
Abstract
The layer-by-layer (LbL) technology was used to adsorb polyelectrolyte multilayers consisting of cationic polyethylenimine (PEI) and anionic sodium hexametaphosphate (SHMP) onto cellulose fibers in order to enhance the flame-retardancy and tensile strength of paper sheets made from these fibers. The fundamental effect of PEI molecular mass on the build-up of the multilayer film was investigated using model cellulose surfaces and a quartz crystal microbalance technique. The adsorption of a low (LMw) and a high molecular weight (HMw) PEI onto cellulose fibers and carboxymethylated (CM) cellulose fibers was investigated using polyelectrolyte titration. The fibers were consecutively treated with PEI and SHMP to deposit 3.5 bilayers (BL) on the fiber surfaces, and the treated fibers were then used to prepare sheets. In addition, a wet-strength paper sheet was prepared and treated with the same LbL coatings. Thermal gravimetric analysis of LbL-treated fibers showed that the onset temperature for cellulose degradation was lowered and that the amount of residue at 800 °C increased. A horizontal flame test and a vertical flame test were used to evaluate the combustion behavior of the paper sheets. Papers prepared from both cellulose fibers and CM-cellulose fibers treated with HMw-PEI/SHMP LbL-combination self-extinguished in a horizontal configuration despite the rather low amounts of adsorbed polymer which form very thin films (wet thickness of ca. 17 nm). The tensile properties of handsheets showed that 3.5 BL of HMw-PEI and SHMP increased the stress at break by 100% compared to sheets prepared from untreated cellulose fibers.
Similar content being viewed by others

Introduction
Cationic polyelectrolytes play an important role in papermaking as they improve the retention of fines and added mineral fillers. They have also been used to increase fiber interactions in fiber/fiber joints to increase paper strength. In earlier investigations, cationic starch (Lindström and Florén 1984; Lindström et al. 2005), polyallylamine (Rathi and Biermann 2000), and polyamideamine epichlorohydrin (Enarsson and Wågberg 2007) resins were used to enhance the paper strength by adsorbing these polyelectrolytes onto the fibers before sheet preparation. The negative charges located on fibers originate from residual hemicelluloses and the oxidation of cellulose or lignin during cooking, and bleaching of the fibers promotes a good interaction between the fibers and cationic polyelectrolytes (Wågberg and Annergren 1997). The adsorption of cationic polyelectrolytes on anionic surfaces is due to an entropically driven ion-exchange process (Fleer et al. 1993; Stuart et al. 1991; Trout 1951; Winter et al. 1986; Wågberg et al. 1987) where the counter-ions to the charges on the polyelectrolytes and the fibers are released upon adsorption. Wågberg et al. investigated the adsorption of cationic polyelectrolytes onto cellulose-rich fibers and the effects of the porous structure of the cell wall (with pores ranging from 1 to 30 nm) (Wågberg and Hägglund 2001), and it has also been shown that the adsorbed amount (i.e., mg/g fiber) is highly dependent on the molecular mass of the polymer (Alince 1990; Haggkvist et al. 1998; Stone et al. 1968; van de Ven and Alince 1997; Wågberg and Hägglund 2001). Earlier works have shown that polyelectrolytes with different molecular weights can penetrate the fiber wall either through a reptation process or through simple diffusion (van de Ven 2000; Wågberg and Hägglund 2001; Wågberg et al. 1988). Strong polyelectrolytes with medium to high molecular mass adsorbed onto the external fiber surface with a flat conformation at low to medium salt concentrations and do not obstruct further adsorption into the fiber wall (Wågberg and Hägglund 2001). It has also been shown that the saturation adsorption of weak cationic polyelectrolytes used in paper making, such as polyethylenimine (PEI), onto cellulose fibers and silica is significantly affected by the molecular weight, concentration of solution, pH and salt concentration used during the adsorption (Alince et al. 1996; van de Ven 2000). HMw-PEI exhibits a two-step adsorption onto porous cellulose fibers in which PEI is first adsorbed to the external surface of the fiber and then in the second step, the adsorption of the low molecular mass fraction of PEI continues to the internal parts of the fiber wall through diffusion. At high PEI addition level (i.e., 40 mg/g fiber) the amount of LMw fraction is also high which as a consequence leads to an increased adsorbed amount of PEI. Van de Ven showed that at pH 10 the percentage of PEI that reached into macro pores of the fiber wall significantly increased compared to the adsorption at pH 6 (van de Ven 2000). This behavior can be explained by the decrease in PEI size at high pH and also by the lower interaction between the cellulose and the PEI due to the decreased charge of the PEI at pH 10 (Horvath et al. 2008). The addition of electrolyte reduces the repulsion between the charged groups of PEI and allows for a contraction of the polymer chain which can also allow for a better access to the interior of the fiber for the polyelectrolyte. The increased electrolyte concentration also decreases the repulsion between adsorbed polymers on the fiber surface which hence also allow for an increased adsorbed amount at higher salt concentrations (Xie et al. 2016). It is also well documented that the adsorption of PEI onto nanoporous structure cellulose-rich fibers and microporous substrates decreases with increasing the molar mass of the polymer (Alince 1990). It has been suggested that such behavior is due to the saturation of the available charges throughout the fiber wall and due to the penetration of the low molar mass fraction PEI at the lower addition levels (Alince 1990). It has been shown that the external part of the fiber wall plays a very important role in creating strong joints between adjacent fibers and that treatment of the external part of the fibers is therefore essential for optimizing the use of modifying chemicals (Lindström et al. 2005). In addition to the treatment of fiber surfaces with single layers of polyelectrolytes and chemical modification of fiber wall (e.g., carboxymethylation (Wågberg et al. 2008)), polyelectrolyte multilayers have also been used to alter the chemistry of the external region of the fiber wall. Polyelectrolyte multilayers are formed by a technique called layer-by-layer (LbL) assembly, where the substrate is treated by consecutively alternating the adsorption of cationic and anionic polyelectrolytes with an intermediate rinsing step between each adsorption (Decher 1997). Wågberg et al. (2002) have studied how such multilayer formation on fibers enhances the dry and wet strength properties of paper made from the modified fibers. Further studies have shown that LbL treatment of fibers can enhance the specific joint strength by increasing the molecular interaction in the fiber–fiber joint (Eriksson et al. 2006; Torgnysdotter and Wågberg 2004). Marais et al. (2014) investigated the build-up of cationic PEI and colloidally stable anionic cellulose nanofibrils in order to study the mechanical properties of paper sheets prepared from treated fibers and showed that the outer layer of the thin film was an important parameter. The LbL deposition onto fibers has also been exploited to impart functionalities such as hydrophobicity (Gustafsson et al. 2012) and electrical conductivity, (Agarwal et al. 2006) and it has been shown that modified fibers can indeed be used for the preparation of new and advanced products. In addition to the poor wet strength of paper and the moisture sensitivity of the cellulose fibers, the highly flammable character of cellulosic fibers obtained from wood is a significant drawback which limits the application of paper-based materials in advanced applications. Paper-based materials are nevertheless used in a large variety of applications such as packaging products and wallpaper, although their flammable character is a possible risk for ignition and/or propagation of fire. It has however been suggested that multilayer formation of polyelectrolytes and/or nanoparticles using the LbL assembly technique may be an alternative flame-retardant treatment for substrates such as cotton (Li et al. 2009, 2010; Malucelli et al. 2014), polyurethane foam (Laufer et al. 2012), plastic thin films (Alongi et al. 2014; Apaydin et al. 2013), polyethylene terephthalate (PET) fabrics (Carosio et al. 2011, 2012) and recently also for cellulose rich fibers (Koklukaya et al. 2015) and cellulose nanofibril-based wet-stable aerogels (Koklukaya et al. 2017). We have recently demonstrated that it is also possible to use the LbL technique to form multilayers on cellulose-rich fibers and reduce their inherent flammability. The use of intumescent thin films (20BL) consisting of chitosan and poly(vinylphosphonic acid) resulted in self-extinguishing behavior in horizontal flame testing (Koklukaya et al. 2015). However, the large number of layers needed to impart flame-retardant properties to the fibers with this chemical strategy would negatively affect the up-scalability of this strategy. It was, therefore, essential to develop an alternative strategy to achieve similar properties with much fewer layers.
