen-dependence in vivo, 17β-estradiol (E2) pellets were removed from tumor-bearing animals (estrogen withdrawal, EWD) and bone lesions were monitored in vivo by microCT (Fig. 1A). In control animals, ovariectomy by itself resulted in a moderate reduction in bone mass (Fig. 1B). In E2-supplemented animals, tumor growth in the tibia was associated with a much more dramatic loss of bone mass (Fig. 1B). However, in response to EWD, these tumors regressed and the tibiae progressively regained bone mass until it reached the same level as in the control animals (Fig. 1B). As shown in parallel experiments using MCF-7-ERE-Fluc cells, EWD was associated with a decline in ERα signaling and regression of regional metastases (Fig. 1C, D). To address the mechanism whereby estrogen might be driving locoregional dissemination, we examined the effects of 17β-estradiol on collective migration of luminal breast cancer cells in vitro. As can be seen in Supplemental Fig. 4, treatment with 17β-estradiol significantly accelerated migration of luminal breast cancer cells in vitro. Thus, estrogen-driven cell migration may be contributing to dissemination of luminal breast cancer cells in vivo.

Bone-tropic luminal breast cancer cell lines retain estrogen-dependence in vivo. MCF-7-derived bone tropic MCF-7-5624 cells were injected into the tibiae of ovariectomized female nude mice supplemented with E2. a Tumor growth was monitored in vivo using microCT (Day 0: Start of EWD). b In animals that were ovariectomized and then supplemented with estrogen, development of tumors in the tibiae was associated with a significant loss of bone mass. Once bone destruction became evident by CT imaging, removing the E2 pellets induced EWD (Day 0: Start of EWD). In control non tumor-bearing animals (n = 5), ovariectomy alone without E2 supplementation resulted in a moderate reduction in bone mass (−Tumor). On the other hand, in tumor-bearing animals (n = 5), EWD induced tumor regression and tibiae progressively regained bone mass until they reached the same level as in the control animals (+ Tumor). In parallel experiments conducted using MCF-7-ERE-FLuc cells, EWD led to a gradual decrease in ERE-luciferase reporter gene activity (c, d)
Primary cultures were established from each of the tibial tumors (Fig. 2A). These secondary cell lines were used to develop systemic metastasis models. Previous attempts at generating in vivo metastasis models of ERα-positive (luminal) breast cancer have used orthotopic or intracardiac (IC) injection to give rise to bone- or visceral metastases [24, 25]. These models have been of limited utility, largely because of the unpredictable and delayed tumor formation and the relatively insensitive in vivo imaging modalities available at the time. Therefore, we decided to test whether luciferase-expressing isolates from tibial tumors (Fig. 2A) had acquired a bone-tropic metastatic phenotype when injected systemically. Female mice that were at least 3 weeks old underwent bilateral ovariectomy. 17β-estradiol supplementation was provided in the form of slow-release E2 pellets. The two independently in vivo selected cell lines, MCF-7-5624A-GF and MCF-7-6012-ERE-FLuc cells, were then injected directly into the systemic arterial circulation via the left cardiac ventricle. Serial in vivo BLI confirmed that both cell lines gave rise to metastatic lesions (Fig. 2B). Lesions were detectable by BLI in over half the mice by 7 days following IC injection, and in all mice by day 35 (Fig. 2C). In both models, mice developed an average of five metastatic lesions each, suggesting that the frequency of metastasis-initiating cells is approximately 1:100,000 (Fig. 2D). In addition, both MCF-7-5624A-GF and MCF-7-6012-ERE-FLuc cells gave rise to remarkably similar patterns of metastasis (Fig. 2E). The predominant metastatic sites included the skeleton (upper- and lower extremities, spine and pelvic bones), the floor of the mouth and mandible, and the adrenal glands. Less common sites included the liver, lungs, brain and the mammary fat pad (MFP) (Fig. 2E). Each of the sites detected by BLI was confirmed histologically (Supplementary Fig. 5). Most strikingly, metastatic lesions that appeared following IC inoculation expanded very rapidly, with a doubling time of approximately 2 days. This was in sharp contrast with tumors that formed following local injection of the same cell lines into the tibia, which had a doubling time of approximately 10 days (Fig. 3A). Moreover, the development of systemic metastases was associated with significant weight loss (Fig. 3A).

