
Abstract
We report the catalytic activity of colloid platinum nanoparticles synthesized with different organic capping layers. On the molecular scale, the porous organic layers have open spaces that permit the reactant and product molecules to reach the metal surface. We carried out CO oxidation on several platinum nanoparticle systems capped with various organic molecules to investigate the role of the capping agent on catalytic activity. Platinum colloid nanoparticles with four types of capping layer have been used: TTAB (Tetradecyltrimethylammonium Bromide), HDA (hexadecylamine), HDT (hexadecylthiol), and PVP (poly(vinylpyrrolidone)). The reactivity of the Pt nanoparticles varied by 30%, with higher activity on TTAB coated nanoparticles and lower activity on HDT, while the activation energy remained between 27 and 28 kcal/mol. In separate experiments, the organic capping layers were partially removed using ultraviolet light-ozone generation techniques, which resulted in increased catalytic activity due to the removal of some of the organic layers. These results indicate that the nature of chemical bonding between organic capping layers and nanoparticle surfaces plays a role in determining the catalytic activity of platinum colloid nanoparticles for carbon monoxide oxidation.
Similar content being viewed by others


1 Introduction
Recent advances in colloidal chemistry have enabled catalytic nanoparticles to be synthesized with tunable particle sizes, shapes, and compositions [1–5]. Consequently, the model catalytic systems generated from colloidal nanoparticles enable quantitative investigations into the effects of these factors on catalytic activity and selectivity [6]. The colloid nanoparticles are capped with stabilizing agents in order to prevent aggregation while in the liquid suspension. On the molecular scale, the porous organic capping layers have open spaces that permit reactant and product molecules to reach the metal catalysts. The degree of access of reactant and product molecules to the active sites in the metal catalyst depends on geometric factors such as length, compactness of the organic layers, and the nature of chemical bonding between the organic molecules and metal sites. It is also known that some elements, such as sulfur, block reactive sites when bonded to the catalyst, thus reducing the overall catalytic activity [7]. Overall, the role of the capping agent in catalytic activity and selectivity is not yet well-understood in spite of its importance to model nanoparticle systems for studying heterogeneous catalysis.
One approach to address the influence of capping layers on the catalytic reaction involves the measurement of catalytic activity after the removal of capping layers. It was found that the organic capping molecules can be decomposed and removed using a UV light irradiation that generates ozone in air (or simply UV/ozone treatment) [8]. Aliaga et al. [9] found that sum frequency generation (SFG) vibrational spectrum under ethylene hydrogenation measured on colloid Pt nanoparticles after UV-ozone treatment revealed the presence of adsorbed ethylidyne and di-σ-bonded reaction intermediate species, suggesting that UV-ozone effectively removes the capping layers.
In this study, we carried out CO oxidation on several platinum nanoparticle systems capped with various organic molecules to elucidate the role of the capping agent on catalytic activity. Platinum colloid nanoparticles with four types of capping layers have been used: TTAB (Tetradecyltrimethylammonium Bromide), HDA (hexadecylamine), HDT (hexadecylthiol), and PVP (poly(vinylpyrrolidone)). We show that the catalytic activity under CO oxidation with partial pressures of 100 Torr O2 and 40 Torr CO for UV-ozone treated Pt nanoparticles is higher compared to untreated nanoparticles, which suggests that some portion of the capping layers block the reactive sites and influence the catalytic turnover rate.
