
Abstract
Redox transitions induced by seasonal changes in water column O2 concentration can have important effects on solutes exchange across the sediment–water interface in systems polluted with acid mine drainage (AMD), thus influencing natural attenuation and bioremediation processes. The effect of such transitions was studied in a mesocosm experiment with water and sediment cores from an acidic reservoir (El Sancho, SW Spain). Rates of aerobic organic matter mineralization and oxidation of reduced inorganic compounds increased under oxic conditions (OX). Anaerobic process, like Fe(III) and sulfate reduction, also increased due to higher O2 availability and penetration depth in the sediment, resulting in higher regeneration rates of their corresponding anaerobic e− acceptors. The contribution of the different processes to oxygen uptake changed considerably over time. pH decreased due to the precipitation of schwertmannite and the release of H+ from the sediment, favouring the dissolution of Al-hydroxides and hydroxysulfates at the sediment surface. The increase in dissolved Al was the main contributor to water column acidity during OX. Changes in organic matter degradation rates and co-precipitation and dissolution of dissolved organic carbon and nitrogen with redox-sensitive Fe(III) compounds affected considerably C and N cycling at the sediment–water interface during redox transitions. The release of NO2− and NO3− during the hypoxic period could be attributed to ammonium oxidation coupled to ferric iron reduction (Feammox). Considering the multiple effects of redox transitions at the sediment–water interface is critical for the successful outcome of natural attenuation and bioremediation of AMD impacted aquatic environments.
Similar content being viewed by others
Introduction
Acid mine drainage (AMD) is the result of the oxidation of sulfide minerals exposed to the atmosphere as a side effect of mining. This oxidation is mediated by a combination of abiotic and microbial reactions and produces waters with a very low pH and high concentrations of SO42−, Fe, and other metals and metalloids. AMD causes high environmental impact, producing serious and long-persisting problems for ecosystem functioning and human health (Geller et al. 2009). Enhancing natural neutralization by favoring microbial and biogeochemical processes that consume acidity and increase the pH is one of the most promising approaches to treat AMD effluents and restore affected ecosystems (Geller et al. 2009; Koschorreck et al. 2007a). However, seasonal changes in the O2 availability and pH affect the long term success and reversibility of such bioremediation approaches in acid lakes and reservoirs (Geller et al. 2009).
Oxygen availability affects microbial metabolism, organic matter mineralization pathways and numerous abiotic redox-dependent physicochemical and mineralogical processes (Froelich et al. 1978; Torres et al. 2014). Under oxic conditions, aerobic oxidation of organic matter (Table 1; R1), abiotic and biological oxidation of H2S and Fe(II) by O2 (Table 1; R5 and R6), and the precipitation of Fe(III) oxyhydroxides (Table 1; R12) contribute to lowering the pH (Jourabchi et al. 2005; Soetaert et al. 2007). In contrast, under anoxic conditions, organic matter is mineralized anaerobically by bacteria utilizing an array of electron acceptors (e.g. SO42−, Fe(III), NO3−) (Table 1; R2, R4 and R3) which consume H+, leading to an increase in pH during stratification (Jourabchi et al. 2005; Soetaert et al. 2007). Therefore, redox transitions, through the changes in O2 availability and pH, can potentially affect many different biogeochemical processes in aquatic environments, which in turn determine the fluxes between the sediment and water column and the mobility of elements, nutrients and contaminants within the sediment. Changes in chemical speciation, bioavailability, toxicity and mobility of many metal and non-metal elements (Borch et al. 2010), bioavailability of limiting nutrients like phosphate (Gunnars and Blomqvist 1997; Hupfer and Lewandowski 2008), and the concentration, speciation and susceptibility to degradation of dissolved organic matter (Sinninghe Damste and De Leeuw 1990; Zonneveld et al. 2010; Arndt et al. 2013) are a few examples of the impact of redox transitions on key processes of aquatic ecosystems. Most likely many other effects remain unknown or poorly studied. In addition, redox oscillations also affect the microbial community (Frindte et al. 2013, 2016).