In the present work, the LbL assembly technique has been employed to build up polyelectrolyte multilayers of PEI and sodium hexametaphosphate (SHMP) on cellulose fibers in an effort to enhance both the flame-retardant properties and strength properties of paper with a small number of deposition steps. Considering earlier work with PEI, it was essential to study the effects of two different molecular mass fractions of PEI adsorbed onto porous fibers. As far as the flame-retardant properties are concerned since PEI acts as a carbon source at elevated temperatures and generates a carbon-rich char layer. The anionic counterpart, SHMP acts as an acid source generating in situ phosphoric acid and favors char formation of both PEI and cellulose (Li et al. 2010, 2011; Zhang et al. 2013). To identify the most effective LbL system, it was necessary to conduct model experiments with well-defined model cellulose surfaces. The deposition conditions identified were then used to treat cellulose fibers prior to paper sheet preparation and to treat specially prepared wet-strength paper sheets after their preparation. The effect of molecular LbL treatment was, on a macroscopic scale, evaluated in terms of both the tensile strength properties of the papers formed from the treated fibers through mechanical testing and the flame-retardant properties of the papers in a set of flammability tests.
Experimental
Materials
Fibers
The fibers used in this investigation were dried, fully bleached, totally chlorine free (TCF) softwood kraft fibers from Östrand Mill, SCA, Sundsvall, Sweden. It was delivered as dry sheets which were disintegrated according to ISO 5263-1:2004 in deionized water. The disintegrated fibers were washed according to a previously described procedure in order to remove any unwanted metal ions and the carboxyl groups present on the fibers were then converted to the sodium form prior to LbL treatment (Wågberg and Björklund 1993). Domsjö dissolving grade never-dried fibers were also carboxymethylated according to a method described earlier (Wågberg et al. 2008). The degree of substitution (D.S) was calculated by conductometric titration (Katz and Beatson 1984) to be 0.026, which corresponds to a charge of 157 µeq/g. Following the carboxymethylation, the fibers were washed, the charges were converted to their sodium form and the dewatered fibers were stored in a refrigerator until use. The surface charges and total charges of the two fibers are shown in Table 1.
Polyelectrolytes
High molecular weight polyethylenimine (HMw-PEI) with a mass average molecular weight Mw = 750 000 Da and a number average molecular weight Mn = 60 000 Da and low molecular weight (LMw-PEI) with Mw = 2 000 Da and Mn = 1 800 Da were obtained from Sigma-Aldrich, Stockholm, Sweden. The HMw-PEI had a large polydispersity (i.e., PDI = 12.5). The charge densities of the polyelectrolyte solutions were determined by polyelectrolyte titration according to the previously described method (Terayama 1952). The charge densities of PEI at the chosen adsorption pH and salt concentration (i.e., pH 9 and 10 mM NaCl) were determined by polyelectrolyte titration and are reported in Table 2. Sodium hexametaphosphate (SHMP) (crystalline, +200 mesh) was obtained from Sigma-Aldrich, Stockholm, Sweden. The charge density of SHMP at the adsorption pH and salt concentration (i.e., pH 4 and 10 mM NaCl) was determined by polyelectrolyte titration and is reported in Table 2. HMw and LMw PEI were delivered as concentrated solutions and SHMP was delivered as a powder. All polyelectrolytes were used without further purification. The polyelectrolytes were dissolved in Milli-Q water (18.2 MΩ cm Milli-Q grade water Synergy 185, Millipore Bellerica, USA) overnight before use. A polyvinylamine (PVAm) with a molecular weight of 340 000 Da was used as anchoring layer for model cellulose surfaces and to prepare wet-strength paper sheets. It was supplied by BASF (Ludwigshafen, Germany) and it was dialyzed and freeze-dried prior to preparation 0.1 wt% solution. The charge density of PVAm at adsorption pH and the salt concentration (i.e., pH 8.5 and 10 mM NaCl) was determined by polyelectrolyte titration and is reported in Table 2. Monochloroacetic acid, methanol, isopropanol, ethanol, hydrochloric acid, sodium hydroxide and sodium chloride were all of analytical grade and were purchased from Merck, Stockholm Sweden.
Methods
Quartz crystal microbalance with dissipation
The multilayer film build-up was investigated using a quartz crystal microbalance equipment with dissipation (QCM-D) supplied by Q-sense AB (Göteborg, Sweden). The substrates used were AT-cut quartz crystals cleaned by sequential rinsing with Milli-Q water, ethanol and Milli-Q water and plasma treated for 3 min. Model cellulose surfaces were prepared on cleaned crystals, using PVAm as anchoring layer prior to spin coating according to the procedure described by Gunnars et al. (2002). LbLs were prepared on the model cellulose surface by consecutive treatment with cationic polymer solution and anionic polymer solution, with an intermediate rinsing step, with a continuous flow of 150 µL/min and the adsorption was monitored in real time in situ by QCM-D. Rodahl et al. (1995) have described the theoretical foundation for the QCM-D procedure, where the change in the normalized frequency is proportional to the total adsorbed mass including the associated solvent. The total mass of the adsorbed film in an estimated flat and uniform conformation can be calculated using the Sauerbrey relation (Sauerbrey 1959).