Development of in vivo luminal breast cancer metastasis models. a In vivo selection and tagging of bone-tropic of MCF-7- and MCF-7-ERE-Fluc-derived cell lines. In most cases, we were able to re-establish cell lines from individual tibia tumors. Second generation bone tropic MCF-7-5624A cells were tagged using pGreenfire1. b Bone tropic MCF-7-5624A-GF or MCF-7-6012-ERE-FLuc tumor cells were injected into the left cardiac ventricle of viral antibody-free 4- to 5-week-old female athymic nude mice (MCF-7-5624A-GF: n = 13; MCF-7-6012-ERE-FLuc: n = 9). All mice had been ovariectomized and implanted with a slow release E2 pellet providing 2 μg 17β-estradiol/day. Tumor development and growth was monitored by serial BLI. c Systemic metastases became detectable in over half the animals after one week and in all animals by week 7. d Using either of the two tumor cell lines, each animal developed an average of five lesions that were detected by BLI and confirmed post-mortem (see Supplemental Fig. 5). e The predominant metastatic sites included the skeleton (upper- and lower extremities, spine and pelvic bones), the floor of the mouth and mandible, and the adrenal glands. Less common sites included the liver, lungs, brain and the mammary fat pad. The distribution of sites was similar between the two models (p = 0.7515, Chi-squared Test for Independence)

Response of luminal breast cancer metastases to estrogen deprivation. Bone tropic MCF-7-5624A-GF tumor cells were injected into the tibia (n = 8) or into the left cardiac ventricle (n = 13) of viral antibody-free 4- to 5-week-old female athymic nude mice to give rise to locoregional or systemic metastases, respectively. All mice had been ovariectomized and implanted with a slow release E2 pellet providing 2 μg 17β-estradiol/day. a MCF-7-5624A-GF metastases that developed following systemic inoculation (blue squares) grew at a rapid rate, which was five times as high as lesions that originated in the tibia (green triangles). Moreover, systemic metastases were associated with significant weight loss (red circles). Following EWD on day 35, tumor growth was arrested and this was associated with weight gain. In addition, response to EWD was associated with prolongation of survival of the animals (Log-Rank p = 0.0012; Wilcoxon p = 0.0032) (b) . c Parallel experiments using systemically injected MCF-7-6012-ERE-Fluc cells (n = 7) confirmed that EWD (day 70) results in a decrease in ERE-luciferase reporter gene activity
Even though a large body of experimental work supports the notion that growth of human ERα-positive breast cancer cell lines is dependent on estrogen both in vitro and as xenografts [26], little is known of the role estrogen signaling plays in systemic metastatic dissemination. We addressed this question in several different ways. First, once systemic metastases had become detectable by BLI, approximately half the animals were treated with EWD by removing the E2 pellets. EWD immediately resulted in stabilization of the BLI signal, reflecting tumor growth arrest (Fig. 3A). Moreover, EWD and clinical tumor response were associated with weight gain (Fig. 3A) and a significant prolongation in survival (Fig. 3B). As expected, EWD was associated with a decrease in estrogen signaling as determined by ERE-Luc activity (Fig. 3C). Thus, growth of established metastatic lesions is highly dependent on the levels of circulating estrogen. These findings are entirely consistent with the clinical experience of treating metastatic luminal breast cancer, indicating that our models phenocopy the human disease [27, 28].
In order to address the role of estrogen in homing to and colonization of secondary sites, we introduced MCF-7-5624A-GF cells via IC injection into 4–5 week old mice that had been either ovariectomized (low estrogen), ovariectomized followed by E2 pellet implantation (high estrogen), or were virgins (intermediate levels of circulating estrogen). As shown in Fig. 4A, the average numbers of metastatic lesions detectable by BLI were significantly lower in ovariectomized mice than in either of the other two treatment groups. Moreover, in mice with the highest levels of circulating estrogen, metastatic lesions grew exponentially from the time they first became detectable, while, in the other two groups, lesions appeared to go through a lag phase prior to entering an exponential growth (Fig. 4B). This finding suggests that metastasis-initiating cells require some time to adapt to different levels of circulating estrogen. Moreover, once metastatic lesions entered an exponential growth phase, the rates of growth were clearly dependent on the level of circulating estrogen (Fig. 4B). In addition, the patterns of metastasis varied considerably as a function of circulating estrogen levels (Fig. 4C). Specifically, the frequency of skeletal and floor of mouth metastases was highest in E2-supplemented animals, while ovariectomized mice developed metastatic lesions only in the adrenal glands and the MFP, two organs that produce endogenous estrogen [29, 30].

Growth of MCF-7-5624A-GF breast cancer metastases is dependent on the level of circulating estrogen. MCF-7-5624A-GF cells were inoculated via the left cardiac ventricle into mice that had been either ovariectomized (low estrogen) (n = 6), ovariectomized followed by E2 pellet implantation (high estrogen) (n = 8), and virgin mice (intermediate estrogen) (n = 15). a The average numbers of metastatic lesions detectable by BLI were significantly lower in ovariectomized mice than in either of the other two treatment groups (Unpaired t test with Welch correction) b Growth of metastatic lesions as a function of circulating estrogen. Lesions grew exponentially from the time they first became detectable in mice that had been supplemented with estrogen, while, in the other two groups, lesions appeared to go through a lag phase prior to entering an exponential growth phase. In addition, growth rates of metastases varied significantly as a function of circulating estrogen levels (BLI doubling times of 1, 2.5 and 4 days in the high, intermediate and low estrogen groups, respectively). c Patterns of metastasis as a function of circulating estrogen levels. The frequency of skeletal and floor of mouth metastases increased as a function of circulating estrogen levels in a dose-dependent manner. The only types of metastatic lesions we detected in ovariectomized mice were in the adrenal glands and the mammary fat pads. Once these animals were supplemented with E2, several additional metastases appeared, indicating that tumor cells had seeded those areas but had remained dormant, presumably because of a lack of estrogen (see Supplemental Fig. 6)
We went on to address the question whether or not homing and establishment of micrometastases were dependent on estrogen in two different ways: First, we introduced E2 pellets into the ovariectomized animals at 10 weeks following MCF-7-5624A-GF inoculation. Several new areas of metastasis appeared, indicating that tumor cells had seeded those areas after the initial IC injection but had remained dormant, presumably because of a lack of estrogen (Supplemental Fig. 6). Similarly, we inoculated tumor cells into the tibiae of ovariectomized animals and introduced E2 pellets 12 weeks later. No tumor growth was observed by microCT over the ensuing 18 weeks. Nonetheless, we were able to isolate viable tumor cells from the tibia post mortem, and propagate these cells in vitro in estrogen-supplemented medium (MCF7-5624-6022) (Fig. 2A). Thus, these tumorigenic cells had remained dormant but viable for a prolonged period of time, even in an estrogen-deficient bone marrow microenvironment. These results are entirely consistent with the clinical observation that micrometastases can remain dormant for many years during anti-estrogen adjuvant therapy, but become manifest as macrometastases once anti-estrogen therapy is discontinued [31, 32].