The effect of hypolimnetic hypoxic and anoxic conditions on degradation and preservation rates of organic matter and the regeneration and release of nutrients from lake sediments has received considerable attention (Maerki et al. 2009; Matzinger et al. 2010; Schwefel et al. 2017). However, the number of studies dealing specifically with the effects of reversible redox oscillations on the biogeochemistry of lake sediments is scarce (Borch et al. 2010; Frindte et al. 2013; Torres et al. 2014; Karimian et al. 2017). More specifically, the effect of redox transitions on spatiotemporal changes of key variables like O2 and pH and their interaction with the biogeochemical cycling of C, Fe, and S at the sediment—water interface have rarely been investigated simultaneously (Elberling and Damgaard 2001, Koschorreck et al. 2003, 2007a). This information is critical to understand and model net fluxes of inorganic nutrients, electron donors and acceptors, metal and non-metal elements, and organic compounds between the sediment and the water column. We hypothesized that redox transitions change the availability and microscale distribution of relevant electron donor and acceptors at the sediment water interface, which ultimately determine the rates and pathways of processes that affect pH and acidity in acidic environments such as organic matter mineralization and the net exchange of iron and aluminium.
The aim of this study was to determine how the hypoxic-oxic and oxic-hypoxic transitions affect the microscale distribution within the sediment and biogeochemical fluxes across the sediment–water interface of the most important electron donors and acceptors, protons, acidity and organic and inorganic nutrients in a model AMD contaminated environment. These redox transitions were studied in a mesocosm experiment with water and sediment cores from the Sancho Reservoir (Iberian Pyrite Belt, SW Spain). Relevant biogeochemical variables (O2, H2S, H+, inorganic N, Fe, Al, acidity, dissolved organic carbon and nitrogen) were monitored at different spatiotemporal scales in the water column, pore water and sediment solid phases. Based on this experiment, Torres et al. (2014) constructed and calibrated a diffusion–reaction model to calculate the net fluxes of metals, taking into account the annual variation of oxic and anoxic conditions of the bottom water, and to predict overall water quality. Here, we focus on the interaction between the cycles of several important elements (organic carbon, nitrogen, oxygen, sulfur, iron and aluminium) and their effect on the exchange of H+, acidity and e− donors and acceptors across the sediment–water interface. The detailed study of redox transitions at the sediment–water interface is not only of theoretical interest but also of a practical one, since it advances our understanding of the biogeochemical and microbial processes behind the immobilization within the sediment or release to the water column of elements affecting the natural neutralization. This is important to increase our predictive capacity on the potential long-term success of current bioremediation ecotechnological treatments.
Materials and methods
Study site, cores and bottom water collection
The Sancho Reservoir (37°27′N, 6°59′O) is located on the Odiel River catchment (Southwest Spain). The reservoir (58 Hm3, 42.7 × 105 m2 and a maximum depth of 40 m close to the dam), with a pH around 3.5, is affected by the discharge of the Meca River. This river is heavily contaminated by AMD, with high concentrations of trace metals, iron and SO42−, and a mean pH of 2.6 (Sarmiento et al. 2009; Cánovas et al. 2016). The Sancho Reservoir is a warm monomictic body of water. During winter, the entire water column is mixed and dissolved oxygen reaches the sediment. During the rest of the year, the water column is stratified and anoxic conditions develop below 15 m (Torres et al. 2013, 2016).
Twelve sediment cores (5.8 cm internal diameter, 60 cm length) were collected using a gravity corer (UWITEC) from a depth of about 30 m close to reservoir dam during the stratification period (November 15th, 2010). The collected cores had around 30 cm of sediment and 30 cm of overlying bottom water. In situ temperature, O2, and pH, measured with a multiparameter probe (Hydrolab MS5), showed characteristic profiles of a late stratification phase (data not shown). Anoxic bottom water was collected with a Niskin bottle approximately 1.5 m above the sediment surface from the same sampling site. Cores and bottom water were protected from light and stored on ice (4 °C) until return to the laboratory on the same day (within 5 h).

Temporal evolution of O2, Fe, SO42−, pH, acidity, Al, dissolved organic carbon (DOC), dissolved organic nitrogen (DON), NH4+ and NO3− in the bulk water phase during the experiment. Data are presented as mean ± standard error (n = 3). Areas shaded in grey indicated initial and final hypoxic conditions periods
Experimental design
In the laboratory, all cores were introduced in a temperature controlled (12–14 °C) tank filled with 25 L of in situ bottom water. Cores were maintained in the dark under hypoxic or oxic conditions as described below. Hypoxic conditions (24 µmol O2 L−1, < 8% saturation) were obtained by vigorous bubbling of the tank water with N2. Although the initial objective was to obtain the same anoxic conditions as observed in situ, we were unable to reduce the O2 concentration to zero in a system open to the laboratory atmosphere. Microsensor measurements also required an open system to allow measurements across the sediment–water interface as described below.