2 Experimental
2.1 Synthesis of TTAB, HDA, HDT and PVP Capped Nanoparticles
In this study, we used platinum colloid nanoparticles with four types of capping layers: TTAB, HDA, HDT, and PVP. TTAB capped nanoparticles were synthesized as reported previously [10]. Briefly, 1 mM aqueous K2PtCl4 in 100 mM TTAB was reduced by 30 mM NaBH4 at 50 °C. Excess H2 evolving from the reacting solution was released by inserting a needle into the septum. After 7 h, the reaction was allowed to cool to room temperature and left overnight to decompose the remaining NaBH4 in water. The Pt nanoparticles were collected and washed by repeated centrifugation and sonication. TTAB stabilized Pt nanoparticles have cubic shapes and an average size of 12.3 (±1.4) nm obtained from the size distribution of 500 nanoparticles [11]. The organic capping layer was exchanged with HDA or HDT. About eight milliliters of TTAB capped nanoparticles were redispersed in 2 mL of deionized water after washing, then 10 mg of HDA or 20 μL of HDT was added to the washed nanoparticles. The solution was refluxed overnight at 50 °C, and the residual HDA or HDT was washed with ethanol. The nanoparticles were further washed by dispersing in chloroform and precipitating with hexane. Finally, the nanoparticles were dispersed in chloroform and deposited on a silicon wafer. Because the exchanging capping layers do not change the size and shape of nanoparticles, HDA and HDT capped nanoparticles had an average size of 12.3 (±1.4) nm with cubic shapes. For PVP capped Pt nanoparticles, 0.1 mmol of chloroplatinic acid hexahydrate (H2PtCl6, ACS reagent, Sigma–Aldrich), 4 mmol of tetramethylammonium bromide ((CH3)4NBr, >98%, Sigma–Aldrich), and 2 mmol of poly(vinylpyrrolidone) (PVP, Mw = 24,000, Sigma–Aldrich) (in terms of the repeating unit), were added to 20 mL of ethylene glycol (>98%, EMD) in a 50 mL three-necked flask at room temperature. PVP capped Pt nanoparticles have an average size of 9.5 (±0.8) nm obtained from the size distribution of 150 particles in TEM images [5].
2.2 Analytic Techniques for Characterization of Platinum Nanoparticles
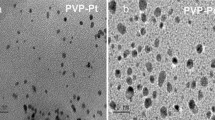
Monolayers of Pt nanoparticles were formed by placing drops of nanocrystal chloroform solution onto the water subphase of a LB trough (Nima Technology, M611) at room temperature. The morphology and chemical composition of the nanoparticles prepared in two-dimensional arrays on silicon wafers were characterized with scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). The nanoparticles were deposited using the Langmuir Blodgett (LB) technique in a 611 Nima Technology LB trough. The surface pressure of the water in the LB trough was set to zero before adding the nanoparticles. The resulting surface monolayer was compressed at a rate of 20 cm2/min, and the deposition of the nanoparticles was carried out by lifting the submerged substrate out of the water. A Zeiss Gemini Ultra-55 analytical scanning electron microscope was used for this study. Figure 1 shows SEM images of Pt nanoparticle arrays on a silicon substrate with each of the four types of capping layers reported in this study. The XPS experiments were conducted on nanoparticle films deposited on silicon wafers. XPS spectra were taken on a 15 kV, 350 W PHI 5400 ESCA/XPS system equipped with an Al anode X-ray source.

Scanning electron microscopy (SEM) images of Pt nanoparticles with capping layers of a HDA, b HDT, c TTAB, and d PVP prepared with LB technique. The scale bars refer to 100 nm
2.3 Catalytic Reactor and UV-ozone Treatment
The reaction studies were performed in an ultrahigh vacuum chamber with a base pressure of 5.0 × 10−8 Torr. CO oxidation studies were carried out in a batch reaction system under 40 Torr CO, 100 Torr O2, and 620 Torr He. The gases were circulated through the reaction line by a Metal Bellows recirculation pump at a rate of 2 L/min. The volume of the reaction loop was 1.0 L. An HP Series II gas chromatograph equipped with a thermal conductivity detector and a 15′, 1/8′′ SS 60/80 Carboxen-1000 (Supelco) was used to separate the products for analysis. The measured reaction rates are reported as turnover frequencies (TOF) and are measured in units of product molecules of CO2 produced per metal surface site per second of reaction time. The irradiation of the nanoparticles by UV-ozone was conducted ex situ using a Bulbtronics 16 W low pressure mercury lamp emitting at 185 and 257 nm inside of a custom made metallic enclosure. The sample was positioned at a distance of 5 mm from the ultraviolet lamp’s quartz tube surface for a determined amount of time, and then removed from the UV chamber. SEM images reveal individual nanoparticles 12 nm in size and indicate that the nanoparticles do not agglomerate due to UV/ozone cleaning.