In order to begin to elucidate the molecular mechanisms that drive the ability of luminal breast cancer cells to metastasize, we characterized the metastatic clones by gene expression profiling using Affymetrix Human 1.0 ST Gene Arrays. One hundred and seventy genes were significantly downregulated by ≥2 fold (p ≤ 0.05) in MCF-7-5624A-GF cells compared to the parental cell line. Conversely, 166 genes were significantly upregulated by ≥2 fold (p ≤ 0.05) (Supplemental Table 1). The first striking observation was that the metastatic cells failed to overexpress any mesenchymal markers or inducers of EMT. The second striking observation was that many of the genes that were upregulated in the metastatic cells are known to be involved in collective migration during development (Table 1).
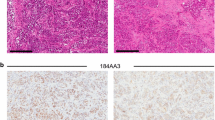
Interestingly, one of the most highly expressed genes was TGF-β2 (Table 1). Given the preeminent role TGF-β plays in driving metastasis of basal-like breast carcinoma cell lines in vivo [8–10], this suggested that this cytokine might play a similar role in luminal breast cancer metastasis. To our surprise, while treatment with TGF-β induced brisk phosphorylation of Smad2 and -3 in ERα-negative human breast cancer cell lines, ERα-positive cell lines display this response either weakly or not at all (Fig. 5A). Consistent with their inability to respond to TGF-β, luminal breast cancer cell lines do not express the TGF-β response gene signature (TBRS), while this is clearly represented within the gene expression profiles of ERα-negative basal-like and HER-2-positive cell lines (Fig. 5B). The inability of the luminal cell lines to activate Smads was apparently due to transcriptional silencing of the TGF-β type II receptor gene (TGFBR2), as we were unable to detect any TGFBR2 mRNA in most of the ERα-positive cell lines, while it was abundantly expressed in each of the ERα-negative cell lines (Fig. 5C). To validate this observation, we examined published gene expression profiles of three large series of human breast cancer cell lines [33–35] (Fig. 5D). Strikingly, human breast cancer cell lines appear to express either ESR1 or TGFBR2 mRNA but rarely or ever both. Moreover, Charafe-Jaufret et al. [36] identified TGFBR2 as one of the 10 genes most strongly differentially expressed between basal-like and luminal breast cancer lines. Thus, by and large, expression of ESR1 and TGFBR2 mRNA appear to be inversely correlated.

TGF-β signaling is inactive in ERα-positive breast cancer cell lines because of transcriptional silencing of the TGFBR2 gene. a TGF-β treatment failed to induce Smad phosphorylation in ERα-positive human breast cancer cell lines, while ERα-negative cell lines responded to exogenous TGF-β by activating Smad2 and -3. b Expression of a 92-gene TBRS [62] in human breast cancer cell lines [33]. Using Gene Set Enrichment Analysis, a score between −1 and +1 was derived, indicating the likelihood that TGF-β signaling is active (>0) or not (<0) in individual cell lines [11]. Cell line scores within each major breast cancer subset are depicted as boxplots. Consistent with their inability to respond to TGF-β, luminal breast cancer cell lines do not express the TBRS, while it is clearly represented within the gene expression profiles of ERα-negative cell lines. c We found a striking inverse correlation between ESR1 and TGFBR2 mRNA transcript levels across our panel of 9 human breast cancer cell lines by qRT-PCR. Thus, unresponsiveness to TGF-β is apparently due to silencing of TGFBR2 gene expression. d Contour plots of ESR1 and TGFBR2 mRNA transcript levels across human breast cancer cell lines reported in three different published studies [33–35]. Strikingly, in each of the three studies, human breast cancer cell lines expressed either ESR1 or TGFBR2 mRNA but rarely both
MCF-7-5624A-GF cells produced much higher amounts of TGF-β2 than parental MCF-7 cells in culture, while production of TGF-β1 was similar in both cell lines (Fig. 6A). In addition, MCF-7-5624A-GF cells strongly induced TGF-β-responsive luciferase activity in co-cultivated mink lung epithelial reporter cells, indicating that the TGF-β2 produced by MCF7-5624A-GF cells is biologically active. Moreover, treatment with the pan-TGF-β neutralizing antibody, 1D11, completely abolished luciferase activation (Fig. 6B). Even though the metastatic MCF7-5624 cells were intrinsically unresponsive to TGF-β because they lack TβR-II receptor expression (Fig. 5), it was possible that TGF-β2 might promote metastasis in a paracrine manner. To address this question, mice were treated with the pan-TGF-β neutralizing antibody 1D11, an isotype control antibody (13C4) or vehicle only and then inoculated systemically with MCF-7-5624A-GF cells. Treatment with 1D11 failed to affect the rate of appearance and growth of metastases (Fig. 6C). Moreover, neither the number of metastases (Fig. 6D) nor the survival of the mice (Fig. 6E) was affected. These experiments demonstrate that the high levels of active TGF-β2 produced by the tumor cells do not contribute significantly to metastasis of MCF-7-5624A-GF cells. Thus, in contrast to basal-like breast cancer models, in which both tumor cell autonomous TGF-β signaling and paracrine effects of tumor cell-associated TGF-β play major roles in driving dissemination and metastasis [8–10], luminal breast cancer cells appear to disseminate independently of auto- or paracrine TGF-β signaling.