The experiment was divided in three different stages: (1) initial hypoxic stage (HI), maintained with a N2 flux during 1 day, (2) oxic stage (OX), where the conditions of the water in the tank were switched to oxic by bubbling air into the tank, and maintained during 50 days, (3) final hypoxic stage (HF), which lasted 10 days, induced by bubbling N2 in the tank. A more detailed description and a sketch of the experimental design can be found elsewhere (Torres et al. 2014).
Water column and sediment analyses
Oxygen (oxygen electrode Crison OXI 45 P), pH, and temperature (pH electrode, Orion) of the bulk water phase of the tank were monitored during the whole experiment. Water from the tank was sampled on days 0, 1, 5, 10, 20, 30, 50, 51, 55 and 60. Water level was constant during stages 1 and 2. A slight evaporation was produced by the dry N2 bubbling during stage 3, and the solute concentrations were corrected assuming Mg conservative. Water samples were filtered through 0.2 µm nylon filters, acidified with 20% HNO3 (final pH < 2) and stored in darkness at −20 °C until analysis. Dissolved organic carbon (DOC) and dissolved total nitrogen (DTN) were measured on a TOC-Analyser (Thermo Finnigan, FlashEA1112) with an analytical error of 1.5 and 3%, respectively. Ammonium (detection limit: 2.38 µM) was analyzed according to Bower and Holm-Hansen (1980), and nitrate (detection limit: 2.15 µM) and nitrite (detection limit: 0.24 µM) were analyzed according to García-Robledo et al. (2014). Dissolved organic nitrogen (DON) was calculated as the difference between DTN and total dissolved inorganic nitrogen (NO3− + NO2− + NH4+). Posphate (detection limit: 2.38 µM) was measured according to Grasshoff (1983) and total P (detection limit: 0.2 mg L−1) by ICP-AES.
At the end of each stage, three cores were removed from the tank and processed for geochemical analyses of the sediment solid phase and pore water. Cores were sliced in several layers, 1-cm slices for the first 8 cm, and 2-cm slices down to 16 cm depth, inside a N2—purged glove box. Total organic carbon (Corg) was measured on a Thermo EA1108 elemental organic analyzer using standard protocols. Total nitrogen and total sulfur contents were measured on a CNHS elemental analyzer (Thermo EA Flash 2000) using standard protocols. The analytical error for Corg, N and S was below 5%. Pore water was extracted from every layer by centrifugation, filtered through 0.2 μm nylon filters. S, Al, Fe, and other metals were analyzed by inductively coupled plasma atomic emission spectrometry (ICP-AES) using a Thermo Jarrel-Ash instrument equipped with a CID detector (analytical error 6%). Fe in solution was always considered to be in the form of ferrous iron (Fe2+) since the pH was generally higher than 4 in the pore water and the water column (Stumn and Morgan 1996). Sulfate was calculated from total S assuming that other intermediate aqueous S species were of minor abundance compared to SO42− in our experimental conditions (Smith and Melville 2004; Torres et al. 2014).
Acidity was estimated from metal and H+ concentrations, as usually considered in in AMD environments (Kirby and Cravotta 2005), according to Eq. (1).
where cx are molal concentrations of element x.