Polyelectrolyte titration
Polyelectrolyte adsorption isotherms for PEI and SHMP on fibers were determined by polyelectrolyte titration of the filtrate after the adsorption. Fiber suspensions with 0.5% w/w concentration were prepared in Milli-Q ultrapure water, the pH of the suspension was adjusted according to the pH of added polyelectrolyte (i.e., pH 9 for PEI, and pH 4 for SHMP). The adsorption time for PEI and SHMP with different initial concentrations was 30 min. The residual amount of PEI in solution was determined by titration with potassium polyvinyl sulphate (KPVS, Wako Pure Chemical Industries, Osaka, Japan) in the presence of ortho-toluidine blue indicator (OTB, phenothiazine-5-ium-3-amino-7-dimethylamino-2-methyl chloride, VWR, Sweden). The pH and salt concentration of the titration solution were adjusted prior to titration (i.e., pH 9 and 10 mM NaCl). The colorimetric end point was detected with the optical two-beam method where the photoelectric detector unit (BASF AG, Ludwigshafen, Germany) was connected to an auto-titration unit (716 DMS Titrino, Metrohm AG, Herisau, Switzerland) for the addition of KPVS. The residual amount of SHMP in the filtrate solution was determined by polyelectrolyte titration using polydiallyldimethylammonium chloride (PDADMAC) (Sigma-Aldrich, Stockholm, Sweden), a rapid particle charge titration unit (Particle Metrix, Stabino, Meerbusch, Germany) being used to detect the end point. The adsorption saturations were calculated by extrapolating the isotherm to zero equilibrium concentrations as described elsewhere (Wågberg et al. 1989).
Atomic force microscopy
Atomic force microscopy (AFM), Nanoscope IIIa (Bruker AXS, Santa Barbara, CA, USA) was used to investigate the surface structure of the multilayer films deposited on model cellulose surfaces. E-type piezoelectric scanners and Scanassyst cantilevers with a nominal resonance frequency of 70 kHz and a 0.4 N/m spring constant were used to scan the surfaces in air. The surface roughness value was calculated from acquired images with an area of 2 × 2 µm2.
Handsheet preparation and mechanical testing
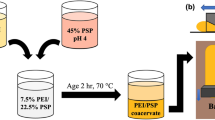
Laboratory sheets were prepared from LbL-treated and untreated fibers in order to investigate the effect of the multilayers on the flammability and mechanical properties of paper. Starting with cationic PEI, polyelectrolytes were added to a fiber suspension (0.3 wt%) in deionized water with 10 mM NaCl background electrolyte concentration. Fibers were washed after every layer deposition with deionized water at the same pH as the previously deposited polyelectrolyte. The polyelectrolyte addition was 30 mg/g of fiber and the adsorption time was 30 min. Paper sheets were prepared with the Rapid Köthen sheet preparation equipment (Paper Testing Instruments (PTI, Pettenbach, Austria) according to ISO 5269-2:2004. The sheets with a target grammage of 100 g/m2 were prepared from fiber dispersions that were vigorously agitated in air before sheet preparation. Sheets were then dried at 93 °C and under a pressure of 100 kPa. The wet-strength paper sheets were prepared in order to be able to handle the paper sheets without possible breakage during the LbL process. The wet-strength additive (PVAm) was used to modify fibers prior to sheet forming. The fiber suspension at 0.3 wt% concentration with 10 mM NaCl electrolyte concentration was adjusted to pH 8.5 and PVAm was added at a concentration of 10 mg/g fiber with an adsorption time of 30 min. After adsorption, sheets were prepared as described previously. The wet-strength papers were LbL-treated by sequential dipping in oppositely charged polyelectrolyte solutions starting with anionic SHMP solution since the PVAm-treated fibers had a positive surface potential. The adsorption time was 30 min and the rinsing time was 10 min. The LbL formation on the fibers and on papers is schematically shown in Fig. 1. The dried and conditioned sheets were tested at 23 °C and a relative humidity (RH) of 50% according to ISO 1924-3:2005. The grammage, thickness, and density were determined in accordance with SCAN C 28:76. The densities of the reference and LbL-treated sheets are shown in the Supplementary material, Fig. S1.

Schematic description of the layer-by-layer assembly on a fibers before sheet preparation, b wet-strength paper sheets
Nitrogen analysis
The nitrogen content due to the PEI adsorbed on the fibers was determined using a nitrogen analyzer ANTEK 7000 (Antek Instruments, Houston, TX, USA). The method is based on combustion of the sample (c.a. 10 mg) at 1050 °C in an oxygen-poor atmosphere where nitrogen is oxidized to NO before being further oxidized to excited NO2 in ozone. The light emitted when the excited NO2 is converted to its standard state is detected by a photomultiplier tube. Calibration with a known amount of PEI is used to calculate the amount of adsorbed PEI (Supplementary material, Fig. S2).
Thermal gravimetric analysis
The thermal degradation of the paper sheets produced from untreated and LbL-treated fibers and LbL-treated paper sheets was investigated by thermogravimetric analysis (TGA) (Mettler Toledo TGA/DSC, Stockholm, Sweden). The samples (10 ± 1 mg) were placed in 70 µL aluminum oxide crucibles and heated at a rate of 10 °C/min from 40 to 800 °C in nitrogen with a flow rate of 50 mL/min.
Flammability
The flame-retardant behavior of paper sheets prepared from LbL-treated and untreated fibers and LbL-treated paper sheets was investigated using flammability tests. The samples (30 × 100 mm2) were placed on a metal frame in horizontal and vertical configurations. The samples were ignited on the short side by a 20 mm methane flame for 3 s following a UL-94 standard for sample orientation and flame parameters.
Limiting oxygen index
The minimum concentration of oxygen in an oxygen/nitrogen mixture that supports a flaming combustion of the specimen was determined by limiting oxygen index (LOI). LOI tests were performed with a FIRE oxygen index apparatus following the ASTMD 2863-06 (2006) standard.
Scanning electron microscopy
A field emission scanning electron microscope (FE-SEM, Hitachi S-4800) was used to investigate the surface morphology of fibers before and after the LbL treatment. The residues from the flammability test were also investigated with FE-SEM to study the change in morphology after flame was applied. Test pieces were coated with a 5 nm thick platinum/palladium layer using a Cressington 208 HR high-resolution sputter coater.
Results
Adsorption of polyelectrolyte multilayers on model cellulose surfaces
To quantitatively determine the build-up of the LbLs, polyelectrolyte multilayers composed of cationic PEI and anionic SHMP were formed on the model cellulose surfaces. Figure 2 shows the results of the QCM-D measurements, where the normalized frequency shift and the change in the dissipation for the third overtone are shown after sequential deposition of two different molecular masses of PEI and SHMP at 10 mM NaCl salt concentration.