Luminal breast cancer metastasis does not require TGF-β signaling. a Conditioned medium of MCF-7-5624A-GF cells (red circles) produced much higher amounts of TGF-β2 than parental MCF-7 cells (blue squares), while production of TGF-β1 was similar in both cell lines. Top two panels represent standard curves. b When MCF-7-5624A-GF cells were co-cultivated with mink lung epithelial cells that express a TGF-β-responsive luciferase reporter, the metastatic cells induced a strong transcriptional response, indicating that the TGF-β2 that was being produced was biologically active. Moreover, treatment with the pan-TGF-β neutralizing antibody, 1D11, abolished luciferase activity in the reporter cells in a dose-dependent manner. c Animals were treated with 1D11 pan-TGF-β neutralizing antibody (n = 10), 13C4 isotype control antibody (n = 8) or vehicle (n = 8). Following systemic inoculation with MCF-7-5624A-GF cells, metastases developed and grew at the same rate in 1D11-treated animals as in those treated with vehicle or 13C4 isotype control antibody. d The number of metastases per mouse as well as the distribution of metastases (not shown) were similar across all three treatment groups. e Survival of the 1D11-treated animals was similar to that of the two control groups. Thus, the high levels of active TGF-β2 produced by the tumor cells does not appear to contribute to metastasis of MCF-7-5624A-GF cells
Discussion
Luminal breast cancer accounts for two-thirds of all breast cancers, and approximately 70 % of all metastatic breast cancers are of the luminal type [1]. Nonetheless, little is known of the molecular underpinnings of this disease. In this report, we describe the first efficient and reproducible regional and systemic metastasis models of human luminal breast cancer. Following systemic injection of tumor cells, distant metastases are detectable by BLI in half the mice after only one week, and in all of the mice by five weeks. Because the metastases can be detected by functional imaging in vivo, these models allow us to conduct long-term treatment experiments. This is a marked improvement over previous models of metastatic luminal breast cancer, which relied on radiography or autopsy to detect metastases, and typically took 2–6 months to complete [25, 37]. The pattern of metastasis in our models is reminiscent but not entirely typical for the human disease [1]. In both cases, the skeleton is by far the most common site of distant metastasis. However, metastases to the lungs and liver are seen more frequently in the human disease than in our MCF-7 models [1]. This raises the question whether the pattern we observed is unique to the MCF-7 models or is a function of the fact that cells were injected directly into the arterial circulation. As we develop additional models using different luminal breast cancer cell lines, we hope to be able to address these questions.
Luminal breast cancer cell metastasis is dependent on estrogen
Our new luminal breast cancer metastasis models are ideally suited to address the molecular mechanisms underlying this process. In this first report, we describe the central role of estrogen in cancer dissemination. The growth of initial lesions in the tibia, dissemination along the retroperitoneal lymph node chain, as well as growth of established distant metastases were all clearly dependent on estrogen supplementation. In this sense, metastases behave similarly to subcutaneous xenografts [24, 26], and to luminal cancers in humans [28, 38]. Indirect evidence that metastasis of MCF-7 cells in nude mice might be dependent on estrogen was first reported by Shafie and Liotta [24]. These investigators found that supplementation of virgin animals with E2 was associated with accelerated growth of MCF-7 xenografts and the occasional appearance of metastases in lungs, liver, or spleen, while no metastases were observed in ovariectomized animals. However, it is difficult to conclude with certainty from these experiments that dissemination and seeding of micrometastases per se were estrogen-dependent, as growth of the “primary” xenografts as well as metastases varied greatly as a function of the available estrogen.
Even though, in our models, tumor growth both in the tibia and at secondary sites was clearly dependent on estrogen signaling, homing and seeding of micrometastases appeared to be independent of estrogen. Thus, we found that MCF-7-5624A GF cells were capable of remaining dormant but viable for prolonged periods of time even in a very low estrogen environment, both after direct injection into the tibia or systemic IC injection. Under low estrogen conditions, no tumor was detectable in the tibia even up to 18 weeks, even though viable tumor cells could be still recovered from the bone. After IC injection, metastases did become detectable but only after a prolonged lag period and only at sites known to produce endogenous estrogen, such as the adrenal glands and the mammary fat pad [29, 30]. Long-term estrogen deprivation of luminal breast cancer cells in vitro has been associated with estrogen hypersensitivity, which is caused by upregulation and membrane localization of ERα and non-genomic estrogen signaling [39]. This phenomenon may account for the lag phase prior to metastatic growth we observed in the ovariectomized and young virgin animals. In addition, supplementing the animals with exogenous estrogen after a prolonged period of estrogen deprivation led to the appearance of additional macrometastases at diverse sites (Supplemental Fig. 6). In aggregate, these results demonstrate unequivocally that, even in the absence of estrogen, metastasis-initiating tumor cells can seed secondary sites and remain viable but dormant. Thus, the establishment of micrometastases per se is not dependent on estrogen.
Besides estrogen, several other mechanisms have been proposed to drive metastases of luminal breast cancer cells. For example, endothelin-1 has been implicated in playing a key role in osteoblastic metastases by T47D and ZR-75-1 luminal breast cancer cells [25, 40, 41]. However, we did not find endothelin-1 mRNA expression to be elevated in the MCF7-5624A-GF model (Supplemental Table 1). Several studies have implicated CD44 as a driver of MCF-7 breast cancer metastasis [42–44]. Even though both studies [42–44] suggested that CD44 might be involved in MCF-7 cell metastasis, neither provided definitive proof for a causal role for CD44 in vivo. Moreover, in our hands, the fraction of CD44+ cells in our metastatic clones was similar to that in the parental lines (data not shown). Thus, it is unlikely that CD44 plays an important role in driving the metastatic phenotype of MCF-7-5624A-GF or MCF-7-6012-ERE-Fluc cells.
