
Abstract
Purpose
Triple-negative breast cancer (TNBC) is associated with worse outcomes relative to other breast cancer subtypes. Chemotherapy remains the standard-of-care systemic therapy for patients with localized or metastatic disease, with few biomarkers to guide benefit.
Methods
We will discuss recent advances in our understanding of two key biological processes in TNBC, homologous recombination (HR) DNA repair deficiency and host anti-tumor immunity, and their intersection.
Results
Recent advances in our understanding of homologous recombination (HR) deficiency, including FDA approval of PARP inhibitor olaparib for BRCA1 or BRCA2 mutation carriers, and host anti-tumor immunity in TNBC offer potential for new and biomarker-driven approaches to treat TNBC. Assays interrogating HR DNA repair capacity may guide treatment with agents inducing or targeting DNA damage repair. Tumor infiltrating lymphocytes (TILs) are associated with improved prognosis in TNBC and recent efforts to characterize infiltrating immune cell subsets and activate host anti-tumor immunity offer promise, yet challenges remain particularly in tumors lacking pre-existing immune infiltrates. Advances in these fields provide potential biomarkers to stratify patients with TNBC and guide therapy: induction of DNA damage in HR-deficient tumors and activation of existing or recruitment of host anti-tumor immune cells. Importantly, these advances provide an opportunity to guide use of existing therapies and development of novel therapies for TNBC. Efforts to combine therapies that exploit HR deficiency to enhance the activity of immune-directed therapies offer promise.
Conclusions
HR deficiency remains an important biomarker target and potentially effective adjunct to enhance immunogenicity of ‘immune cold’ TNBCs.
Similar content being viewed by others


Introduction
Triple-negative breast cancer (TNBC) accounts for approximately 10–15% of all breast cancers and is defined pathologically by the absence of the estrogen receptor (ER), progesterone receptor (PR), and absence of amplification or overexpression of the HER2/neu oncogene [1]. Relative to other subtypes of breast cancer, TNBC is associated with poorer prognosis and disproportionately larger proportion—over one-third—of breast cancer-specific deaths [2, 3]. Despite significant progress over the past decades in the management of breast cancer through development of anti-estrogen and anti-HER2 therapies, the standard-of-care for TNBC in the early and advanced disease settings remains cytotoxic chemotherapy. Recent advances in our understanding of two key biological processes in TNBC, homologous recombination (HR) DNA repair deficiency and host anti-tumor immunity, and the intersection of these processes, offer potential for new and biomarker-driven approaches to treat TNBC.
Homologous recombination DNA repair deficiency in TNBC
Breast cancer that arises in BRCA1 and BRCA2 mutation carriers is characterized by defects in homologous recombination (HR) DNA repair [4]. Loss of functional BRCA1 and BRCA2 in the tumor leads to an increase in genomic instability and increased copy number alterations [5]. HR deficiency has also been implicated in sporadic breast cancer, particularly TNBC, and suggested mechanisms include BRCA1 promoter methylation, mutation in HR-related genes including somatic mutations in BRCA1 and BRCA2, or other epigenetic mechanisms.
Numerous biomarkers of HR deficiency / proficiency have been evaluated with increasing frequency in the clinical setting (Fig. 1). Biomarkers with potential to distinguish HR deficient from HR proficient tumors include (1) germline BRCA1 and BRCA2 mutations, (2) germline mutations in HR pathway genes beyond BRCA1 and BRCA2, (3) somatic HR pathway mutations, (4) genomic instability or “scar” biomarkers and mutational signature-based approaches, (5) gene expression signatures of ‘BRCAness,’ and (6) functional HR assays. Other than germline mutations, none are yet validated.

Homologous recombination (HR) deficiency biomarkers. HR homologous recombination, LOH Loss of heterozygosity, TAI telomeric allelic imbalance, LST large-scale state transitions
In the current clinical management of both early and advanced TNBC, important questions remain regarding the role of platinum chemotherapy and also if PARP inhibitors have a potential role beyond BRCA1 and BRCA2 mutation carriers. Alternative approaches to exploiting HR and other DNA repair deficiencies in TNBC are also being explored, including inhibition of enzymes involved in DNA repair cell cycle checkpoints such as CHEK2, WEE1 and ATR as well as stabilization of G-quadruplex structures in the genome [6,7,8,9]. Given the heterogeneity of TNBC, biomarkers of HR deficiency may have important clinical implications in the future treatment of TNBC.
Germline BRCA1 and BRCA2 mutation status as a HR deficiency biomarker
Reported in 2005, BRCA1- and BRCA2-deficient cells were found to be markedly sensitive to inhibition of PARP in contrast to those cells that were wild-type or heterozygous for BRCA1/2, implicating the potential for synthetic lethality [10, 11]. Based on these observations, proof-of-concept studies tested PARP inhibition in advanced BRCA1 and BRCA2 mutation-associated breast and ovarian cancer. Among 27 heavily pretreated BRCA1/2 mutant breast cancer subjects who received olaparib 400 mg orally twice daily, Tutt and colleagues reported an objective response rate of 41% and a clinical benefit rate of 85% [12]. Responses were seen in both BRCA1 and BRCA2 mutation carriers and were irrespective of breast cancer subtype.
Around the same time, a small proof-of-concept trial explored the activity of single-agent cisplatin 75 mg/m2 IV every 3 weeks for 4 cycles as neoadjuvant therapy for BRCA1 mutation-associated breast cancer and demonstrated 61% pathologic complete response [13]. However, in the neoadjuvant GeparSixto clinical trial of weekly paclitaxel, weekly non-pegylated liposomal doxorubicin and bevacizumab with or without weekly carboplatin, BRCA1/2 mutation carriers did not have higher pCR rates with the addition of carboplatin [14]. Response rates to standard chemotherapy in BRCA1 and BRCA2 mutation carriers are not well known. A retrospective analysis of BRCA1/2 mutation carriers at a single center reported a pCR rate of 46% in BRCA1 mutant versus 31% BRCA1/2 wild-type TNBC patients treated with anthracyclines with or without taxanes [15]. Another retrospective series reported a pCR rate of 67% in BRCA1/2 mutant versus 37% in wild-type TNBC patients [16]. In the frontline metastatic TNBC setting, the TNT trial compared single-agent carboplatin versus single-agent docetaxel for 6 cycles followed by cross-over upon progression [17]. While both agents resulted in similar rates of response in the overall study, objective response rates among BRCA1/2 mutation carriers were 68% with carboplatin compared to 33% with docetaxel (absolute difference 34.7%; 95% CI 6.3–63.1; p = 0.03).