LbL build-up of (PEI/SHMP) 3.5 bilayers on model cellulose and model CM-cellulose surfaces from QCM-D measurements. a Change in normalized frequency, b change in energy dissipation and c total adsorbed mass of the thin film calculated using the Sauerbrey equation, including the immobilized solvent within the adsorbed layer
It has been well documented that the degree of dissociation and charge density of weak polyelectrolytes depend on the pH and salt concentration (Shiratori and Rubner 2000; Yoo et al. 1998). PEI shows an expanded conformation at lower pH values as a consequence of the strong repulsion between protonated amino groups but it has a lower charge density when the pH is increased and becomes uncharged above pH 11 (Alince et al. 1996; Allan et al. 1970). In the present study, PEI was deposited at pH 9 and SHMP at pH 4. The difference in pH of these polyelectrolytes alters the charge density of the previously adsorbed layer due to the sequential adsorption and the intermediate rinsing. Earlier studies showed a super-linear increase in adsorption in a system where polyallylamine hydrochloride (PAH) at pH 7.5 and poly(acrylic acid) (PAA) at pH 3.5 were adsorbed onto silicon oxide surfaces (Eriksson et al. 2005). During the build-up of multilayers on model cellulose and CM-cellulose surfaces, two different growth behaviors were observed. In the case of model cellulose surface, every cationic PEI layer adsorption led to a decrease in normalized frequency and an increase in dissipation which implies the formation of swollen layers with a visco-elastic behavior. On the other hand during the adsorption of anionic counterpart SHMP, except the first SHMP layer, all the SHMP layer additions showed an increase in frequency and a decrease in dissipation. This behavior can possibly be attributed to a partial removal of adsorbed cationic PEI layer but this possibility is very unlikely since the sequential adsorption of polyelectrolytes led to a continuous decrease in frequency. So the most likely explanation is that the change in the structure of the pre-adsorbed layers due to the release of associated water from the adsorbed layer which consequently forms a more collapsed and rigid film shown by the decrease in dissipation. For the CM cellulose surfaces the behavior is generally different. For the LMw-PEI there is an increase in frequency when the PEI is added, apart from the first layer, indicating that some of the SHMP is removed from the preadsorbed layer since no change is observed in the dissipation with this increase in frequency. For the HMw-PEI, the situation is different since the addition of PEI leads to small decreases in frequency but large increases in dissipation. This situation is very similar to what has been shown before for PAH/PAA multilayers which was ascribed to the creation of a less rigid film when the cationic polyelectrolyte was added (Notley et al. 2005). Due to this similarity it is suggested that for the combination of model CM-cellulose surfaces and HMw-PEI, the addition of PEI led to a less ordered film due to a change of the complexation of the polyelectrolytes within the film. The increase in frequency for the last layer of PEI addition indicates desorption of the preadsorbed polyelectrolyte, again showing the complexities of the molecular details for the formation of LbLs on these model surfaces. The results of the QCM measurements also show that for both surfaces the LMw-PEI systems show a very low dissipation regardless of the number of adsorbed layers, which indicates that these layers have a very close association with the surface, i.e. a very compact conformation. A greater adsorbed amount on the more highly charged cellulose surface (Fig. 2c) was also expected, since more PEI is needed to neutralize the charges on the surface. In the HMw-PEI systems, the adsorption follows the same trend as for the LMw-PEI systems, but the adsorption is significantly higher as expected for this polyelectrolyte (Fig. 2c) (Alince 1990; Alince et al. 1996; Allan et al. 1970; Lindquist and Stratton 1976; Petlicki and van de Ven 1994; van de Ven and Alince 1997). The dissipation values (Fig. 2b) show a large dependence on the polyelectrolyte in the external layer of the LbL, as has been reported earlier for the PAH/PAA system (Eriksson et al. 2005). The results in Fig. 2 show that the PEI/SHMP can be used to adsorb significant amounts of polyelectrolytes both on the model cellulose surface and on the model CM-cellulose surface and, with the strategies chosen for adsorption, a steady build-up on the fibers, corresponding to these model surfaces, can therefore be expected. It also shows that the ordering of the HMw-PEI layers is significantly dependent on the polymer in the most external layer.
AFM imaging
AFM, in tapping mode, was used to characterize the morphology and roughness of the LbL-assembled dry thin films on the model cellulose surfaces. The height images of PEI/SHMP thin films are shown in Fig. 3. The multilayer thin films were formed by sequential dipping of the model cellulose surfaces into a cationic PEI solution at pH 9 and an anionic SHMP solution at pH 4, both solutions having polymer concentration 1 g/L and an electrolyte concentration of 10 mM NaCl. The surfaces were rinsed between the layers first with Milli-Q water at the same pH as the previous solution and then with Milli-Q water at the same pH as the following solution. The rinsing solutions also contained 10 mM NaCl.

AFM tapping mode height images of (PEI/SHMP) 3.5 bilayers, i.e. PEI in the external layer, deposited on a a model cellulose surface and b a model CM-cellulose surface. The images are 2 × 2 µm2 and the z-range is indicated in the scale bar to the right of the images
The surface structure of the LbL-assembled multilayers was different for the different cellulose surfaces. When the multilayers were formed on the model cellulose surfaces, the structure of the model surface was basically preserved with LMw-PEI, whereas a slight increase in roughness was detected with the HMw-PEI. On the CM-cellulose surfaces, however, there is a much larger change in structure both for the LMw-PEI and especially for the HMw-PEI where both the size of the aggregates and the roughness on the surface were detected. The difference observed in AFM height images of model cellulose and CM-cellulose surfaces can possibly be attributed to the partial removal of the final layer of PEI which created a surface topography with different appearance and roughness upon consolidation of the thin film after drying. These results are in agreement with the adsorption results shown in Fig. 2. The roughness values calculated from the AFM measurements are summarized in Supplementary material, Table S1.
Adsorption of PEI and SHMP onto cellulose fibers
The adsorption capacity of each polyelectrolyte to the differently charged fibers was determined by polyelectrolyte titration. An equilibration time of 30 min was chosen for the different isotherms. Adsorption isotherms for the sequential addition of high and low molecular mass PEI and SHMP to cellulose fibers and carboxymethylated cellulose fibers were examined for the first two layers and summarized in Fig. 4.

Adsorption isotherms for two consecutive layers of PEI and SHMP adsorbed onto a cellulose fibers and b carboxymethylated cellulose fibers. Each series shows the adsorbed amount of each individual layer
The carboxymethylated cellulose fibers showed a significantly higher adsorption capacity than the cellulose fibers as expected from the model experiments. The amount of HMw-PEI adsorbed onto the cellulose fibers was larger than the amount of LMw-PEI adsorbed. However, in the case of CM-cellulose fibers, the amounts of LMw-PEI and HMw-PEI adsorbed were similar. It should also be noticed that no true plateau was reached for several of the adsorption isotherms and it was not therefore possible to extrapolate each adsorption isotherm to zero residual concentration in a simple way. Figure 5 shows the total amount of PEI adsorbed on the cellulose and CM-cellulose fibers after the adsorption of 3.5 BL thin film determined using nitrogen analysis. About 17 mg/g of LMw-PEI and 33 mg/g HMw-PEI were adsorbed in total when cellulose fibers were treated with 7 layers. In contrast, 54 mg/g of LMw-PEI and 75 mg/g of HMw-PEI were adsorbed with the same number of layers on the CM-cellulose fibers. It can be suggested that there is a direct relationship between the amount of PEI adsorbed and the charge density of the fibers as expected. It is also evident that the LbLs can be steadily built on the fibers and it is, therefore, possible to increase the adsorbed amount by increasing the number of layers if necessary.

The amounts of PEI adsorbed to the cellulose fibers and CM-cellulose fibers after LBL-treatment with 3.5 BL of LMw-PEI/SHMP and HMw-PEI/SHMP determined by nitrogen analysis. The error bars represent the standard deviations based on three measurements
Thermal stability and flame-retardant properties of paper from treated and untreated fibers
The thermal stability of paper sheets prepared from untreated and LbL-treated fibers was investigated by thermogravimetric analysis under nitrogen. Table 3 shows the collected data.