Luminal breast cancer cells maintain their epithelioid phenotype throughout locoregional dissemination
In our MCF-7 model, regional metastasis via lymphatic channels and lymph nodes eventually gave cells access to the bloodstream, resulting in the appearance of lung metastases. Throughout this entire process, the tumor cells retained a cohesive appearance and continued to express epithelioid markers. Moreover, the metastatic cells failed to differentially express any mesenchymal markers or inducers of EMT (Supplemental Table 1). This is consistent with earlier reports that MCF-7 cells do not express vimentin, do not invade into Matrigel and form tight spheres in 3D Matrigel® cultures [12] (Grazioli and Reiss, unpublished observations). Consistent with our findings, Harrell et al. [42, 43] reported that MCF-7 and T47D cells injected orthotopically into inguinal mammary fat pads metastasized to regional lymph nodes and, occasionally, to distant sites. The morphology of the in-transit lymphatic metastases was highly reminiscent of what we observed following intratibia injections. These observations suggest that luminal breast cancer cells may be spreading regionally by recapitulating the collective migration cells utilize during development. Collective migration involves the coordinated movement of a sheet of cells in a single direction (movement directionality), which seems to be driven by planar cell polarity, i.e., the phenomenon that cells move along an axis that is in the plane of the sheet [45, 46]. It is striking that our highly metastatic MCF-7-5624A-GF cells expressed a large number of genes known to be involved in collective migration and planar cell polarity at much higher levels than parental MCF-7 cells (Table 1).
Following inoculation of luminal breast cancer cells into the tibia, we were able to detect regional metastases only in E2-supplemented animals. This suggests that the migration of the tumor cells in vivo may be dependent on estrogen. In support of this idea, we were able to show that treatment of luminal tumor cells with 17β-estradiol stimulated collective migration of the cells in vitro (Supplemental Fig. 4). This is consistent with several previous reports. For example, Gamba et al. [47] recently showed that collective migration of the posterior lateral line of zebrafish is dependent on estrogen signaling. Similarly, several groups [48–53] have reported that estrogen accelerates collective migration of human luminal breast cancer cells in vitro. Sanchez et al. reported that 17β-estradiol induced tumor cell migration via rapid extranuclear signaling of ERα interacting in a multiprotein complex with c-Src, phosphatidylinositol 3-OH kinase, and FAK [51]. Moreover, recent evidence suggests important roles for PELP1 and the integrin-linked kinase in this process [53, 54]. Thus, estrogen effects on cell migration may be largely mediated by non-genomic signaling, a process that involves localization of the ERα to caveolae at the cell membrane. In this regard, it is striking that MCF-7-5624A-GF cells strongly overexpress the two principal components of caveolae, caveolin-1 and -2 (Table 1). We are currently testing the hypothesis that caveolin expression targets the ERα to the cell membrane, thereby enabling its non-genomic signaling and pro-migratory effects.
Luminal breast cancer cells metastasize independently of TGF-β
Our third major finding is that, in sharp contrast to basal-like breast cancer, luminal breast cancer metastasis is independent of TGF-β signaling. Our first clue was the striking inverse correlation between expression of ERα and the TβR-II receptor. Established human breast carcinoma cell lines appear to express either ESR1 or TGFBR2 mRNA but rarely both. In addition, luminal breast cancer cell lines are typically unresponsive to exogenous TGF-β in terms of Smad phosphorylation and activation of the TGF-β responsive gene expression program (Fig. 5). This turns out to be due to transcriptional silencing of the TGFBR2 gene. Consistent with this idea, we and others have reported that the absence of a TGF-β response gene signature was predictive of a poor prognosis across different luminal breast cancer series [11, 55].
In spite of being intrinsically unresponsive to TGF-β, we found that the highly metastatic MCF-7-5624A-GF cells produced very high levels of bioactive TGF-β2, suggesting the possibility that this cytokine might play a paracrine pro-metastatic role. However, neutralization of TGF-βs using the 1D11 antibody failed to lower the overall burden of metastases in vivo. In addition, tumor-derived TGF-β2 did not appear to play a major role in organ tropism as the site distribution of metastases was not significantly affected by 1D11 treatment. Thus, it is unlikely that paracrine TGF-β signaling plays a significant role in driving metastasis in our MCF-7 model. In addition, our results rule out the possibility that, in our model, TGF-β signaling is switched on transiently to allow dissemination as was recently noted in a rat mammary carcinoma model [56–58].
It should be noted that, because our studies were conducted in athymic nude mice, we couldn’t formally exclude the possibility that the increased levels of TGF-β2 might enhance metastasis by repressing anti-tumor immunity. Support for this possibility comes from genetic mouse models in which the TGFBR2 gene was conditionally deleted in the mammary ductal epithelium [59, 60]. When TGFBR2 null mice were mated to polyomavirus middle T antigen transgenic mice, mammary tumor development was greatly accelerated, and even associated with a disproportionate increase in the number of pulmonary metastases. TGFBR2 null mammary carcinomas were associated with an increased production of TGF-β1 [61]. Whether these high levels of TGF-β1 contributed to their highly metastatic phenotype, or represented a compensatory tumor suppressive host cell response is unresolved. In our hands, 1D11 treatment was associated with a slightly greater number and size of metastases and a shorter median survival (37.5 vs. 43.5 days). Even though these differences failed to reach statistical significance, they would be consistent with a protective effect of tumor-associated TGF-β. Based on our findings, we would recommend that treatment of metastatic luminal breast cancer with TGF-β pathway antagonists be avoided until these issues are clarified.