3 Results and Discussion
3.1 Catalytic Activity of Pt Nanoparticles with Different Capping Layers
Figure 2 shows number of CO2 molecules per metal site produced by the CO oxidation reaction on TTAB coated Pt nanoparticles as a function of reaction time. No deactivation occurs during the 5 h of reaction time in which total CO conversion remains below 20%. In hydrogenation reactions, CO tends to block reactive sites, causing deactivation [12, 13]. In the case of CO oxidation, CO on the active sites is catalytically converted to gas phase CO2, and site blocking, and consequently deactivation, is not a significant issue. This implies that CO oxidation on the nanoparticles leads to a reliable and reproducible measurement of turnover rate. Figure 3 shows turnover rates of nanoparticle arrays with four different capping layers (TTAB, HDA, HDT and PVP) as a function of temperature (over the temperature range between 180 and 240 °C). The number of metal sites is calculated from geometric considerations based on scanning electron microscopy (SEM) measurements of the surface area of the nanoparticle array.

Number of CO2 per Pt site as a function of reaction time measured on TTAB capped Pt nanoparticles at 240 °C

Turnover rate as a function of reaction temperature measured on Pt nanoparticles with four capping layers (TTAB, HAD, HDT, and PVP) under CO oxidation
Table 1 shows the comparison of the activation energy obtained from the Arrhenius plot and the turnover rate measured at 240 °C for each sample. Also it illustrates the chemical structures of each of the capping layers tested in this study. As shown in the plot, TTAB coated nanoparticles exhibit highest activity. This could be associated with weak bonding between TTAB and the Pt atoms in the nanoparticles, which opens up the more active sites. PVP capped Pt nanoparticles appear to exhibit the intermediate activity, presumably because of the polymerization of cross-linked PVP molecules under reaction conditions. The catalytic activity of nanoparticles is associated with the interaction between the capping layers and the Pt surface. Borodko et al. [14, 15] revealed the nature of the interaction between PVP and Pt nanoparticles using UV-Raman and FTIR (Fourier transform infrared) spectroscopy and found that the interaction between PVP and Pt at an oxidized surface is stronger than on reduced nanoparticles, based on changes in the intensities of the Raman spectrum of adsorbed PVP. The weak activity observed for HDA and HDT capped nanoparticles may be due to strong NH2 and sulfur bonds, respectively, with Pt, which can block reaction sites and thus lower the reaction activity. Sulfur, in particular, is well known to poison the catalytic activity of platinum surfaces during CO oxidation [7, 16]. The total variation of turnover rate on nanoparticles with different capping layers is within 30%, which suggests that the effect of capping layers on catalytic activity during CO oxidation is not significant. The activation energy remains constant (27–28 kcal/mol) within the error of measurement, which further suggests that changing the capping layer also does not alter the kinetics of catalytic behavior on nanoparticles.
We now compare the catalytic activity of Pt nanoparticles with that of single crystal surfaces. Su et al. [17] carried out CO oxidation on Pt (111) surface at 100 Torr O2 and 40 Torr CO between 540 and 630 K. Below the ignition temperature, the reaction rate is governed by surface reaction kinetics and follows Langmuir–Hinshelwood kinetics [18–21]. The activation energy for this reaction regime was found to be between 30 [22] and 42 kcal/mol [17]. From Su et al’s study, turnover frequency (TOF) for Pt (111) (at 513 K) is ~3 (CO2/metal site/second). This TOF value is lower than those of nanoparticles within a factor of two. The higher activity on Pt nanoparticles compared to the single crystal is most likely associated with the higher number of kinks and edge sites at nanoparticle surfaces, which are well known to be very reactive [23, 24].