Microsensor measurements
Vertical distributions of O2, pH, and H2S were determined at the sediment–water interface using selective microelectrodes after 1 day under HI conditions, and on days 1, 6, 10, 20, and 50 (OX stage), and days 51, 56, and 60 (HF stage) in the same 3 cores during the three stages of the experiment. On each sampling occasion, one core at a time was removed from the tank and maintained under a continuous flow-through system with in situ bottom water at similar physicochemical conditions (T, O2, pH) than those of the tank for the duration of the measurements (~ 1 h). Microelectrodes (Unisense®, Denmark) were used and calibrated according to Unisense manuals (www.unisense.com) and as described before (Corzo et al. 2005). Oxygen and H2S microelectrodes were connected to a picoammeter (PA2000, Unisense®), pH microelectrode to a high impedance mV-meter (MeterLab), and both signals recorded directly to a computer using an A/D converter (ACD-216, Unisense®). Profiles were automated using a motorized micromanipulator connected to a computer and operated by the software Sensor TracePro (Unisense®). In each core, three different vertical profiles of O2 (detection limit: 0.3–1 µM), pH (resolution 0.1 units) and H2S (detection limit: 0.3–1 µM) were measured in random positions on every sampling date. These repeated non-destructive measurements over time in the same cores are possible due to the slim diameter of the microsensors (25 µm at the tip).
Speciation of hydrogen sulfide in solution depends on pH, salinity and temperature (Millero et al. 1988) and since, microsensors can only measure H2S, the amount of S2− total (\({\text{S}}^{ 2- }_{\text{tot}}\) = H2S + HS− + S2−) was calculated from parallel pH measurements in given experimental conditions. For a pH < 9, \({\text{S}}^{ 2- }_{\text{tot}}\) can be calculated from Eq. (2):
where K1 is the first dissociation constant of H2S, and [H2S] and [H+] are the concentrations of H2S and protons measured with the H2S and pH microsensors, respectively. K1 can be calculated from pK1 according to Eq. (3):
where T is temperature (Kelvin), Ln is Napierian Logarithm and S is salinity (Millero et al. 1988). In our experimental conditions with a salinity of 0.33 and a temperature of 13ºC, pK1 of the first dissociation reaction was 7.11. Given this pK1 and the range of pH measured in the water column and the sediment, H2S always accounted for almost 100% of \({\text{S}}^{ 2- }_{\text{tot}}\). HS− was only scarcely relevant in the deepest layer of the sediment where pH was higher than 6.
Biogeochemical modelling and acidity net production calculations
Depth profiles of production/consumption and areal integrated rates for O2, H2S, H+, Fe and SO42− were calculated by biogeochemical modelling based on measured concentration profiles using the PROFILE software (Berg et al. 1998). Free solution molecular diffusion coefficients for O2, H2S, H+, Fe2+, and SO42− were calculated using the R package MARELAC (Soetaert et al. 2010). To ensure that the calculus domain could be extended to the sediment surface for Fe2+ and SO42−, their concentrations at the sediment surface were extrapolated by setting a diffusive boundary layer thickness equal to that obtained from microscale O2 profiles (i.e. 400–500 µm).
Net production rates of acidity within the sediment can be assumed to nearly equal the fluxes from sediment to water column since the downward directed flux was minimal (about 5% of net production). Net production rates of acidity were estimated by the addition of the contribution of each acidity component according to:
where Racidity is the areal integrated rate of net production or consumption of acidity (meq m−2 h−1), N is the total number of acidity components, ni is the number of equivalents per mole of the ith species and Ri is the areal integrated rate of production/consumption of the ith species (µmol m−2 h−1). Ri was previously calculated by numerical modelling the concentration profiles of each component (Berg et al. 1998). Previous studies on acid lakes used Eq. (4), but iron transformation rates were the only source of acidity (Peine et al. 2000). However, according to Eq. (1) more chemical species should be considered to explain acidity fluxes in the Sancho reservoir (i.e. Al, Fe(II), Mn, Zn, Cu, Co, and pH). Thus, Eq. (4) involves a multicomponent budget analogous to those carried out for the estimation of alkalinity fluxes in similar systems (Carignan 1985).
Statistical analysis
Data for the different variables are presented as mean ± standard error. A three-way permutational analysis of variance (PERMANOVA) using Euclidean distances was applied to test for differences between Corg, N, S, and C:N ratio (mol:mol) in the upper 2 cm at the end of every stage (HI, OX and HF) with stage and depth as fixed factors and core identity as a random factor nested within stage. A post hoc pairwise comparison was performed where a factor was significant at a level of p < 0.05.