The weight loss and the derivative of the weight loss are shown as functions of temperature in Figs. 6 and 7, respectively. It is well documented that cellulose undergoes thermal degradation by a single step under nitrogen atmosphere. Pyrolysis results in two competitive paths: depolymerization leading to low molecular weight, flammable volatiles (mainly levoglucosan) and decomposition of the glycosyl units leading to thermally stable char (Alongi et al. 2013). As shown in Fig. 6a, all employed reference show this degradation behavior with nearly identical final residues. The paper sheets prepared from LbL-treated cellulose fibers and the LbL-treated wet-strength paper sheet all showed a reduction in the cellulose degradation temperature, as shown by the Tonset10% values in Table 3. The reduction was greater for the paper sheet prepared with HMw-PEI treated cellulose fibers. The final residue at the end of the degradation was significantly higher for the paper sheets prepared from LbL-treated cellulose fibers than for these from the LbL-treated paper sheet.

Weight loss of a different reference samples, b paper prepared from (PEI/SHMP) 3.5 BL treated cellulose fibers, c paper prepared from (PEI/SHMP) 3.5 BL treated carboxymethylated fibers, and d (PEI/SHMP) 3.5 BL treated wet-strength paper. The TGA measurements were performed under N2 atmosphere at a heating rate of 10 °C/min

Derivative of weight loss of a different reference samples, b paper prepared from (PEI/SHMP) 3.5 BL treated cellulose fibers, c paper prepared from (PEI/SHMP) 3.5 BL treated carboxymethylated cellulose fibers, and d (PEI/SHMP) 3.5 BL treated wet-strength paper. The TGA measurements were performed under N2 atmosphere at a heating rate of 10 °C/min
Paper sheets prepared from untreated fibers, LbL-treated fibers and LbL-treated carboxymethylated fibers were compared with the wet-strength paper sheets prepared from PVAm-treated fibers which were further LbL-treated by sequential dipping into the respective polyelectrolyte solutions. Paper sheets were subjected to flammability tests in both horizontal and vertical configurations in order to investigate their flame-retardant properties. This flammability test is one of the most usual tools for fire safety evaluation and provides information on the ability of the material to propagate a fire by studying the reaction to a direct flame. In addition, LOI tests have been performed. This test evaluates the minimum oxygen concentration needed to sustain combustion; an increase in the LOI value indicates an FR effect. Figure 8 shows the residue of each sample and flammability data is shown in Table 4. Untreated paper sheets immediately ignited and they were completely consumed by the advancing flame and eventually by flameless combustion (normally defined as afterglow) in less than 1 min. On the other hand, all the LbL-treated samples were able to suppress the afterglow phenomenon and increase the amount of residue at the end of the test. Paper sheets prepared from HMw-PEI treated fibers were also able to self-extinguish in the horizontal flame test. This behavior is reflected by three points increase in LOI values as reported in Table 4. On the other hand, LbL-treated wet-strength paper did not show similar improvements regardless of the adopted molecular weight.

Photographs taken after the flammability tests of paper sheets prepared from (PEI/SHMP) 3.5 BL treated cellulose fibers, CM-cellulose fibers and (PEI/SHMP) 3.5 BL treated wet-strength paper. a Vertical flame test and b horizontal flame test
Morphological analysis
Scanning electron microscopy (SEM) was used to investigate the surface morphology of the fibers before and after both carboxymethylation and LbL assembly. Micrographs of untreated and LbL-treated fibers are shown in Fig. 9 and Fig. S3 shows the SEM images of cellulose fibers and CM-cellulose fibers. The external microstructure of fibers showed no significant difference after carboxymethylation. The high magnification images of untreated and LbL-treated fibers show an almost unaltered morphology thus point out the deposition of a very thin film considering the rather low adsorbed amounts.

SEM images of fibers before and after LbL treatment with 3.5 BL of (PEI/SHMP). a Cellulose fibers, b CM-cellulose fibers and c wet-strength paper. Inset images show the high magnification of corresponding fibers (The scale bar of inset is 10 µm)
The residues from the flammability tests were also investigated by SEM in order to monitor structural changes of fibers upon exposure to flame and the images are shown in Fig. 10. Untreated fibers were completely consumed by a flame that left no intact material to be investigated. Paper sheets prepared from LbL-treated fibers with LMw-PEI showed an apparent shrinkage of the fiber network, but the original texture and shape of the fibers were preserved for the fibers treated with HMw-PEI. The formation of micrometer-size bubbles typical for intumescent coatings was observed for the fibers treated with HMw-PEI but no such structures were detected in the case of the LMw-PEI-treated fibers. LbL-treated wet-strength paper sheets showed slight dimensional change after the flame exposure, but this change was less pronounced for the papers coated with HMw-PEI.

SEM images of residues of (PEI/SHMP) 3.5 BL treated paper sheets after the horizontal flame test. a Cellulose fibers, b CM-cellulose fibers and c wet-strength paper. LMw-PEI/SHMP treated (left) and HMw-PEI/SHMP treated (right) fibers
Mechanical properties of hand sheets
The tensile strength properties of sheets made of untreated cellulose fibers and CM-cellulose fibers were compared to those of sheets made from the LbL-treated fibers, and the strength properties of sheets made of LbL-treated fibers were also compared to those of LbL-treated wet-strength paper sheets. Figure 11 shows the tensile index and strain at break values. The sheet with LbL-treated fibers containing HMw-PEI showed the greatest increase in both tensile index and strain at break, but the LbL-treated fibers containing LMw-PEI showed no increase. PVAm treatment increased the tensile index of the sheet and LbL-treatment of wet-strength paper sheets showed a similar increase in the tensile index for both high and low molecular mass PEI. The tensile index increased from 18 Nm/g to 36 Nm/g and the strain at break from 1.1 to 3% when the paper sheet was prepared from LbL-treated cellulose fibers containing HMw-PEI. For the paper sheets prepared from HMw-PEI treated CM-cellulose fibers the tensile index increased from 13 Nm/g to 20 Nm/g and the strain at break from 0.9 to 1.6%. It is also worth noticing that in order to avoid the influence of fines on the LbL treatment, no beating of the fibers was used in the present work.