More recently, we have seen Phase III results comparing the PARP inhibitor olaparib or talazoparib to non-DNA-damaging chemotherapy treatment of physician’s choice in BRCA1 and BRCA2 mutation carriers with advanced HER2-negative breast cancer. Both studies showed improvement of median PFS with the PARP inhibitor of ~ 3 months and greater tolerability, with no overall survival benefit evident as yet. Among patients with measurable disease, the objective response rates were approximately 60% with the PARP inhibitor compared to approximately 30% in physician choice (need to check talazaparib) [18, 19]. The FDA approved olaparib for the treatment of germline BRCA1/2 metastatic breast cancer in January 2018.
Germline HR pathway mutations beyond BRCA1 and BRCA2 as HR deficiency biomarkers
Of the genes implicated in familial breast cancer beyond BRCA1 and BRCA2, many moderate penetrance genes are likewise involved in the HR DNA repair pathway [20]. In women testing negative for BRCA1 and BRCA2 mutations, multi-gene germline sequencing identifies up to another 10% of patients with pathogenic mutations [21]. Recently, a large study of multi-gene germline testing in over 1,800 unselected patients with TNBC demonstrated that overall mutation rate was 14.6% with 8.5% having mutations in BRCA1, 2.7% in BRCA2 and an additional 3.7% with mutations in other genes such as PALB2 (1.2%), BRIP1, BARD1, and RAD51C among others [22]. Cancers arising in these genetic backgrounds are hypothesized to have HR DNA repair defects and therefore may have similar chemosensitivity to DNA-damaging therapies as BRCA1/2 mutation carriers [4, 23]. Clinical trials are currently underway to assess this hypothesis [24].
Genomic scars and mutational signatures as HR deficiency biomarkers
An important outstanding question is whether evidence of HR deficiency in TNBC patients without germline alterations is also a biomarker for treatment. The Homologous Recombination Deficiency (‘HRD’) assay measures levels of genomic instability or ‘scarring’ caused by any number of alterations in DNA repair capacity. At present, the HRD assay incorporates three measures of genomic instability: telomeric allelic imbalance (TAI; the number of regions with allelic imbalance that extend to the subtelomere, but do not cross the centromere), loss of heterozygosity (LOH; the number of regions > 15 Mb and less than one chromosome lost across the genome), and large-scale state transitions (LST; the number of chromosomal breaks between adjacent genomic regions longer than 10 Mb after filtering regions < 3 Mb) [25,26,27]. The HRD score is currently calculated by adding the LOH, TAI, and LST scores and is a continuous score from 0 to 100 with a score of < 41 (previously < 10) as HR proficient and ≥ 42 (previously ≥ 10) as HR deficient. Using this cutoff, it is estimated that approximately 50% of TNBC patients will be classified as HR deficient. Tumors with BRCA1/2 mutations are classified as HR deficient regardless of HRD score.
Recently, multiple groups have reported that the HRD assay can be used to identify both BRCA1/2 mutant and wild-type TNBC patients more likely to achieve a favorable response to platinum-based neoadjuvant chemotherapy [27]. In the neoadjuvant PrECOG 0105 trial, responders to this platinum-based therapy had significantly higher mean HRD-LOH scores compared to non-responders; this was true for both BRCA1/2 wild-type and mutant responders [28]. Overall, 66% of patients with an HRD-LOH score of ≥ 10 or BRCA1/2 mutation responded compared with 8% of patients with an HRD-LOH score of < 10 and intact BRCA1/2.
The combined HRD score has been assessed in a number of clinical trials of platinum-based therapy. In a pooled analysis of six phase 2 neoadjuvant platinum-based TNBC trials (n = 267), the adjusted odds ratio for pathological response in HR deficient compared to non-deficient tumors was 4.64 (95% CI 2.32–9.27; p = < 0.0001) [29]. In addition to assessments in multiple single-arm studies of neoadjuvant platinum, in the randomized phase 2 GeparSixto trial, patients with high HRD score or tumor BRCA mutation were more likely to achieve pCR (55.9% vs. 29.8%), odds ratio 2.51 (p = 0.009) in multivariate analyses [30]. The addition of carboplatin numerically improved pCR rates with a non-significant interaction between HR deficiency and carboplatin benefit [31]. These data suggest that the HRD assay is promising in concept but whether it can be used to identify germline BRCA1 and BRCA2 wild-type patients who may benefit from platinum-based therapy remains to be seen.
While the results in the neoadjuvant setting have been relatively consistent, results in the metastatic setting are more difficult to interpret. In the metastatic TBCRC009 clinical trial of platinum in the 1st and 2nd line setting, mean HRD scores were significantly higher in patients achieving an objective response [32]. In the larger TNT phase 3 study of docetaxel versus carboplatin, however, platinum sensitivity was not associated with higher HRD scores [17]. Subsequent analyses suggested greater numerical (but not significant) response rates to docetaxel than carboplatin for both BRCA1 promoter methylation and silencing [33]. It is hypothesized that a genomic scar once induced in a tumor will persist, but secondary events may lead to a restoration of HR repair capacity. While genomic scars certainly have significant potential to be clinically useful particularly in the newly diagnosed setting, questions surrounding the specificity of the test and whether the assay predicts sensitivity to chemotherapy in general or specifically platinum and other DNA repair-targeted therapies require additional clarification. Ultimately, prospective validation of the predictive value of HRD in both platinum and PARP inhibitor benefit is needed.
An alternative approach to the genomic ‘scar’ phenotype detected by the HRD assay is to use mutational signatures derived from exome- or genome-level sequencing. Polak and colleagues investigated presence of mutational signature 3 [34] in whole exome sequencing data from nearly 1000 breast cancer patients [35]. They found that the presence of this signature could accurately classify BRCA1/2 alterations, while also demonstrating that epigenetic silencing of RAD51C and BRCA1 and germline variants in PALB2 were associated with signature 3 [35]. A second approach defined six new mutational signatures from whole genome sequencing of breast cancer patients that were predictive of BRCA1/2 deficiency. They integrated these in a weighted model, termed ‘HRDetect,’ which identifies BRCA1/BRCA2-deficient tumors with high (98.7%) sensitivity and also identifies tumors with somatic loss or functional BRCA1/BRCA2 deficiency [36, 37]. HRDetect was validated in an independent dataset and evaluation for clinical utility is ongoing [37].