Results
Oxygen
Mean O2 concentration in the mesocosm water column was about 7% of saturation (24 µmol O2 L−1) during the initial hypoxic conditions (HI) (Fig. 1a). Oxygen microprofiles at the sediment–water interface indicated that O2 was consumed within the upper 1 mm sediment layer (Fig. 2a). During the oxic stage (OX) of the experiment, O2 concentration in the water column rose to about 100% saturation (307–327 µmol O2 L−1). Oxygen penetration depth (zox) in this period increased with time; from ~ 2 mm the day after oxygenation to ~ 6 mm on day 50 (Fig. 2a). Oxygen profiles and zox similar to the initial ones were measured at the final hypoxic stage (HF).

Mean O2 (a), H2S (b), pH (c) and H+ (d) microprofiles at the sediment–water interface at different times during the three experimental stages: initial hypoxic (HI), oxic (OX) and final hypoxic (HF) conditions. Every plotted profile is a mean of 3–9 profiles. Notice the different depth scales in A compared with B, C, and D panels
Biogeochemical modeling of O2 profiles allowed detecting sediment layers with different net O2 consumption rate (Fig. 3a–c). In HI, maximum rates of net O2 consumption occurred in a narrow upper layer slightly below the sediment–water interface, whereas at the end of OX, net O2 consumption was maximum at the bottom of the oxic layer. The integrated net O2 consumption rate, which represents the diffusive flux of O2 to the sediment (Diffusive oxygen uptake, DOU), experienced a steep increase from HI (0.21 ± 0.07 mmol O2 m−2 h−1) to OX (2.20 ± 0.69 mmol O2 m−2 h−1) remaining at similar levels during the first 10 days and decreasing to values 4 times lower at the end of OX. During HF, DOU returned to levels similar to HI (Fig. 4a).

Representative profiles of O2 (a–c), \({\text{S}}^{ 2- }_{\text{tot}}\) (d–f) and H+ (g–i) concentrations in the initial hypoxic conditions (first column), after 10 days in oxic conditions (middle column) and after 50 days in oxic conditions (last column). Dots are experimental data. Black solid lines are the modeled profile (model). Vertical grey lines indicate net production/consumption rates (bottom axis) calculated for different sediment layers by the model

Temporal evolution of diffusive O2 uptake rate (a), net rates of \({\text{S}}^{ 2- }_{\text{tot}}\) production and oxidation within the sediment (b), and net H+ production and consumption within the sediment and net H+ flux across the sediment–water interface (b). Values were estimated from modelled profiles as shown in Fig. 3. Negative and positive values indicate net consumption and net production, respectively. Data are means ± standard errors of 6 different profiles (2 profiles × 3 cores). Areas shaded in grey indicated initial and final hypoxic conditions periods
Sulfide, sulfate, and total sulfur
Hydrogen sulfide was never detected in the water column during the experiment. However, microsensor measurement detected variable amounts of H2S within the sediment, up to 50 µmol L−1, under both hypoxic and oxic conditions (Fig. 2b). Vertical profiles of H2S were variable, although they presented typically a subsurface peak between 5 and 10 mm below the sediment surface (Fig. 2b), indicating the presence of a narrow production layer at this depth. Although, H2S represented almost 100% of total sulphide (\({\text{S}}^{ 2- }_{\text{tot}}\)), H2S profiles were transformed to \({\text{S}}^{ 2- }_{\text{tot}}\) profiles to calculate production and consumption rates. Produced \({\text{S}}^{ 2- }_{\text{tot}}\) was quickly consumed in the upper sediment layer (about 500 µm thick) and more slowly toward deeper sediment layers (Fig. 3d–f).
Net \({\text{S}}^{ 2- }_{\text{tot}}\) production rates increased from 50 to 233 µmol m−2 h−1 after 50 days in OX, decreasing immediately in HF to levels similar to those measured in HI (Fig. 4b). Net \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation rates above the production layer were 26.0 and 1.5 µmol m−2 h−1 in HI and HF respectively, and up to 139.1 µmol m−2 h−1 after 50 days in OX. Usually, the bottom consumption layer tended to be wider than the upper oxic layer and with lower volumetric H2S oxidation rates (Figs. 3d–f, 4b). Hydrogen sulfide was found significantly deeper when zox increased (r = 0.89, α = 0.05) and generally a small overlap existed between the final part of O2 profiles and the beginning of H2S or \({\text{S}}^{ 2- }_{\text{tot}}\) profiles (Figs. 2, 3).