a Tensile index and b strain at break values for papers prepared from untreated and (PEI/SHMP) 3.5 BL treated cellulose fibers, CM-cellulose fibers, and wet-strength paper
Discussion
Adsorption of multilayers onto model cellulose surfaces and fibers
The adsorption of multilayers onto model cellulose surfaces, as investigated by monitoring the change in frequency of the oscillating QCM-D crystal, was compared with the amount adsorbed onto fibers, as determined from adsorption saturation curves obtained by polyelectrolyte titration. In this respect, it is important to note that, although model studies provide molecular information about the adsorption process and the properties of the adsorbed layers, a direct quantitative comparison with the adsorption to fibers cannot be made and the results are hence complementary to each other. Indeed, while the model surface can mimic the surface chemistry of the real fiber, it cannot represent the internal porous structure that plays an important role for polyelectrolyte adsorption to the fibers, even though the properties of the formed layers are probably the same regardless of where they are situated inside the fiber wall. Together with the AFM results in Fig. 3, these QCM-D measurements, i.e. higher adsorbed amounts/m2 and clear changes in dissipation upon the formation of the different layers, show that only the HMw-PEI is able to form a continuous film on the cellulose surfaces with the wet thickness of c.a 17 nm which is calculated with the aid of Sauerbrey equation (Sauerbrey 1959) under the assumption of a layer density of 1000 kg/m3(Enarsson and Wågberg 2008). The location of the polyelectrolyte film on the fibers is highly dependent on the molecular mass of the polyelectrolyte and on the swelling and pore structure of the fiber wall. In Fig. 4, it is clear that the HMw-PEI and LMw-PEI exhibit a very similar adsorption on the two types of porous fibers, despite the large difference in molecular mass and, for the non-modified fibers, the adsorption of the HMw is larger than that of the LMw-PEI, which is in agreement with the QCM adsorption measurements. These results thus indicate that, despite the low molecular mass of the PEI, it is not able to penetrate into the porous fiber wall of the non-modified fibers and that the LbLs are formed only on the external layers of these non-beaten fibers for both the LMw and the HMw. In the case of the carboxymethylated fibers, the adsorbed amount of PEI is similar for the two molecular mass polymers and this indicates that the LMw polymer can penetrate into the fiber wall to give a higher adsorbed amount. The adsorption of the HMw is also much larger that on the non-modified fibers but, considering that both types of polymers have similar adsorbed amounts, it appears that the HMw is mostly restricted to the external surfaces of the fibers. This also means that the LbLs formed with the HMw will be thicker than the non-modified fibers and that the LbLs containing LMw-PEI will be partly found inside the fibers due to the higher degree of swelling of CM-cellulose fibers. The adsorption of the SHMP onto the preadsorbed layers of PEI shows that a significant amount of this anionic analogue can be adsorbed onto the fibers and, especially in the case of the HMw, the increase compared to the non-modified fibers is large, which is in agreement with the data in Fig. 2 showing the formation of thicker wet layers of the polyelectrolytes on the external surface of the fibers. These conclusions regarding the properties and location of the LbLs are based on a combination of QCM-D measurements and adsorption to fibers. All the adsorption isotherms have a rounded shape, which is typically observed for polydisperse polymers and usually attributed to competitive adsorption of the high and low molecular mass polymers (Stuart et al. 1980). This makes the evaluation of the saturation adsorption more difficult but we consider the general conclusions drawn from a combination of the QCM and fiber adsorption to be adequate to describe how and where the LbLs are deposited on or within the fibers.
Thermal stability and flame-retardant properties
PEI/SHMP treated fibers had a lower decomposition temperature than paper sheet prepared with untreated fibers, as shown by the Tonset10% and Tmax temperatures where 10% of the substrate was degraded and the maximum weight loss was observed respectively. This reduction in degradation temperature can be ascribed to the presence of phosphorus containing groups within the multilayer coating. It is well documented that such polymers promote the in situ formation of phosphoric acid that leads to an earlier thermal degradation with a substantial increase in the formation of a thermally stable carbonaceous structure (Horrocks 2011). A greater reduction was observed with HMw-PEI treatment on both cellulose fibers and CM-cellulose fibers. This can be expected since the adsorbed amount of SHMP is higher in the case of HMw-PEI and since the film formation with this LbL combination is much more complete than with the other combinations. The residue at 800 °C was greater for HMw-PEI/SHMP treated samples than for the untreated samples and a stable char was formed. However, despite the decrease in the Tonset10% and Tmax temperatures, the LbL-treated wet-strength paper did not show an increase in the amount of residue, probably due to an insufficient coverage of the substrate, since the interfiber contacts could not be covered by the surface treatment approach. In the flammability tests, the LbL treatment of fibers and wet-strength paper showed a reduction in the burning time of the samples, and the LbL treatment on both fibers and wet-strength papers was able to eliminate the after-glow phenomenon. During VFT, untreated samples were completely consumed but the LbL-treated samples, prepared from LbL-treated fibers, formed a coherent residue. The presence of SHMP favored the char-forming ability of cellulose and this effect was greater for the coating consisting of HMw-PEI/SHMP which generated a char barrier capable of suppressing and extinguishing the propagating flame during HFT. High magnification SEM images showed the formation of micro-bubbles on the fiber surface as a result of carbonaceous layer formation and the complete film formation on the fibers in the case of the HMw-PEI/SHMP combination. The fibers maintained their original texture and structure after the HFT. In contrast, fibers treated with LMw-PEI/SHMP appeared to be partially protected since a shrinkage and dimensional change of the fibers were observed. Cellulose fibers and CM-cellulose fibers showed similar behavior in flammability tests. On the other hand, no self-extinguishing was observed with the same number of bilayers on the wet-strength paper sheets. These results are linked to the better film formation of PEI and SHMP with HMw-PEI and a larger amount of SHMP adsorbed within the film, as demonstrated by the model adsorption studies and by the thermal analysis. LOI values showed an increase of 20–25% for HMw-PEI treated cellulose fibers and of 21–25% for HMw-PEI treated CM-cellulose fibers. The fibers treated with LMw-PEI showed no improvement in LOI value. These results suggest that the coating efficiency is greater when HMw-PEI is used, as observed in the flammability tests, whereas LbL-treated wet-strength paper showed no increase in the LOI test. LbL treatment of the fibers in suspension prior to sheet making resulted in a better overall film formation on the entire fibers than LbL treatment of the paper sheet.
Influence of PEI/SHMP multilayers on strength properties of paper
Figure 11 shows that the tensile index was higher when HMw-PEI was the outermost layer on both the cellulose fibers and the CM-cellulose fibers, probably because the low molecular mass polymer chains of PEI penetrate into the fiber wall, at least when the fibers are more highly charged. The polymer entanglements between the LMw-PEI and SHMP polymers are lower, giving no improvements in the fiber joint strength. On the other hand, in the high molecular mass PEI/SHMP system, the high molecular mass polymer chains are adsorbed onto the fiber wall giving both an anchoring layer on the fiber and an excellent possibility for intermixing of polyelectrolytes across the adsorbed layers. The effect on the final paper material of an LbL treatment of fibers has previously been investigated, and this showed that fiber joint strength was responsible for the increase in the mechanical properties (Eriksson et al. 2006). Figure 2b shows the change in dissipation upon PEI/SHMP multilayer formation. Previous studies have shown that having an anionic polyelectrolyte in the outer layer of thin film results in a compact and rigid film (Notley et al. 2005), but having HMw-PEI in the outer layer results in a film which is associated with more coupled water. In contrast, LMw-PEI showed no increase in dissipation and the increase in mechanical properties was also very small. Notley et al. (Notley et al. 2005) demonstrated that viscoelastic thin films associated with water make it easier for the adjacent fibers to come into close contact, and that this increases the probability of the molecules migrating across the interface between the fibers, enhancing the fiber–fiber joint strength and the strength of the paper. Thus the presence of HMw-PEI as the outermost layer and the associated water content can be considered to be the main factors that plasticize the multilayer and hence contribute to the formation of stronger fiber–fiber joints in the wet state so that the LbL-assembled coatings were stronger after drying and consolidation of the paper. McKenzie and Pelton et al. investigated the formation of fiber–fiber joints as a diffusion/interpenetration mechanism (McKenzie 1984; Pelton et al. 2000). It can thus be suggested that the intermixing of polymer at the fiber–fiber interface increases the molecular interaction between the fibers upon drying.