Additional potential HR deficiency biomarkers: expression signatures and functional assays
Non-genomic approaches to characterize ‘BRCAness’ have also been explored. The ‘DDR deficiency assay’ is a 44-gene expression signature derived to identify loss of the Fanconi anemia/BRCA DNA repair pathways [38]. In two independent datasets of patients treated with (neo)adjuvant fluorouracil, anthracycline and cyclophosphamide, the DDR deficiency assay was significantly associated with pCR and relapse-free survival. In the neoadjuvant I-SPY2 trial evaluating the addition of carboplatin and PARP inhibitor veliparib to standard anthracycline and taxane-based therapy, a 77-gene BRCAness signature was associated with response to veliparib/carboplatin relative to control [39]. Methods to functionally characterize HR deficiency in fresh tumor samples offer a ‘gold standard’ for assessing HR, but are technically difficult and at this time not feasible to scale in a clinical setting. Powell and colleagues demonstrated that lack of RAD51 foci after ex vivo ionizing radiation on fresh breast cancer samples was significantly associated with genomic scars (LOH, LST, TAI) and biallelic inactivation of DNA repair genes [40]. In a prospective series, 16/56 (29%) primary breast tumors revealed defective RAD51 recruitment following irradiation [41]. Both gene expression signatures and functional characterization of HR require further validation in prospective studies yet could help to distinguish between functional HR capacity in the context of a genomic scar.
Host anti-tumor immunity in TNBC
In breast cancer, several studies of large cohorts over the past decade have demonstrated that infiltration of tumors with lymphocytes is associated with improved outcomes [42,43,44,45]. Tumor infiltrating lymphocytes (TILs) in pretreatment tumor samples are associated with improved response to neoadjuvant chemotherapy in TNBC and survival among those patients with residual disease after neoadjuvant chemotherapy [46,47,48,49]. Lymphocytes can intercalate between tumor cells (intratumoral TILs) or infiltrate the surrounding stromal tissue (stromal TILs). Stromal TIL assessment has been more reproducible and of greater predictive/prognostic value [50]. Only a subset of breast cancers demonstrate a significant number of stromal TILs, with median percent of stroma infiltrated with TILs of 10% for ER-positive/HER2-negative, 15% for HER2-positive, and 20% for TNBCs [42].
Evasion of host anti-tumor response and T-cell ‘exhaustion’ phenotype in breast cancer
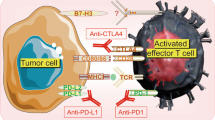
Tumor cells evade host anti-tumor immune responses through a variety of mechanisms, including tumor cell-intrinsic avoidance of immune detection (loss of tumor antigens or MHC proteins), elaboration of immunosuppressive cytokines, immunosuppressive cell recruitment, and immunoediting—the process by which tumors evolve or are selected to escape host anti-tumor immunity [51,52,53]. Failure of TILs to kill adjacent tumor cells offers both a challenge and potential avenue for therapeutic intervention. TILs are primarily T-cells that express checkpoints such as PD-1 and CTLA-4, implying chronic antigen exposure but progressive loss of T-cell functions, elevated inhibitory receptors, and impaired cytokine production resulting in the inability to target tumor cells even in close proximity [46, 54, 55].
The ‘exhausted’ T-cell phenotype could reflect the end stage of immunoediting. Immunoediting is thought to begin early in cancer development as highly immunogenic tumor subclones are eliminated or adapt a less immunogenic profile in response to anti-tumor T-cells [55, 56]. (Fig. 2). As the tumor develops, induction of repressive molecules and decrease in co-stimulatory molecules in the infiltrating T-cells, among other processes, leads to the exhaustion phenotype [57]. Perturbations to this balance between host anti-tumor immunity and tumor immunosuppressive signals—such as surgery, chemotherapy, or radiation—may temporarily reinvigorate memory T-cells until immunogenic tumor cells are eliminated or another round of immunoediting occurs [55]. In this way, the subset of tumors with substantial numbers of TILs locked in this ‘stalemate’ may be primed for reactivation by immune checkpoint inhibitors.

Exhausted T-cells and cancer result in a stalemate. We hypothesize that during the development of some primary breast cancers (TNBC, HER2+), the activated T-cells progressively lose T-cell function, increase inhibitory receptors, and demonstrate impaired cytokine production, a process termed ‘T-cell exhaustion.’ This steady-state function eventually fails and is not sufficient enough to control cancer. The level and duration of chronic antigen stimulation (e.g., long history of metastatic disease, larger tumor burden, secretion of other tumor immunosuppressive molecules i.e., VEGF) are perhaps key factors that lead to T-cell exhaustion and immunosuppression. It is possible that only a subset of exhausted T-cells can be reinvigorated by checkpoint blockade
Targeting the PD-1/PD-L1 checkpoint in TNBC
Programmed death receptor 1 (PD-1) is an inhibitory immune checkpoint receptor expressed on activated T-cells and other immune cells and plays a key role in regulating the activity of effector T-cells. The PD-1 ligand, PD-L1, is expressed by multiple cancer types and upon binding to PD-1 exerts an immunosuppressive signal. The expression of PD-L1 has emerged as an important mechanism by which multiple cancer types are able to evade destruction by anti-tumor effector cells. Monoclonal antibodies targeting PD-1 (pembrolizumab, nivolumab) or PD-L1 (atezolizumab, avelumab, durvalumab) have garnered FDA approvals for the treatment of multiple malignancies. In TNBC, multiple studies are exploring the activity of these agents alone and in combination with cytotoxic chemotherapy, targeted therapy or radiation. As yet, none of these drugs have been FDA approved in breast cancer.