3.2 XPS and Catalytic Activity of UV-ozone Cleaned Platinum Nanoparticles
In order to study the role of capping layers on surface chemical processes, we investigated the catalytic activity after UV-ozone treatment. Transmission electron microscopy (TEM) characterization before and after the treatment carried out by Aliaga et al. [9] reveal a partial decomposition of the capping layers while the shape and size of the nanoparticles are preserved. Figure 4 shows the plot of the Pt 4f peaks of TTAB capped nanoparticles (a) before and (b) after UV-ozone cleaning for 2 h. The binding energies of each spectrum are referenced to the C1s core level (284.5 eV). The Pt 4f7/2 core level is visible at 70.9 eV, which is slightly shifted from the value of Pt (71.2 eV) reported in the literature [25, 26]. The C1s peak is reduced after 2 h of UV-ozone cleaning, which increased the ratio of Pt4f/C1s by a factor of 4.4. The natural oxidation states of Pt are 2+ and 4+, i.e., PtO and PtO2. The chemical shifts of the Pt 4f core level of PtO and PtO2 relative to Pure Pt are about 1.2 and 3.0 eV [27, 28]. As shown in Fig. 4a, the Pt 4f peak of bare TTAB capped nanoparticles exhibits an oxidation state of Pt 2+. After UV-ozone cleaning, the XPS Pt4f peak reveals higher oxidation states, which indicate the formation of Pt 4+. Interestingly, both PtO (marked in red line) and PtO2 (blue line) are visible, as shown in Fig. 4b.

XPS plots of Pt 4f of Pt nanoparticles with TTAB capping measured a before and b after UV-ozone cleaning technique for 2 h. The 2+ (PtO) and 4+ (PtO2) oxidation peaks are represented with red and blue curves. The chemical shift of Pt 4f core level of PtO and PtO2 relative to Pure Pt are about 1.2 and 3.0 eV
UV-ozone cleaning also increased the turnover rate, as shown in Fig. 5 for both TTAB and HDT coated Pt nanoparticles. While the turnover rate increased by 60–100% for TTAB and HDT coated Pt nanoparticles, the activation energy remains the same (between 27 and 28 kcal/mol), within the error of measurement (1 kcal/mol). This higher catalytic activity may be associated with the removal of capping layers by UV-ozone cleaning, which leads to more reactive sites. It is interesting to note that TTAB and HDT coated nanoparticles exhibit different turnover rate after UV-ozone cleaning. This result implies that UV/ozone treatment does not seem to completely remove all organic adsorbates, which is consistent with the recent Diffuse Reflectance Infrared Fourier Transform Spectroscopy measurements performed by Aliaga et al. [9].

Turnover rate and activation energy measured on TTAB and HDT before and after UV-ozone cleaning
After UV-ozone cleaning, the Pt surface exhibits higher oxidation states. Whether the oxidized Pt surface is catalytically active, however, is of some debate [29, 30]. In our study, the turnover rate of Pt nanoparticles after UV-ozone cleaning is higher than that of the bare Pt nanoparticle surface with capping layers, as well as clean Pt (111) surface. This result implies that the oxidized Pt surface is not a catalytically inactive species. This argument is also consistent with recent CO oxidation results on core-shell Pt/mesoporous silica nanoparticles which exhibited partial oxidation of Pt. The turnover rate of CO oxidation on these core-shell Pt/mesoporous silica nanoparticles was found to be as high as that on bare Pt nanoparticles [31]. However, the role of Pt oxide in catalytic activity is beyond the scope of this study, and in addition, the removal of capping layers that occurs during the UV-ozone treatment makes a direct comparison of catalytic activity of Pt oxide with the bare Pt surface difficult. One approach to rule out the contribution of capping layers would be to synthesize Pt nanoparticles without capping layers and then compare their catalytic activity before and after UV-ozone treatment. The preparation of nanoclusters with UHV deposition [32] can be an approach to prepare capping layer-free nanoparticles and to study the role of platinum oxide on the catalytic activity exclusively.
4 Conclusion
Carbon monoxide oxidation was carried out on platinum nanoparticle systems capped with various organic molecules to study the influence of chemical bonding between the molecules and metal atoms on the catalytic activity. Four types of capping layers—TTAB, HDA, HDT, and PVP—were tested. The turnover rate of TTAB capped nanoparticles exhibited the highest activity presumably because of weak bonding between TTAB and the Pt atoms. Weak activity was observed using HDA and HDT capped nanoparticles, which can be attributed to strong NH2 and sulfur bonds. For all the capping layers, the activation energy remained the same within the error of the measurement. After partial removal of capping layers with UV-ozone treatment, the activity of CO oxidation increased by 60–100%, depending on the type of capping layer, while the activation energy remained the same.