Sulfate concentration in the pore water decreased exponentially with depth, reaching levels below detection limit at 5 cm depth. Sulfate increased reversibly from 0.4 to 2.1 mmol L−1 in the pore water of the first cm of sediment and from 1.2 to 1.6 mmol L−1 in the water phase during OX, decreasing both in the sediment and the water column in HF (Figs. 1b, 5a). Biogeochemical modelling of SO42− profiles confirmed the net uptake of SO42− by sediment (44 µmol SO42− m−2 h−1) in HI, while in OX, due to the strong and reversible increase of net SO42− production, the sediment became a net source of SO42− for the water column (70 µmol SO42− m−2 h−1) (Fig. 6a). The change to HF reversed the direction of the net flux and almost 95% of the SO42− net consumption within the sediment was supported by SO42− uptake from the water column (Fig. 6a).

Vertical pore water profiles of sulfate, Fe and acidity at the end of each experimental stages: initial hypoxic (HI), oxic (OX) and final hypoxic (HF) conditions. Every plotted profile is a mean ± SE of 3 cores

Depth integrated SO42− (a), Fe (b), and acidity (c) production, consumption and net rates in the sediment at the end of each experimental stage: initial hypoxic conditions (HI), 50 days of oxic conditions (OX) and 10 days of hypoxic conditions (HF). Values were estimated from modelled profiles as shown in Fig. 3
Total S content varied only in upper 2 cm between the three experimental stages, whereas deeper sediment layers remained basically unaffected (result not shown, see Torres et al. 2014). Thus, mean concentrations of S at the end of every stage were calculated for the upper 2 cm layer only (Table 2). Total S decreased significantly (2 mg cm−3) after 50 days in OX, while no change was observed after 10 days in HF (Table 2).
pH and proton flux
The pH measured in the bulk water phase in HI was 4.6 (Figs. 1, 2), similar to that measured in situ close to the sediment surface when cores were collected (data not shown). The pH in the sediment increased with depth, reaching a maximum pH of 6.5 (Fig. 2c). In OX, the pH near the sediment–water interface decreased respect to HI and the depth at which the maximum pH was observed increased during OX. Bulk water column pH decreased to 3.7 in the first 10 days in OX.
Proton concentration profiles were calculated from pH profiles to determine the changes in H+ fluxes at the sediment–water interface during the oxic-hypoxic transitions (Figs. 2d, 3). Consumption of H+ was restricted to a very narrow upper layer coincident with zox in HI (Fig. 3g). After the transition to OX, the H+ consumption profile changed reversibly to production profile and a clear net H+ production layer developed in the upper oxic sediment layer. Below the net H+ production layer, a strong net consumption of H+ was observed in OX (Fig. 3f). Maximum increase in integrated H+ production occurred in the first 20 days in OX, but since H+ consumption increased similarly as well, the net H+ efflux from the sediment to the water column (138.35 ± 28.03 µmol H+ m−2 h−1) was important only from day 20 in OX (Fig. 4c).
Organic carbon and nitrogen
Dissolved organic carbon (DOC) and dissolved organic nitrogen (DON) increased initially from 300 to 375 µmol DOC L−1 and from 39 to 51 µmol DON L−1 µmol L−1 in OX, decreasing slowly until the end of this stage (Fig. 1f). The transition from OX to HF induced the immediate release of DOC and DON from the sediment as well. Differences in the Corg and N content of the sediment were observed only in the upper 2 cm of the sediment column (Table 2). Mean Corg decreased only 1 mg cm−3 after 50 days in OX, however, this difference was not statistically significant. However, Corg decreased 4 mg cm−3 after 10 days in HF, being the decrease in the sediment coincident with an increase in DOC (60 µmol L−1) in the water column (Fig. 1f). Sediment N content followed generally the same pattern as Corg during the experiment, whereas the C:N ratio did not change significantly during redox transitions (Table 2).
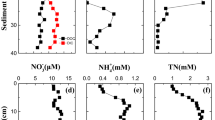
The dominant form of inorganic N in the water phase was NH4+ (more than 80%) during the entire experiment. Concentrations of NH4+ increased in OX, showing a peak after 6 days that coincided with a peak in DOC (Fig. 1e). Thereafter, concentrations increased steadily up to a maximum of 175 μmol L−1 at the end of OX, followed by a sharp decrease after the change to HF. NO2− and NO3− were almost undetectable during OX, but the sediment released small amounts of NO2− (result not shown) and relatively larger amounts of NO3− after the change to HF. Phosphate and total dissolved P remained always below detection limits.