To date in metastatic TNBC, responses to single-agent anti-PD-1/anti-PD-L1 monoclonal antibodies have been modest and biomarkers of response have been actively pursued. Nanda and colleagues reported initial results from the KEYNOTE 0–12 Phase Ib study of pembrolizumab in patients with metastatic triple-negative breast cancer with PD-L1-positive tumor status (expression in stroma or in ≥ 1% tumor cells). Among 27 evaluable patients with a median of 2 prior therapies for metastatic disease, the objective response rate was 18.5% [58]. The subsequent KEYNOTE-086 Phase II trial evaluated pembrolizumab in metastatic TNBC patients that were previously treated with any level of PD-L1 expression (Cohort A) and as 1st line treatment in patients with PD-L1+ status (Cohort B) [59]. Among 170 patients enrolled in cohort A, 105 (61.8%) had PD-L1+ status and 43.5% had received 3 or more prior lines of therapy. Objective responses were seen in 4.7% with no significant difference in ORR based on PD-L1 expression status. Patients enrolled in Cohort B (n = 52) had an ORR of 23.1% suggesting greater benefit in patients with less heavily pretreated disease.
A Phase Ia study of atezolizumab enrolled 115 patients with metastatic TNBC. Patients had heavily pretreated disease with a median of 7 prior lines of therapy and 63% of patients were PD-L1+, 33% PD-L1- and 4% had unknown PD-L1 status [60]. The overall ORR was 10% with a 13% ORR in PD-L1+ patients and 5% ORR in PD-L1− patients. Among 1st line patients, the ORR was higher at 26%. Avelumab was evaluated in the Phase I JAVELIN study and among 58 patients with TNBC, an ORR of 8.6% was observed [61]. Patients with PD-L1+ status (n = 9) had an ORR of 44.4% and those with PD-L1− status had an ORR of 2.4%. Taken together, these early phase results in metastatic TNBC suggest higher activity in 1st line treatment and in those with PD-L1+ status. Multiple phase II and III trials of PD-1/PD-L1 checkpoint inhibitors in metastatic TNBC are ongoing and are evaluating monotherapy as well as combinations with chemotherapy, other immunotherapeutic agents and targeted therapies (Table 1).
In addition, the role of PD-1/PD-L1 blockade in TNBC is also being actively assessed in the adjuvant and neoadjuvant settings. Recent data from the I-SPY 2 trial demonstrated that adding pembrolizumab to neoadjuvant anthracycline and taxane-based chemotherapy results in an estimated pCR rate of 60% versus 20% in TNBC patients [62]. Multiple phase II and II studies are currently ongoing in the curable setting assessing these agents in combination with standard chemotherapy and also as monotherapy following completion of standard combination chemotherapy in high-risk patients.
Enhancing breast cancer immunogenicity
Many TNBC tumors have relatively few infiltrating lymphocytes and, in the absence of T-cells to be activated, checkpoint blockade is unlikely to be effective. For the poor-prognosis basal-like, non-immune infiltrated tumors, inducing lymphocyte infiltration in immune ‘quiet’ tumors poses a greater challenge than activating lymphocytes in tumors with existing TILs. Although tumor immunogenicity is hypothesized to relate to the number of ‘neoantigens,’ or tumor-specific antigens recognized as foreign, and neoantigen load has been associated with response to immune checkpoint therapies [63, 64], breast cancers have relatively few somatic mutations and inferred neoantigen load [65]. Analyses to date do not show a positive correlation between mutational or neoantigen load and gene expression evidence of immune cell infiltration in TNBCs [66].
Common strategies to enhance tumor immunogenicity
Many of the strategies to enhance tumor immunogenicity focus broadly on the induction of DNA damage and neoantigens, for example through radiation or chemotherapy. However, certain—but not all—chemotherapeutics (taxanes, anthracyclines, cyclophosphamide) appear to induce ‘immunogenic cell death’ by activating type I IFN-receptor ligand systems resulting in recruitment of T-cells, among other mechanisms [67, 68]. Radiation enhances the diversity of the T-cell receptor repertoire of intratumoral T-cells but optimal dose, schedule, and immunotherapy combination is not known [69]. Monoclonal antibodies trigger antibody-dependent cellular cytotoxicity that subsequently activates antigen-presenting cells, a phenomenon observed with trastuzumab in HER2-positive breast cancers [70]. Certain oncogenic drivers, such as the Ras/MAPK pathway, may specifically downregulate MHC proteins and TILs, implying that inhibition may synergize with checkpoint inhibition [49]. A variety of other approaches to augment T-cell infiltration, including standard or personalized/neoantigen vaccines, chemotherapy or radiotherapy, immune agonists targeting a variety of molecules including STING, adoptive T-cell transfer, and others are under investigation.
Targeting homologous recombination deficiency to enhance breast cancer immunogenicity
Recent clinical trials demonstrate that adding PARP inhibitors may enhance activity of immunotherapy in breast cancer [71, 72]. Several recent pre-clinical studies suggest that PARP inhibition enhances immunogenicity in models of ovarian and breast cancer and that combining the two results in synergistic effects [73,74,75]. TOPACIO, a multicenter, open-label phase 1/2 study evaluated the PARP inhibitor niraparib plus pembrolizumab in metastatic TNBC and advanced ovarian cancer and during the dose-finding portion of the trial, none of the eight evaluable patients progressed: 4/8 had objective response and 4/8 had stable disease [71]. The MEDIOLA trial of olaparib plus durvalumab included a cohort of BRCA1/2 mutant metastatic TNBCs and showed an objective response rate of 52% [72]. Collectively, these pre-clinical and early phase clinical data suggest that targeting or enhancing HR defects may enhance tumor immunogenicity and potentially sensitize to immune checkpoint inhibitors.
Conclusions
In summary, HR deficiency remains an important biomarker target and potentially effective adjunct to enhance immunogenicity of ‘immune cold’ TNBCs. There is now an FDA-approved non-chemotherapeutic agent for patients with germline BRCA1/2 mutations and we are moving closer to integrating biomarkers of HR deficiency to individualize therapy in patients with TNBC beyond BRCA1/2. Genomic scar biomarkers may identify HR deficiency beyond BRCA1/2 in TNBC, but require prospective validation to determine clinical utility. Harnessing host anti-tumor immunity provides a promising approach for TNBC, including PD-1/PD-L1 checkpoint inhibitors and combinations of standard plus immune therapies. A greater understanding of the underlying biology of T-cells, T-cell exhaustion, and immunoediting may be crucial to understanding how to leverage anti-tumor immunity in TNBC.