In summary, we have developed novel in vivo metastasis models of human luminal breast cancer that are both reproducible and efficient and share many of the features of the human disease. We show that, throughout the process of dissemination from a primary lesion to regional and then systemic metastases, the tumor cells maintain their epithelioid phenotype. Estrogen is required for tumor cell growth, both at primary and secondary sites, as well as for locoregional metastasis, while seeding of distant micrometastases appears to be an estrogen-independent process. Micrometastases can remain dormant and viable for prolonged periods of time in the absence of estrogen. This may be a major reason that anti-estrogens have proven to be very effective in the adjuvant setting. Elucidating the molecular mechanisms underlying the establishment of micrometastatic disease is likely to provide novel targets for therapy of luminal breast cancer.
Abbreviations
- EMT:
-
Epithelial-to-mesenchymal transitions
- TGF-β:
-
Transforming growth factor-β
- ERα:
-
Estrogen receptor α
- TGFBR2 :
-
TGF-β type II receptor gene
- Ara-C:
-
Cytosine-β-d-arabinofuranoside-hydrochloride
- ESR1 :
-
Estrogen receptor α gene
- PGR :
-
Progesterone receptor gene
- GAPDH :
-
Glyceraldehyde 3-phosphate dehydrogenase gene
- BLI:
-
Bioluminescence imaging
- IC:
-
Intracardiac
- TRAP:
-
Tartrate resistant acid phosphatase
- ANOVA:
-
Analysis of variance
- E2:
-
17β-estradiol
- EWD:
-
Estrogen withdrawal
- MFP:
-
Mammary fat pad
- TBRS :
-
TGF-β response gene signature
References
Kennecke H, Yerushalmi R, Woods R et al (2010) Metastatic behavior of breast cancer subtypes. J Clin Oncol 28(20):3271–3277
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331(6024):1559–1564
Kang Y, Siegel PM, Shu W et al (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3(6):537–549
Gupta GP, Minn AJ, Kang Y et al (2005) Identifying site-specific metastasis genes and functions. Cold Spring Harb Symp Quant Biol 70:149–158
Kang Y (2006) New tricks against an old foe: molecular dissection of metastasis tissue tropism in breast cancer. Breast Dis 26:129–138
Acloque H, Adams MS, Fishwick K et al (2009) Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 119(6):1438–1449
Ganapathy V, Ge R, Grazioli A et al (2010) Targeting the transforming growth factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer 9(1):122
Ge R, Rajeev V, Ray P et al (2006) Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-beta type I receptor kinase in vivo. Clin Cancer Res 12(14 Pt 1):4315–4330
Korpal M, Yan J, Lu X et al (2009) Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med 15(8):960–966
Tan AR, Alexe G, Reiss M (2009) Transforming growth factor-beta signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res Treat 115(3):453–495
Bae SN, Arand G, Azzam H et al (1993) Molecular and cellular analysis of basement membrane invasion by human breast cancer cells in matrigel-based in vitro assays. Breast Cancer Res Treat 24(3):241–255
Blick T, Widodo E, Hugo H et al (2008) Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis 25(6):629–642
Blick T, Hugo H, Widodo E et al (2010) Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/−) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia 15(2):235–252
Kowalski PJ, Rubin MA, Kleer CG (2003) E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res 5(6):R217–R222
Bukholm IK, Nesland JM, Borresen-Dale AL (2000) Re-expression of e-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients [seecomments]. J Pathol 190(1):15–19
Weigelt B, Hu Z, He X et al (2005) Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res 65(20):9155–9158
Friedl P, Gilmour D (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10(7):445–457
Leu YW, Yan PS, Fan M et al (2004) Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res 64(22):8184–8192
Fan M, Yan PS, Hartman-Frey C et al (2006) Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res 66(24):11954–11966
Fan M, Long X, Bailey JA et al (2002) The activating enzyme of NEDD8 inhibits steroid receptor function. Mol Endocrinol 16(2):315–330
Parfitt AM, Drezner MK, Glorieux FH et al (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR histomorphometry nomenclature committee. J Bone Miner Res 2(6):595–610
Campbell FC, Blamey RW, Elston CW et al (1981) Oestrogen-receptor status and sites of metastasis in breast cancer. Br J Cancer 44(3):456–459
Shafie SM, Liotta LA (1980) Formation of metastasis by human breast carcinoma cells (MCF-7) in nude mice. Cancer Lett 11(2):81–87
Guise TA, Yin JJ, Mohammad KS (2003) Role of endothelin-1 in osteoblastic bone metastases. Cancer 97(3 Suppl):779–784
Osborne CK, Hobbs K, Clark GM (1985) Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res 45(2):584–590
Allegra JC, Lippman ME, Thompson EB et al (1979) Relationship between the progesterone, androgen, and glucocorticoid receptor and response rate to endocrine therapy in metastatic breast cancer. Cancer Res 39(6 Pt 1):1973–1979
Clark GM, Sledge GW Jr, Osborne CK et al (1987) Survival from first recurrence: relative importance of prognostic factors in 1,015 breast cancer patients. J Clin Oncol 5(1):55–61
Edery M, Carreau S, Drosdowsky A (1980) In vitro pregnenolone metabolism by mouse adrenal gland: I-estrogen synthesis. Steroids 35(4):381–388
Silberstein GB, Van Horn K, Shyamala G et al (1994) Essential role of endogenous estrogen in directly stimulating mammary growth demonstrated by implants containing pure antiestrogens. Endocrinology 134(1):84–90