Conclusions
This investigation has shown a clear positive effect of the adsorption of intumescent flame-retardant constituents (i.e., PEI and SHMP) onto lignocellulosic fibers on the fire-retardant properties of papers formed from the LbL-treated fibers. Thermogravimetric analysis in nitrogen atmosphere revealed an increased amount of residue at 800 °C suggesting the enhanced char forming ability of LbL-treated fibers. The LbL treatment significantly altered the flammability of the paper sheet. During the horizontal flame test, a paper sheet prepared from HMw-PEI and SHMP treated fibers showed self-extinguishing behavior 15 s after the flame application, in contrast to the untreated paper sheet which was completely consumed by the flame in 56 s. After the vertical flame test, all the LbL-treated paper sheets showed the formation of a coherent and intact char layer on the fiber. This effect was more distinct with HMw-PEI/SHMP treated carboxymethylated fibers. The increase in LOI value also showed the enhanced flame-retardant properties after LbL treatment, where more oxygen is needed to support the flame propagation. This was attributed to the complete film formation with this LbL combination, especially for the HMw-PEI/SHMP combination, as demonstrated by the model QCM-D studies and the AFM investigations, when the amount of SHMP in the adsorbed layer was higher. The mechanical strength properties of paper sheets prepared from LbL-treated fibers was significantly higher, particularly in the case of cellulose fibers with HMw-PEI. In all cases, HMw-PEI/SHMP led to a greater improvement than LMw-PEI/SHMP. These findings, achieved with only 3.5 BL deposited, suggest that the LbL technique is an effective and simple method of modifying the surface properties of fibers to reduce the flammable character of wood fibers while strengthening the mechanical properties of the paper material.
References
Agarwal M, Lvov Y, Varahramyan K (2006) Conductive wood microfibres for smart paper through layer-by-layer nanocoating. Nanotechnology 17:5319-5325
Alince B (1990) The role of porosity in polyethylenimine adsorption onto cellulosic fibers. J Appl Polym Sci 39:355–362
Alince B, Vanerek A, van de Ven T (1996) Effects of surface topography, pH and salt on the adsorption of polydisperse polyethylenimine onto pulp fibers Berichte der Bunsengesellschaft für physikalische. Chemie 100:954–962
Allan G, Akagane K, Neogi A, Reif W, Mattila T (1970) Physical entrapment of polyelectrolytes within microporous solids: the “jack-in-the-box” effect. Nature 225:175–176
Alongi J, Camino G, Malucelli G (2013) Heating rate effect on char yield from cotton, poly (ethylene terephthalate) and blend fabrics. Carbohydr Polym 92:1327–1334
Alongi J, Di Blasio A, Carosio F, Malucelli G (2014) UV-cured hybrid organic–inorganic layer by layer assemblies: effect on the flame retardancy of polycarbonate films. Polym Degrad Stab 107:74–81
Apaydin K, Laachachi A, Ball V, Jimenez M, Bourbigot S, Toniazzo V, Ruch D (2013) Polyallylamine–montmorillonite as super flame retardant coating assemblies by layer-by layer deposition on polyamide. Polym Degrad Stab 98:627–634
Carosio F, Laufer G, Alongi J, Camino G, Grunlan JC (2011) Layer-by-layer assembly of silica-based flame retardant thin film on PET fabric. Polym Degrad Stab 96:745–750
Carosio F, Alongi J, Malucelli G (2012) Layer by layer ammonium polyphosphate-based coatings for flame retardancy of polyester–cotton blends. Carbohydr Polym 88:1460–1469
Decher G (1997) Fuzzy nanoassemblies: toward layered polymeric multicomposites. Science 277:1232–1237
Enarsson L-E, Wågberg L (2007) Kinetics of sequential adsorption of polyelectrolyte multilayers on pulp fibres and their effect on paper strength. Nord Pulp Pap Res J 22:258–266
Enarsson L-E, Wågberg L (2008) Adsorption kinetics of cationic polyelectrolytes studied with stagnation point adsorption reflectometry and quartz crystal microgravimetry. Langmuir 24:7329–7337
Eriksson M, Notley SM, Wågberg L (2005) The influence on paper strength properties when building multilayers of weak polyelectrolytes onto wood fibres. J Colloid Interface Sci 292:38–45
Eriksson M, Torgnysdotter A, Wågberg L (2006) Surface modification of wood fibers using the polyelectrolyte multilayer technique: effects on fiber joint and paper strength properties. Ind Eng Chem Res 45:5279–5286
Fleer G, Stuart MC, Scheutjens J, Cosgrove T, Vincent B (1993) Polymers at interfaces. Springer, Berlin, pp 354-367
Gunnars S, Wågberg L, Cohen Stuart M (2002) Model films of cellulose: I. Method development and initial results. Cellulose 9:239–249
Gustafsson E, Larsson PA, Wågberg L (2012) Treatment of cellulose fibres with polyelectrolytes and wax colloids to create tailored highly hydrophobic fibrous networks. Colloids Surf A 414:415–421
Haggkvist M, Li T-Q, Odberg L (1998) Effects of drying and pressing on the pore structure in the cellulose fibre wall studied by 1H and 2H NMR relaxation. Cellulose 5:33–49
Horrocks AR (2011) Flame retardant challenges for textiles and fibres: new chemistry versus innovatory solutions. Polym Degrad Stab 96:377–392
Horvath AT, Horvath AE, Lindström T, Wågberg L (2008) Diffusion of cationic polyelectrolytes into cellulosic fibers. Langmuir 24:10797–10806
Katz S, Beatson RP (1984) The determination of strong and weak acidic groups in sulfite pulps. Svensk Papperstidning 87:48–53
Koklukaya O, Carosio F, Grunlan JC, Wagberg L (2015) Flame-retardant paper from wood fibers functionalized via layer-by-layer assembly. ACS Appl Mater Interfaces 7:23750–23759
Koklukaya O, Carosio F, Wågberg L (2017) Superior flame-resistant cellulose nanofibril aerogels modified with hybrid layer-by-layer coatings. ACS Appl Mater Interfaces 9:29082–29092
Laufer G, Kirkland C, Cain AA, Grunlan JC (2012) Clay–chitosan nanobrick walls: completely renewable gas barrier and flame-retardant nanocoatings. ACS Appl Mater Interfaces 4:1643–1649
Li Y-C, Schulz J, Grunlan JC (2009) Polyelectrolyte/nanosilicate thin-film assemblies: influence of pH on growth, mechanical behavior, and flammability. ACS Appl Mater Interfaces 1:2338–2347
Li Y-C, Schulz J, Mannen S, Delhom C, Condon B, Chang S, Zammarano M, Grunlan JC (2010) Flame retardant behavior of polyelectrolyte-clay thin film assemblies on cotton fabric. ACS Nano 4:3325–3337