References
Carey L, Winer E, Viale G, Cameron D, Gianni L (2010) Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 7(12):683–692. https://doi.org/10.1038/nrclinonc.2010.154
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13(15 Pt 1):4429–4434. https://doi.org/10.1158/1078-0432.CCR-06-3045
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V (2007) Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer 109(9):1721–1728. https://doi.org/10.1002/cncr.22618
Lord CJ, Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16(2):110–120. https://doi.org/10.1038/nrc.2015.21
Roy R, Chun J, Powell SN (2011) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 12(1):68–78. https://doi.org/10.1038/nrc3181
McLuckie KI, Di Antonio M, Zecchini H, Xian J, Caldas C, Krippendorff BF, Tannahill D, Lowe C, Balasubramanian S (2013) G-quadruplex DNA as a molecular target for induced synthetic lethality in cancer cells. J Am Chem Soc 135(26):9640–9643. https://doi.org/10.1021/ja404868t
Xu H, Di Antonio M, McKinney S, Mathew V, Ho B, O’Neil NJ, Santos ND, Silvester J, Wei V, Garcia J, Kabeer F, Lai D, Soriano P, Banath J, Chiu DS, Yap D, Le DD, Ye FB, Zhang A, Thu K, Soong J, Lin SC, Tsai AH, Osako T, Algara T, Saunders DN, Wong J, Xian J, Bally MB, Brenton JD, Brown GW, Shah SP, Cescon D, Mak TW, Caldas C, Stirling PC, Hieter P, Balasubramanian S, Aparicio S (2017) CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun 8:14432. https://doi.org/10.1038/ncomms14432
Zimmer J, Tacconi EMC, Folio C, Badie S, Porru M, Klare K, Tumiati M, Markkanen E, Halder S, Ryan A, Jackson SP, Ramadan K, Kuznetsov SG, Biroccio A, Sale JE, Tarsounas M (2016) Targeting BRCA1 and BRCA2 deficiencies with G-quadruplex-interacting compounds. Mol Cell 61(3):449–460. https://doi.org/10.1016/j.molcel.2015.12.004
Telli ML, Lord S, Dean E, Abramson V, Arkenau HT, Becerra C, Tolaney SM, Tang R, Penney MS, Pollard J, Conboy G, Fields SZ, Shapiro G (2017) 242PDInitial results of a phase 1 dose expansion cohort of M6620 (formerly VX-970), an ATR inhibitor, in combination with cisplatin in patients with advanced triple-negative breast cancer NCT02157792). Ann Oncol. https://doi.org/10.1093/annonc/mdx365.005
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035):917–921. https://doi.org/10.1038/nature03445
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434(7035):913–917. https://doi.org/10.1038/nature03443
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376(9737):235–244. https://doi.org/10.1016/S0140-6736(10)60892-6
Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J, Jakubowicz J, Cybulski C, Wisniowski R, Godlewski D, Lubinski J, Narod SA (2014) Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat 147(2):401–405. https://doi.org/10.1007/s10549-014-3100-x
von Minckwitz G, Loibl S, Schneeweiss A, Salat C, Rezai M, Zahm D-M, Klare P, Blohmer J-U, Tesch H, Khandan F, Fasching P, Jakisch C, Nekljudova V, Untch M (2015) Early survival analysis of the randomized phase II trial investigating the addition of carboplatin to neoadjuvant therapy for triple-negative and HER2-positive early breast cancer (GeparSixto). San Antonio Breast Cancer Symposium
Arun B, Bayraktar S, Liu DD, Gutierrez Barrera AM, Atchley D, Pusztai L, Litton JK, Valero V, Meric-Bernstam F, Hortobagyi GN, Albarracin C (2011) Response to neoadjuvant systemic therapy for breast cancer in BRCA mutation carriers and noncarriers: a single-institution experience. J Clin Oncol 29(28):3739–3746. https://doi.org/10.1200/jco.2011.35.2682
Paluch-Simon S, Friedman E, Berger R, Papa MZ, Dadiani M, Friedman N, Shabtai M, Zippel D, Gutman M, Golan T, Catane R, Yosepovich A, Modiano TM, Kaufman B (2014) Does pathologic complete response predict for outcome in BRCA mutation carriers with triple-negative breast cancer? J Clin Oncol 32(5S):Abstract 1023
Tutt A, Ellis PA, Kilburn L, Gilett C, Pinder S, Abraham J, Barrett S, Barrett-Lee P, Chan S, Cheang M, Fox L, Grigoriadis A, Harper-Wynne C, Hatton M, Kernaghan S, Owen J, Parker P, Rahman N, Roylance R, Smith I, Thompson R, Tovey H, Wardley A, Wilson G, Harries M, Bliss J 2014 The TNT trial: a randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer (CRUK/07/012). In: Proceedings of AACR San Antonio Breast Cancer Symposium, San Antonio, TX, pp Abstract S3–01
Litton J, Rugo H, Ettl J, Hurvitz S, Gonçalves A, Lee K-H, Fehrenbacher L, Yerushalmi R, Mina L, Martin M, Roché H, Im Y-H, Quek R, Tudor I, Hannah A, Eiermann W, Blum J (2018) Abstract GS6-07: EMBRACA: A phase 3 trial comparing talazoparib, an oral PARP inhibitor, to physician’s choice of therapy in patients with advanced breast cancer and a germline < em> BRCA</em> mutation. Cancer Res. https://doi.org/10.1158/1538-7445.sabcs17-gs6-07
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P (2017) Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 377(6):523–533. https://doi.org/10.1056/NEJMoa1706450
Foulkes WD (2008) Inherited susceptibility to common cancers. N Engl J Med 359(20):2143–2153. https://doi.org/10.1056/NEJMra0802968
Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM (2014) Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol 32(19):2001–2009. https://doi.org/10.1200/JCO.2013.53.6607
Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, Slettedahl S, Hallberg E, Guidugli L, Davila JI, Beckmann MW, Janni W, Rack B, Ekici AB, Slamon DJ, Konstantopoulou I, Fostira F, Vratimos A, Fountzilas G, Pelttari LM, Tapper WJ, Durcan L, Cross SS, Pilarski R, Shapiro CL, Klemp J, Yao S, Garber J, Cox A, Brauch H, Ambrosone C, Nevanlinna H, Yannoukakos D, Slager SL, Vachon CM, Eccles DM, Fasching PA (2015) Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol 33(4):304–311. https://doi.org/10.1200/JCO.2014.57.1414