Iron, aluminium and acidity
Iron concentration in the pore water and water column were similar in HI but changed considerably in OX. Fe content in the water column initially increased slightly but became undetectable after 10 days in OX (Fig. 1b), while Fe increased in the pore water, from 0.13 to 0.77 mmol Fe L−1 at the sediment surface after 50 days in OX (Fig. 5b). Both Fe production rate in the pore water (245 µmol Fe m−2 h−1) and net flux towards the water column increased in OX compared to HI (Fig. 6b). The Fe released to the oxic water column immediately precipitated as schwertmannite in OX (result not shown, see Torres et al. 2014). Fe content in the pore water decreased immediately in HF due to the decrease in Fe net production (50%), whereas at the same time a strong increase of Fe in the water column was observed, despite of a lower efflux rate, most likely because the released Fe remained in solution in the hypoxic water column.
Aluminium concentration in the water column was also notably affected by O2, increasing in OX and decreasing in HF (Fig. 1d). Acidity in the water column followed a trend similar to Al, after an initial transient decrease, increased strongly (0.75 meq m−2 h−1) up to 1.05 meq L−1 at day 30 and remained constant the last 20 days of OX (Fig. 1d). Acidity within the sediment was similar to that of water column and increased considerably in the upper 5 cm layer in OX (1.64 meq L−1), being higher than the water column (Fig. 5c). The reestablishment of hypoxic conditions did not change significantly the acidity in the water column, while Al decreased clearly. On the contrary, acidity in the sediment reversed back to initial conditions with the exception of the surface layer.
The contribution of Al, Fe, H+ and other elements to acidity in the water column and sediment changed in different ways during redox transitions. In the water column, Al, Fe and H+ contributions to acidity, estimated from Eq. (1), were 44, 36 and 1.5% respectively in HI. However, since Fe disappeared from the water column in OX, the increase in acidity in this stage was mainly a consequence of the increase in Al, increasing its contribution to acidity as well (68.9%), the rest being attributed to increases in H+ (about 6%) and Zn, Co, Ni, and Cu in the water column (results not shown, see Torres et al. 2014). In contrast to water column, Fe was always the main contributor (65–98%) to pore water acidity, with the contributions of Al and pH being minimal under hypoxic and oxic conditions. The flux of acidity indicated a net sediment consumption (0.08 meq m−2 h−1) in HI that changed to production of acidity within the upper sediment layers in OX, being partially exported to the water column (0.52 meq m−2 h−1) after 50 days in OX. These changes reversed in HF (Fig. 6c).
Discussion
Oxygen uptake at the sediment–water interface
Benthic oxygen flux has been shown to depend on several factors including organic carbon content, upward flux of reduced inorganic species, O2 concentration, temperature, water turbulence and bioirrigation (Rabouille and Gaillard 1991; Cai and Reimers 1995; Lorke et al. 2003; Glud 2008). In our experimental conditions, temperature and water turbulence can be considered constant whereas bioirrigation does not exist since sediment cores were collected in anoxic conditions. Therefore, only diffusive fluxes will be considered.
Diffusive oxygen uptake (DOU) rates at the sediment water interface were in the range or slightly higher than those available in the literature for acid lakes (Kühl et al. 1998; Koschorreck et al. 2003; Lagauzère et al. 2011) and neutral lakes (Sweerts et al. 1991; Maerki et al. 2009; Matzinger et al. 2010; Schwefel et al. 2017). The instantaneous increase in O2 concentration in the HI-OX transition probably induced an increase in the oxidation rates of organic matter and reduced compounds like Fe2+, \({\text{S}}^{ 2- }_{\text{tot}}\) (mainly as H2S), and metallic sulfides, among others (Table 1). However, DOU decreased during OX likely due to a concurrent decrease of labile organic matter and reduced inorganic compounds. In addition, the relative contribution of these compounds to DOU likely changed during the experiment.