References
El-Sayed MA (2004) Acc Chem Res 37:326
Somorjai GA, Park JY (2008) Top Catal 49:126
Demir MM, Gulgun MA, Menceloglu YZ, Erman B, Abramchuk SS, Makhaeva EE, Khokhlov AR, Matveeva VG, Sulman MG (2004) Macromolecules 37:1787
Narayanan R, El-Sayed MA (2004) Nano Lett 4:1343
Park JY, Zhang Y, Grass M, Zhang T, Somorjai GA (2008) Nano Lett 8:673
Somorjai GA, Park JY (2008) Angew Chem 47:9212
Bartholomew CH, Agrawal PK, Katzer JR (1982) Adv Catal 31:135
Vig JR (1985) J Vac Sci Technol A 3:1027
Aliaga C, Park JY, Yamada Y, Lee HS, Tsung CK, Yang P, Somorjai GA (2008) (submitted)
Lee H, Habas SE, Kweskin SJ, Butcher D, Somorjai GA, Yang P (2006) Angew Chem 45:7824
Bratlie KM, Lee H, Komvopoulos K, Yang P, Somorjai GA (2007) Nano Lett 7:3097
Twigg MV, Spencer MS (2001) Appl Catal A Gen 212:161
Englisch M, Jentys A, Lercher JA (1997) J Catal 166:25
Borodko Y, Habas SE, Koebel M, Yang P, Frei H, Somorjai GA (2006) J Phys Chem B 110:23052
Borodko Y, Humphrey SM, Tilley TD, Frei H, Somorjai GA (2007) J Phys Chem C 111:6288
Park JY, Lee H, Renzas JR, Zhang YW, Somorjai GA (2008) Nano Lett 8:2388
Su XC, Cremer PS, Shen YR, Somorjai GA (1997) J Am Chem Soc 119:3994
Alavi A, Hu PJ, Deutsch T, Silvestrelli PL, Hutter J (1998) Phys Rev Lett 80:3650
Campbell CT, Ertl G, Kuipers H, Segner J (1980) J Chem Phys 73:5862
Imbihl R, Cox MP, Ertl G (1986) J Chem Phys 84:3519
McCrea KR, Parker JS, Somorjai GA (2002) J Phys Chem B 106:10854
Berlowitz PJ, Peden CHF, Goodman DW (1988) J Phys Chem 92:5213
Salmeron M, Gale RJ, Somorjai GA (1977) J Chem Phys 67:5324
Gambardella P, Sljivancanin Z, Hammer B, Blanc M, Kuhnke K, Kern K (2001) Phys Rev Lett 8705:4
Moulder JF, Stickle WF, Sobol PE, Bomben KD (1995) In: Chastain J, King RC Jr (eds) Handbook of X-ray photoelectron spectroscopy. Physical Electronics, Eden Prairie, p 181
Jung MC, Kim HD, Han M, Jo W, Kim DC (1999) Jpn J Appl Phys Part 1-Regul Pap Short Notes Rev Pap 38:4872
Bancroft GM, Adams I, Coatsworth LL, Bennewitz CD, Brown JD, Westwood WD (1975) Anal Chem 47:586
Barr TL (1978) J Phys Chem 82:1801
Hendriksen BLM, Frenken JWM (2002) Phys Rev Lett 89
Chen MS, Cal Y, Yan Z, Gath KK, Axnanda S, Goodman DW (2007) Surf Sci 601:5326
Joo SH, Park JY, Tsung CK, Yamada Y, Yang P, Somorjai GA (2009) Nat Mater (in press)
Valden M, Lai X, Goodman DW (1998) Science 281:1647
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geological and Biosciences and Division of Materials Sciences and Engineering of the US Department of Energy under contract No. DE-AC02-05CH11231.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Park, J.Y., Aliaga, C., Renzas, J.R. et al. The Role of Organic Capping Layers of Platinum Nanoparticles in Catalytic Activity of CO Oxidation. Catal Lett 129, 1–6 (2009). https://doi.org/10.1007/s10562-009-9871-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-009-9871-8