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A (2006) Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66(16):8109–8115. https://doi.org/10.1158/0008-5472.CAN-06-0140
Afghahi A, Chang P-J, Ford JM, Telli ML (2016) The Talazoparib Beyond BRCA (TBB) trial: A phase II clinical trial of talazoparib (BMN 673) in BRCA1 and BRCA2 wild-type patients with (i) advanced triple-negative breast cancer (TNBC) and homologous recombination deficiency (HRD) as assessed by myriad genetics HRD assay, and (ii) advanced HER2-negative breast cancer (BC) with either a germline or somatic mutation in homologous recombination (HR) pathway genes. Cancer Res 76 (4): OT2–05 Abstract OT2-05-04
Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, Houdayer C, Sastre-Garau X, Vincent-Salomon A, Stoppa-Lyonnet D, Stern MH (2012) Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res 72(21):5454–5462. https://doi.org/10.1158/0008-5472.CAN-12-1470
Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, Tung N, Ryan PD, Garber JE, Silver DP, Szallasi Z, Richardson AL (2012) Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov 2(4):366–375. https://doi.org/10.1158/2159-8290.CD-11-0206
Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, Kalva S, Potter J, Tran TV, Chen J, Iliev D, Sangale Z, Tikishvili E, Perry M, Zharkikh A, Gutin A, Lanchbury JS (2014) Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res 16(6):475
Telli ML, Jensen KC, Vinayak S, Kurian AW, Lipson JA, Flaherty P, Timms K, Abkevich V, Schackmann EA, Wapnir IL, Carlson RW, Sparano JA, Head B, Goldstein LJ, Haley B, Dakhil SR, Reid JE, Hartman AR, Manola J, Ford JM (2015) A phase II study of gemcitabine, carboplatin and iniparib as neoadjuvant therapy for triple-negative and BRCA1/2 mutation-associated breast cancer with assessment of a tumor-based measure of genomic instability (PrECOG 0105). J Clin Oncol. 33:1895–1901
Telli ML, McMillan A, Ford JM, Richardson AL, Silver DP, Isakoff SJ, Kaklamani VG, Gradishar W, Stearns V, Connolly RM, Loibl S, Elkin EP, Timms K, Hartman A-R, von Minckwitz G (2016) Homologous recombination deficiency (HRD) as a predictive biomarker of response to neoadjuvant platinum-based therapy in patients with triple negative breast cancer (TNBC): a pooled analysis. Cancer Res 76 (4): P3–07 Abstract P3-07-12
von Minckwitz G, Timms K, Untch M, Elkin EP, Fasching PA, Schneeweiss A, Salat C, Rezai M, Blohmer JU, Zahm DM, Jackisch C, Gerber B, Klare P, Kummel S, Eidtmann H, Paepke S, Reid JE, MNekljudova V, Hartman A-R, Loibl S (2015) Prediction of pathological complete response (pCR) by homologous recombination deficiency (HRD) after carboplatin-containing neoadjuvant chemotherapy in patients with TNBC: results from GeparSixto. J Clin Oncol 33:1004
von Minckwitz G, Timms K, Untch M, Elkin EP, Fasching PA, Schneeweiss A, Salat C, Rezai M, Blohmer JU, Zahm DM, Jackisch C, Gerber B, Klare P, Kümmel S, Eidtmann H, Paepke S, Reid JE, Nekljudova V, Hartman A-R, Loibl S (2015) Prediction of pathological complete response (pCR) by homologous recombination deficiency (HRD) after carboplatin-containing neoadjuvant chemotherapy in patients with TNBC: results from GeparSixto. J Clin Oncol 33(15_suppl):1004–1004. https://doi.org/10.1200/jco.2015.33.15_suppl.1004 doi
Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, Timms KM, Hartman AR, Borger DR, Finkelstein DM, Garber JE, Ryan PD, Winer EP, Goss PE, Ellisen LW (2015) TBCRC009: A multicenter Phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J Clin Oncol 33(17):1902–1909. https://doi.org/10.1200/JCO.2014.57.6660
Tutt A, Cheang M, Kilburn L, Tovey H, Gillett C, Pinder S, Lanchbury J, Abraham J, Barrett S, Barrett-Lee P, Chan S, Gazinska P, Grigoriadis A, Kernaghan S, Hoadley K, Gutin A, Harper-Wynne C, Hatton M, Owen J, Parker P, Roylance R, Shaw A, Smith I, Thompson R, Timms K, Wardley A, Wilson G, Harries M, Ellis P, Ashworth A, Perou C, Bliss J, Rahman N, Brown R (2016) BRCA1 methylation status, silencing and treatment effect in the TNT trial: A randomized phase III trial of carboplatin compared with docetaxel for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer (CRUK/07/012). Paper presented at the San Antonio Breast Cancer Symposium, San Antonio, TX
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome I, Consortium IBC, Consortium IM-S, PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR (2013) Signatures of mutational processes in human cancer. Nature 500(7463):415–421. https://doi.org/10.1038/nature12477
Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kubler K, Mouw KW, Kamburov A, Maruvka YE, Leshchiner I, Lander ES, Golub TR, Zick A, Orthwein A, Lawrence MS, Batra RN, Caldas C, Haber DA, Laird PW, Shen H, Ellisen LW, D’Andrea AD, Chanock SJ, Foulkes WD, Getz G (2017) A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49(10):1476–1486. https://doi.org/10.1038/ng.3934
Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, Simpson PT, King TA, Raine K, Eyfjord JE, Kong G, Borg A, Birney E, Stunnenberg HG, van de Vijver MJ, Borresen-Dale AL, Martens JW, Span PN, Lakhani SR, Vincent-Salomon A, Sotiriou C, Tutt A, Thompson AM, Van Laere S, Richardson AL, Viari A, Campbell PJ, Stratton MR, Nik-Zainal S (2017) HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23(4):517–525. https://doi.org/10.1038/nm.4292
Zhao EY, Shen Y, Pleasance E, Kasaian K, Leelakumari S, Jones M, Bose P, Ch’ng C, Reisle C, Eirew P, Corbett R, Mungall KL, Thiessen N, Ma Y, Schein JE, Mungall AJ, Zhao Y, Moore RA, Den Brok W, Wilson S, Villa D, Shenkier T, Lohrisch C, Chia S, Yip S, Gelmon K, Lim H, Renouf D, Sun S, Schrader KA, Young S, Bosdet I, Karsan A, Laskin J, Marra MA, Jones SJM (2017) Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin Cancer Res 23(24):7521–7530. https://doi.org/10.1158/1078-0432.CCR-17-1941