Goss P, Allan AL, Rodenhiser DI et al (2008) New clinical and experimental approaches for studying tumor dormancy: does tumor dormancy offer a therapeutic target? APMIS 116(7–8):552–568
Goss PE, Chambers AF (2010) Does tumour dormancy offer a therapeutic target? Nat Rev Cancer 10(12):871–877
Neve RM, Chin K, Fridlyand J et al (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10(6):515–527
Kao J, Salari K, Bocanegra M et al (2009) Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE 4(7):e6146
Hollestelle A, Nagel JH, Smid M et al (2010) Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res Treat 121(1):53–64
Charafe-Jauffret E, Ginestier C, Monville F et al (2006) Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 25(15):2273–2284
Thomas RJ, Guise TA, Yin JJ et al (1999) Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology 140(10):4451–4458
Allegra JC, Lippman ME, Thompson EB et al (1980) Estrogen receptor status: an important variable in predicting response to endocrine therapy in metastatic breast cancer. Eur J Cancer 16(3):323–331
Santen RJ, Song RX, Masamura S et al (2008) Adaptation to estradiol deprivation causes up-regulation of growth factor pathways and hypersensitivity to estradiol in breast cancer cells. Adv Exp Med Biol 630:19–34
Mohammad KS, Guise TA (2003) Mechanisms of osteoblastic metastases: role of endothelin-1. Clin Orthop Relat Res (415 Supp):S67–S74
Yin JJ, Mohammad KS, Kakonen SM et al (2003) A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Nat Acad Sci USA 100(19):10954–10959
Harrell JC, Dye WW, Allred DC et al (2006) Estrogen receptor positive breast cancer metastasis: altered hormonal sensitivity and tumor aggressiveness in lymphatic vessels and lymph nodes. Cancer Res 66(18):9308–9315
Harrell JC, Dye WW, Harvell DM et al (2007) Estrogen insensitivity in a model of estrogen receptor positive breast cancer lymph node metastasis. Cancer Res 67(21):10582–10591
Uchino M, Kojima H, Wada K et al (2010) Nuclear beta-catenin and CD44 upregulation characterize invasive cell populations in non-aggressive MCF-7 breast cancer cells. BMC Cancer 10:414
Rorth P (2009) Collective cell migration. Annu Rev Cell Dev Biol 25:407–429
Wang Y (2009) Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther 8(8):2103–2109
Gamba L, Cubedo N, Ghysen A et al (2010) Estrogen receptor ESR1 controls cell migration by repressing chemokine receptor CXCR4 in the zebrafish posterior lateral line system. Proc Nat Acad Sci USA 107(14):6358–6363
Planas-Silva MD, Bruggeman RD, Grenko RT et al (2006) Role of c-Src and focal adhesion kinase in progression and metastasis of estrogen receptor-positive breast cancer. Biochem Biophys Res Commun 341(1):73–81
Planas-Silva MD, Waltz PK (2007) Estrogen promotes reversible epithelial-to-mesenchymal-like transition and collective motility in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol 104(1–2):11–21
Li Y, Wang JP, Santen RJ et al (2010) Estrogen stimulation of cell migration involves multiple signaling pathway interactions. Endocrinology 151(11):5146–5156
Sanchez AM, Flamini MI, Baldacci C et al (2010) Estrogen receptor-alpha promotes breast cancer cell motility and invasion via focal adhesion kinase and N-WASP. Mol Endocrinol 24(11):2114–2125
Giretti MS, Fu XD, De Rosa G et al (2008) Extra-nuclear signalling of estrogen receptor to breast cancer cytoskeletal remodelling, migration and invasion. PLoS ONE 3(5):e2238
Zheng S, Huang J, Zhou K et al (2011) 17beta-estradiol enhances breast cancer cell motility and invasion via extra-nuclear activation of actin-binding protein ezrin. PLoS ONE 6(7):e22439
Chakravarty D, Nair SS, Santhamma B et al (2010) Extranuclear functions of ER impact invasive migration and metastasis by breast cancer cells. Cancer Res 70(10):4092–4101
Bierie B, Chung CH, Parker JS et al (2009) Abrogation of TGF-beta signaling enhances chemokine production and correlates with prognosis in human breast cancer. J Clin Invest 119(6):1571–1582
Sahai E (2007) Illuminating the metastatic process. Nat Rev Cancer 7(10):737–749
Giampieri S, Manning C, Hooper S et al (2009) Localized and reversible TGF beta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol 11(11):1287–1296
Giampieri S, Pinner S, Sahai E (2010) Intravital imaging illuminates transforming growth factor beta signaling switches during metastasis. Cancer Res 70(9):3435–3439
Forrester E, Chytil A, Bierie B et al (2005) Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res 65(6):2296–2302
Bierie B, Stover DG, Abel TW et al (2008) Transforming growth factor-beta regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res 68(6):1809–1819
Yang L, Huang J, Ren X et al (2008) Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1 + CD11b + myeloid cells that promote metastasis. Cancer Cell 13(1):23–35
Kareddula A, Zachariah E, Notterman D et al (2008) Transforming growth factor-β signaling strength determines target gene expression profile in human keratinocytes. J Epithel Biol Pharmacol 1:40–94
Acknowledgments
This study was supported by PHS R01 CA120623 award from the National Cancer Institute, National Institutes of Health, US to M.R and by the Histology & Imaging, Bioinformatics and Preclinical Imaging Shared Resources of The Cancer Institute of New Jersey (P30 CA 72720). We wish to express our gratitude to Dr. Kenneth Nephew (Indiana University) for generously sharing his MCF-7 derived cell lines, and to Dr. Yibin Kang (Princeton University) for MDA-MB-231 and SCP2 cells.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10585_2012_9466_MOESM1_ESM.tif