Li Y-C, Mannen S, Morgan AB, Chang S, Yang YH, Condon B, Grunlan JC (2011) Intumescent all-polymer multilayer nanocoating capable of extinguishing flame on fabric. Adv Mater 23:3926–3931
Lindquist GM, Stratton RA (1976) The role of polyelectrolyte charge density and molecular weight on the adsorption and flocculation of colloidal silica with polyethylenimine. J Colloid Interface Sci 55:45–59
Lindström T, Florén T (1984) The effects of cationic starch wet end addition on the properties of clay filled papers [retention, polyacrylics, acrylamide, potato starch]. Svensk Papperstidning 87:R99–R104
Lindström T, Wågberg L, Larsson T (2005) On the nature of joint strength in paper-a review of dry and wet strength resins used in paper manufacturing. In: 13th fundamental research symposium. The Pulp and Paper Fundamental Research Society Cambridge, UK, pp 457–562
Malucelli G, Carosio F, Alongi J, Fina A, Frache A, Camino G (2014) Materials engineering for surface-confined flame retardancy. Mater Sci Eng, R 84:1–20
Marais A, Utsel S, Gustafsson E, Wågberg L (2014) Towards a super-strainable paper using the layer-by-layer technique. Carbohydr Polym 100:218–224
McKenzie A (1984) The structure and properties of paper. XXI: the diffusion theory of adhesion applied to interfibre bonding. Appita 37:580–583
Notley SM, Eriksson M, Wågberg L (2005) Visco-elastic and adhesive properties of adsorbed polyelectrolyte multilayers determined in situ with QCM-D and AFM measurements. J Colloid Interface Sci 292:29–37
Pelton R, Zhang J, Wågberg L, Rundlöf M (2000) The role of surface polymer compatibility in the formation of fiber/fiber bonds in paper. Nord Pulp Pap Res J 15:400–406
Petlicki J, van de Ven T (1994) Adsorption of polyethylenimine onto cellulose fibers. Colloids Surf A 83:9–23
Rathi MS, Biermann CJ (2000) Application of polyallylamine as a dry strength agent for paper. Tappi J 83:62
Rodahl M, Höök F, Krozer A, Brzezinski P, Kasemo B (1995) Quartz crystal microbalance setup for frequency and Q-factor measurements in gaseous and liquid environments. Rev Sci Instrum 66:3924–3930
Sauerbrey G (1959) Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Z Physik 155:206–222
Shiratori SS, Rubner MF (2000) pH-dependent thickness behavior of sequentially adsorbed layers of weak polyelectrolytes. Macromolecules 33:4213–4219
Stone J, Scallan A (1968) The effect of component removal upon the porous structure of the cell wall of wood Part III. A comparison between the sulphite and kraft processes. Pulp Paper Mag Can 69:T288
Stuart MC, Scheutjens JM, Fleer GJ (1980) Polydispersity effects and interpretation of polymer adsorption isotherms. J Polym Sci Polym Phys Ed 18:559-573
Stuart MC, Fleer G, Lyklema J, Norde W, Scheutjens J (1991) Adsorption of ions, polyelectrolytes and proteins. Adv Colloid Interface Sci 34:477–535
Terayama H (1952) Method of colloid titration (a new titration between polymer ions). J Polym Sci 8:243–253
Torgnysdotter A, Wågberg L (2004) Influence of electrostatic interactions on fibre/fibre joint and paper strength. Nord Pulp Pap Res J 19:440–447
Trout PE (1951) The mechanism of the improvement of the wet strength of paper by polyethylenimine. Tappi 34:539
van de Ven TGM (2000) A model for the adsorption of polyelectrolytes on pulp fibers: relation between fiber structure and polyelectrolyte properties. Nord Pulp Pap Res J 15:494–501
van de Ven TGM, Alince B (1997) Porosity of swollen pulp fibers evaluated by polymer adsorption, Paper presented at the Fundamentals of Papermaking, Transactions of the 11th Research Symposium held at Cambridge, Leatherhead, U.K., pp 771-788
Wågberg L, Annergren G (1997) Physicochemical characterization of papermaking fibres, Fundamentals of Papermaking. Paper presented at the Transactions of the 11th Research Symposium held at Cambridge, Leatherhead, UK, pp 1-82
Wågberg L, Björklund M (1993) Adsorption of cationic potato starch on cellulosic fibres. Nord Pulp Paper Res J 8:399-404
Wågberg L, Hägglund R (2001) Kinetics of polyelectrolyte adsorption on cellulosic fibers. Langmuir 17:1096–1103
Wågberg L, Forsberg S, Johansson A, Juntti P (2002) Engineering of fibre surface properties by application of the polyelectrolyte multilayer concept. Part I: modification of paper strength. J Pulp Pap Sci 28:222–228
Wågberg L, Winter L, Ödberg L, Lindström T (1987) On the charge stoichiometry upon adsorption of a cationic polyelectrolyte on cellulosic materials. Colloids Surf 27:163–173
Wågberg L, Ödberg L, Lindström T, Aksberg R (1988) Kinetics of adsorpton and ion-exchange rections during adsorption of cationic polyelectrolytes onto cellulosic fibers. J Colloid Interface Sci 123:287–295
Wågberg L, Ödberg L, Glad-Nordmark G (1989) Charge determination of porous substrates by polyelectrolyte adsorption. Nord Pulp Pap Res J 4:71-76
Wågberg L, Decher G, Norgren M, Lindström T, Ankerfors M, Axnäs K (2008) The build-up of polyelectrolyte multilayers of microfibrillated cellulose and cationic polyelectrolytes. Langmuir 24:784–795
Winter L, Wågberg L, Ödberg L, Lindström T (1986) Polyelectrolytes adsorbed on the surface of cellulosic materials. J Colloid Interface Sci 111:537–543
Xie F, Lu H, Nylander T, Wågberg L, Forsman J (2016) Theoretical and experimental investigations of polyelectrolyte adsorption dependence on molecular weight. Langmuir 32:5721–5730
Yoo D, Shiratori SS, Rubner MF (1998) Controlling bilayer composition and surface wettability of sequentially adsorbed multilayers of weak polyelectrolytes. Macromolecules 31:4309–4318
Zhang T, Yan H, Wang L, Fang Z (2013) Controlled formation of self-extinguishing intumescent coating on ramie fabric via layer-by-layer assembly. Ind Eng Chem Res 52:6138–6146
Acknowledgements
Lars Wågberg, Oruç Köklükaya and Federico Carosio gratefully acknowledge financial support from The Swedish Foundation for Strategic Research (SSF) and Lars Wågberg also acknowledges The Wallenberg Wood Science Centre for financial support.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Köklükaya, O., Carosio, F. & Wågberg, L. Tailoring flame-retardancy and strength of papers via layer-by-layer treatment of cellulose fibers. Cellulose 25, 2691–2709 (2018). https://doi.org/10.1007/s10570-018-1749-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-1749-8