Mulligan JM, Hill LA, Deharo S, Irwin G, Boyle D, Keating KE, Raji OY, McDyer FA, O’Brien E, Bylesjo M, Quinn JE, Lindor NM, Mullan PB, James CR, Walker SM, Kerr P, James J, Davison TS, Proutski V, Salto-Tellez M, Johnston PG, Couch FJ, Paul Harkin D, Kennedy RD (2014) Identification and validation of an anthracycline/cyclophosphamide-based chemotherapy response assay in breast cancer. J Natl Cancer Inst 106(1):djt335. https://doi.org/10.1093/jnci/djt335
Glas AM, Peeters J, Yau C, Wolf DM, Sanil A, Li Y, Severson T, Linn S (2014) Evaluation of a BRCAness signature as a predictive biomarker of response to veliparib/carboplatin plus standard neoadjuvant therapy in high-risk breast cancer: results from the I-SPY 2 TRIAL. Eur J Cancer 50:(173)
Willers H, Taghian AG, Luo CM, Treszezamsky A, Sgroi DC, Powell SN (2009) Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res 7(8):1304–1309. https://doi.org/10.1158/1541-7786.MCR-09-0149
Powell SN, Riaz N, Mutter RW, Ng CKU, Delsite R, Riscuoglio S, King TA, Martelotto L, Sakr R, Brogi E, Edelweiss M, Lim R, Higginson D, Weigelt B, Lee W, Reis-Filho JS (2016) A functional assay for homologous recombination (HR) DNA repair and whole exome sequencing reveal that HR-defective sporadic breast cancers are enriched for genetic alterations in DNA repair genes. Cancer Res 76 (4):Abstract S4-03
Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, Rouas G, Francis P, Crown JP, Hitre E, de Azambuja E, Quinaux E, Di Leo A, Michiels S, Piccart MJ, Sotiriou C (2013) Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02–98. J Clin Oncol 31(7):860–867. https://doi.org/10.1200/JCO.2011.41.0902
Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, Wolff AC, Wood WC, Davidson NE, Sledge GW, Sparano JA, Badve SS (2014) Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol 32(27):2959–2966. https://doi.org/10.1200/JCO.2013.55.0491
Ali HR, Provenzano E, Dawson SJ, Blows FM, Liu B, Shah M, Earl HM, Poole CJ, Hiller L, Dunn JA, Bowden SJ, Twelves C, Bartlett JM, Mahmoud SM, Rakha E, Ellis IO, Liu S, Gao D, Nielsen TO, Pharoah PD, Caldas C (2014) Association between CD8 + T-cell infiltration and breast cancer survival in 12,439 patients. Ann Oncol 25(8):1536–1543. https://doi.org/10.1093/annonc/mdu191
Dieci MV, Mathieu MC, Guarneri V, Conte P, Delaloge S, Andre F, Goubar A (2015) Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann Oncol 26(8):1698–1704. https://doi.org/10.1093/annonc/mdv239
Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Torne C, Weichert W, Engels K, Solbach C, Schrader I, Dietel M, von Minckwitz G (2010) Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 28(1):105–113. https://doi.org/10.1200/JCO.2009.23.7370
Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, Pfitzner BM, Salat C, Loi S, Schmitt WD, Schem C, Fisch K, Darb-Esfahani S, Mehta K, Sotiriou C, Wienert S, Klare P, Andre F, Klauschen F, Blohmer JU, Krappmann K, Schmidt M, Tesch H, Kummel S, Sinn P, Jackisch C, Dietel M, Reimer T, Untch M, Loibl S (2015) Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 33(9):983–991. https://doi.org/10.1200/JCO.2014.58.1967
Dieci MV, Criscitiello C, Goubar A, Viale G, Conte P, Guarneri V, Ficarra G, Mathieu MC, Delaloge S, Curigliano G, Andre F (2014) Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: a retrospective multicenter study. Ann Oncol 25(3):611–618. https://doi.org/10.1093/annonc/mdt556
Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, Combs S, Rimm DL, Giltnane JM, Estrada MV, Sanchez V, Sanders ME, Cook RS, Pilkinton MA, Mallal SA, Wang K, Miller VA, Stephens PJ, Yelensky R, Doimi FD, Gomez H, Ryzhov SV, Darcy PK, Arteaga CL, Balko JM (2016) RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res 22(6):1499–1509. https://doi.org/10.1158/1078-0432.CCR-15-1125
Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, Wienert S, Van den Eynden G, Baehner FL, Penault-Llorca F, Perez EA, Thompson EA, Symmans WF, Richardson AL, Brock J, Criscitiello C, Bailey H, Ignatiadis M, Floris G, Sparano J, Kos Z, Nielsen T, Rimm DL, Allison KH, Reis-Filho JS, Loibl S, Sotiriou C, Viale G, Badve S, Adams S, Willard-Gallo K, Loi S, International TWG (2015) The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol 26(2):259–271. https://doi.org/10.1093/annonc/mdu450
van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ (2016) Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer 16(4):219–233. https://doi.org/10.1038/nrc.2016.16
Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, Loi S (2016) Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol 13(4):228–241. https://doi.org/10.1038/nrclinonc.2015.215
Dunn GP, Koebel CM, Schreiber RD (2006) Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6(11):836–848. https://doi.org/10.1038/nri1961
Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM (2012) Leukocyte composition of human breast cancer. Proc Natl Acad Sci USA 109(8):2796–2801. https://doi.org/10.1073/pnas.1104303108
Wherry EJ, Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15(8):486–499. https://doi.org/10.1038/nri3862
Teng MW, Galon J, Fridman WH, Smyth MJ (2015) From mice to humans: developments in cancer immunoediting. J clin invest 125(9):3338–3346. https://doi.org/10.1172/JCI80004
Zarour HM (2016) Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin Cancer Res 22(8):1856–1864. https://doi.org/10.1158/1078-0432.CCR-15-1849
Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L (2016) Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol 34(21):2460–2467. https://doi.org/10.1200/JCO.2015.64.8931
Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, Cescon DW, Iwata H, Campone M, Nanda R, Hui R, Curigliano G, Toppmeyer D, O’Shaughnessy J, Loi S, Paluch-Shimon S, Card D, Zhao J, Karantza V, Cortes J (2017) Phase 2 study of pembrolizumab (pembro) monotherapy for previously treated metastatic triple-negative breast cancer (mTNBC): KEYNOTE-086 cohort A. J Clin Oncol 35(15_suppl):1008–1008. https://doi.org/10.1200/JCO.2017.35.15_suppl.1008