Supplemental Fig. 1. Local tumor growth following intratibia tumor cell injection. MCF-7-derived bone tropic MCF-7-5624 cells were injected into tibiae of nude mice. Tumor growth was monitored in vivo using microCT and BLI. Tumor lesions retained ERα expression, and were characterized by a highly epithelioid phenotype, as demonstrated by expression of cytokeratins (pan-CK) and membrane associated E-cadherin. Moreover, these lesions induced a predominantly osteoblastic response of the surrounding bone, as shown by orange G and phloxine positivity (a measure of new bone formation), while there was little to no evidence of osteolytic activity (TRAP negativity). Comparison was made with the SCP2 bone tropic subclone of the basal-like ERα-negative human breast cancer line, MDA-MB-231. When injected into the tibia, SCP2 cells also gave rise to bone lesions with a phenotype that was quite distinct from that of MCF-7-5624: None of the cells expressed ERα or PR (not shown). In addition, SCP2-induced lesions were associated with significant osteolysis, as evidenced by strong TRAP positivity. Thirdly, SCP2-derived tumors were distinctly less epithelioid as pan-cytokeratin expression was significantly weaker than in MCF-7-5624 lesions and they failed to express E-cadherin on. Supplementary material 1 (TIFF 93765 kb)
10585_2012_9466_MOESM2_ESM.tif
Supplementary Fig. 2. Locoregional tumor cell dissemination via retroperitoneal lymphatic channels. A. BLI demonstrated that tumors that arose following intra-tibia inoculation of MCF-7-5624A-GF cells disseminated regionally via the iliac and retroperitoneal lymph node chain. B. Solid cords of tumor cells could be seen in local lymphatic channels in the vicinity of the initial tibia lesions. C. Solid cords of tumor cells are also seen to fill retroperitoneal lymphatic channels and lymph nodes. Ly Ch: Lymphatic channel; Ly No: Lymph node. Supplementary material 2 (TIFF 201227 kb)
10585_2012_9466_MOESM3_ESM.tif
Supplementary Fig. 3. Luminal breast cancer cells disseminate as cohesive epithelial clusters. Metastatic MCF-7-5624A-GF cell deposits in the lymphatic vessels, heart and lungs all strongly expressed E-cadherin at the cell membrane, indicating that these tumor cells retain cohesiveness throughout the process of dissemination. Supplementary material 3 (TIFF 92846 kb)
10585_2012_9466_MOESM4_ESM.tif
Supplementary Fig. 4. 17β-estradiol stimulates migration of luminal breast cancer cells. Confluent cultures of ERα-positive luminal human breast cancer cells were wounded using a sterile plastic pipet tip. A. The resulting wound was photographed at multiple sites and the wound area calculated using Image J. Tumor cells migrate collectively to close the gap. B. Treatment with 17β-estradiol resulted in a marked acceleration of wound closure compared to treatment with vehicle only (p=0.0045, t test with Welch correction). Supplementary material 4 (TIFF 87528 kb)
10585_2012_9466_MOESM5_ESM.tif
Supplementary Fig. 5. Histopathology of systemic metastatic lesions. Representative examples of MCF-7-5624A-GF-derived metastases to the skeleton, adrenal glands, central nervous system, lungs, muscle, lymph nodes and mammary gland (Photomicrographs 200x). Supplementary material 5 (TIFF 92705 kb)
10585_2012_9466_MOESM6_ESM.tif
Supplementary Fig. 6. Tumor dormancy in estrogen-deficient animals. Ovariectomized mice were inoculated systemically with MCF-7-5624A-GF cells. At 10 weeks post-inoculation, a small number of metastatic lesions in adrenal glands and mammary fatpads were detectable by BLI (Baseline) (see Figure 4). We then introduced E2 pellets into these animals (Figure 4B). Subsequently, several additional metastases appeared in the skeleton and adrenal glands, indicating that tumor cells had initially seeded those areas and had remained viable but dormant, presumably because of a lack of estrogen. Supplementary material 6 (TIFF 92933 kb)
10585_2012_9466_MOESM7_ESM.xls
Supplemental Table 1. Genes differentially expressed between MCF-7 parental cells and metastatic MCF-7-5624A-GF cells. Gene expression profiling experiments were run in triplicate on the Affymetrix Human Exon 1.0 ST exon microarray platform (1.4 million probes). Using GeneSpring GX 11.5.1 (Agilent Technologies, Inc., Santa Clara, CA, USA), raw exon expression signals were combined and summarized with ExonRMA16 (RMA) using all transcripts (28,829 transcript clusters from RefSeq and full-length GenBank mRNAs). The data were quantile normalized with baseline transformation by the median of all samples. Furthermore, the normalized expression signals were averaged between biological replicates. Gene expression data were first filtered by 10th percentile cut-off, resulting in removal of genes with low signal. The 336 genes shown to be differentially expressed between MCF-7-5624A-GF and MCF-7 parental cells were identified by looking for a significant fold change of >2.0 and an unpaired t test p value with Benjamini Hochberg FDR correction of <0.05. Supplementary material 7 (XLS 124 kb)
Rights and permissions
About this article
Cite this article
Ganapathy, V., Banach-Petrosky, W., Xie, W. et al. Luminal breast cancer metastasis is dependent on estrogen signaling. Clin Exp Metastasis 29, 493–509 (2012). https://doi.org/10.1007/s10585-012-9466-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-012-9466-4