Schmid P, Cruz C, Braiteh FS, Eder JP, Tolaney S, Kuter I, Nanda R, Chung C, Cassier P, Delord J-P, Gordon M, Li Y, Liu B, O’Hear C, Fasso M, Molinero L, Emens LA (2017) Abstract 2986: Atezolizumab in metastatic TNBC (mTNBC): Long-term clinical outcomes and biomarker analyses. Cancer Res 77(13 Supplement):2986–2986. https://doi.org/10.1158/1538-7445.am2017-2986
Dirix L, Takacs I, Nikolinakos P, Jerusalem G, Arkenau H-T, Hamilton E, von Heydebreck A, Grote H-J, Chin K, Lippman M (2016) Abstract S1-04: Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase Ib JAVELIN solid tumor trial. Cancer Res 76(4 Supplement):S1-04. https://doi.org/10.1158/1538-7445.sabcs15-s1-04
Nanda R, Liu MC, Yau C, Asare S, Hylton N, Veer LVt, Perlmutter J, Wallace AM, Chien AJ, Forero-Torres A, Ellis E, Han H, Clark AS, Albain KS, Boughey JC, Elias AD, Berry DA, Yee D, DeMichele A, Esserman L (2017) Pembrolizumab plus standard neoadjuvant therapy for high-risk breast cancer (BC): Results from I-SPY 2. J Clin Oncol 35(15_suppl):506–506. https://doi.org/10.1200/JCO.2017.35.15_suppl.506
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA (2015) Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Sci 350(6257):207–211. https://doi.org/10.1126/science.aad0095
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348(6230):124–128. https://doi.org/10.1126/science.aaa1348
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160(1–2):48–61. https://doi.org/10.1016/j.cell.2014.12.033
Safonov AM, Jiang T, Bianchini G, Platt J, Shi W, Rimm DL, Pusztai L, Hatzis C (2016) Association between DNA level aberrations and immune cell infiltration in breast cancer. ASCO Meeting Abstracts 34(15_suppl):3078
Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, Muggia F, Symmans WF (2001) Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin Cancer Res 7(10):3025–3030
Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G (2015) Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 28(6):690–714. https://doi.org/10.1016/j.ccell.2015.10.012
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, Mick R, Pryma DA, Xu X, Feldman MD, Gangadhar TC, Hahn SM, Wherry EJ, Vonderheide RH, Minn AJ (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520(7547):373–377. https://doi.org/10.1038/nature14292
Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, Liu Y, Tang J, Wang S, Fu YX (2010) The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 18(2):160–170. https://doi.org/10.1016/j.ccr.2010.06.014
Konstantinopoulos P, Sachdev J, Schwartzberg L, Matulonis U, Sun P, Wang J, Guo W, Bobilev D, Aktan G, Karantza V (2017) 1143PDDose-finding combination study of niraparib and pembrolizumab in patients (pts) with metastatic triple-negative breast cancer (TNBC) or recurrent platinum-resistant epithelial ovarian cancer (OC)(TOPACIO/Keynote-162). Ann Oncol. https://doi.org/10.1093/annonc/mdx376
Domchek S, Postel-Vinay S, Bang Y-J, Park Y, Alexandre J, Delord J-P, Italiano A, You B, Bastian S, Krebs M, Wang D, Waqar S, Angell H, Learoyd M, Chang S-C, Gresty C, Herbolsheimer P, Kaufman B (2018) Abstract PD6-11: An open-label, multitumor, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline < em> BRCA</em>-mutated (g < em> BRCA</em> m) HER2-negative metastatic breast cancer (MBC). Cancer Res. https://doi.org/10.1158/1538-7445.sabcs17-pd6-11
Huang J, Wang L, Cong Z, Amoozgar Z, Kiner E, Xing D, Orsulic S, Matulonis U, Goldberg MS (2015) The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem Biophys Res Commun 463(4):551–556. https://doi.org/10.1016/j.bbrc.2015.05.083
Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, Adams SF (2015) CTLA-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol Res 3(11):1257–1268. https://doi.org/10.1158/2326-6066.CIR-15-0044
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, Li CW, Chou CK, Lim SO, Chang SS, Litton J, Arun B, Hortobagyi GN, Hung MC (2017) PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res 23(14):3711–3720. https://doi.org/10.1158/1078-0432.CCR-16-3215
Acknowledgements
On December 7, 2015, the Triple Negative Breast Cancer Foundation convened a meeting in San Antonio, Texas of advocates, clinical, translational and basic investigators to review the state of current research on triple-negative breast cancer and make recommendations regarding opportunities for research in this breast cancer subtype. This motivation for this review developed from that meeting led by organizing committee Lisa A. Carey, Susan M. Domchek, Lisa A. Newman, George W. Sledge, Jr., and Eric P. Winer. We acknowledge and sincerely thank the Triple Negative Breast Cancer Foundation for their support of this symposium. Members who attended this symposium include Melinda Telli, Sherene Loi, Sam Aparicio, Daniel Stover, Sunil Badve, Judith Balmaña, Powel Brown, Fergus J. Couch, Massimo Cristofanilli, Rebecca Dent, Hayley Dinerman, Leisha Ann Emens, Adres Forero, Elizabeth Frank, Shridar Ganesan, Judy E. Garber, Lyndsay N. Harris, Steven J. Isakoff, Steve Kandel, Ian E. Krop, Elgene Lim, Olufunmilayo I. Olopade, Ben Ho Park, Lajos Pusztai, Andrea Richardson, Hope Rugo, William Sikov, Daniel Silver, Sandra M. Swain, Tiffany Traina, Naoto Tada Ueno, Christine Wilson, and Antonio Wolff.
Research Support
Financial support provided by Susan G. Komen for the Cure CCR17480903 (D.G.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Telli, M.L., Stover, D.G., Loi, S. et al. Homologous recombination deficiency and host anti-tumor immunity in triple-negative breast cancer. Breast Cancer Res Treat 171, 21–31 (2018). https://doi.org/10.1007/s10549-018-4807-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-018-4807-x