The changes in the partition of the DOU between different process (organic matter mineralization, sulfide and Fe2+ oxidation) during the redox transitions was estimated through several complementary approaches (Supplementary Tables S1, S2). The fraction of DOU due to the oxidation of \({\text{S}}^{ 2- }_{\text{tot}}\) (DOUS) can be estimated from the \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation rates calculated from the modeling of \({\text{S}}^{ 2- }_{\text{tot}}\) profiles in the oxic layer (up oxidation, Fig. 4) and the stoichiometry of reaction 6 (Table 1). DOUS consumed 0.054 mmol O2 m−2 h−1 in HI, increasing to 0.281 mmol O2 m−2 h−1 after 50 days in OX. These calculations indicate that in HI about 26% of DOU was due to the oxidation of \({\text{S}}^{ 2- }_{\text{tot}}\), while in OX the contribution of DOUS to DOU changed from a minimal contribution during the first 10 days (4%) to 46% after 50 days (Table S2). Similar seasonal variability in H2S oxidation rates has been found in field studies (Sweerts et al. 1991; Urban et al. 1994). Our calculations assume that \({\text{S}}^{ 2- }_{\text{tot}}\) is oxidized in the upper oxidation layer only by O2. However, \({\text{S}}^{ 2- }_{\text{tot}}\) can be oxidized by FeOOH(s) as well, so the estimation of O2 consumption by \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation represents maximal values. In addition, oxidation of \({\text{S}}^{ 2- }_{\text{tot}}\) to S° might decouple the temporal changes of \({\text{S}}^{ 2- }_{\text{tot}}\) and O2. However, this process was unlikely in the Sancho sediments because we detected a clear decrease of total S during OX (Table 2).
The fraction of DOU consumed in the mineralization of OM (DOUCorg) was calculated in several ways. Three out of the four approaches used here (Supplementary Table S1) to calculate DOUCorg, i.e. a mechanistic model based on the Monod equation (Supplementary material, Eq. (1); Rabouille and Gaillard 1991; Canavan et al. 2006), an empirical equation (Cai and Reimers 1995) and the difference in Corg during OX, gave very similar estimates. DOUCorg was 0.08–0.12 and mmol O2 m−2 h−1 in HI and increased to 0.40–0.63 mmol O2 m−2 h−1 in OX (Supplementary Table S1). In the contrast, the calculation of the OM mineralization based on the NH4+ release rate to the water column during OX produced DOUCorg values 2–3 times higher than those calculated with the other approaches. This approach likely overestimated DOUCorg since part of the NH4+ released to the water column might originate from the exchangeable NH4+ fraction, which is known to be large in lake sediments and is released in OX (Mackin and Aller 1984; Morse and Morin, 2005). In summary, the estimated DOUCorg in HI was about 49% of the total DOU measured with O2 microsensors. Of the remaining DOU, 26% was attributed to \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation and the rest 25% to the oxidation of free Fe2+ and metallic sulfides (Table S2).
Consumption of O2 by the oxidation of Fe and other metallic sulfides (DOUMS) can be estimated as: DOUMS = DOU − (DOUS + DOUCorg). The transition to OX increased DOUCorg in absolute terms, but this rate was only 20–26% of the measured transient increase in total DOU during the first 10 days in OX. These calculations indicated that the DOUMS represented 68–79% of total O2 uptake during the first 10 days of OX. However, after 50 days in OX no oxygen consumption could be attributed to metallic sulfides oxidation; the sum of aerobic oxidation of organic matter plus \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation was similar to the total O2 consumption rate measured by O2 microsensors. Therefore, these estimates suggest that oxidation of FeS, FeS2 and other metal sulfides by O2 (Table 1, R10) was very important at the beginning of OX. Its importance decreases or even disappears completely after a prolonged oxic period, once these compounds are exhausted in the oxic layer. The biogeochemical analysis of spatiotemporal changes presented here represent an important improvement of aerobic mineralization of organic matter contribution estimates with respect to previous ones (Torres et al. 2014) and emphasize the importance of C and N cycling at the sediment–water interface in AMD contaminated reservoirs.
Sulfur cycling at the sediment–water interface
Net \({\text{S}}^{ 2- }_{\text{tot}}\) production occurred mainly in a narrow layer (200–500 µm) located immediately below zox in both oxic and hypoxic conditions (Figs. 2b, 3d–f). Sulfide concentrations within the sediments were similar or lower than those measured in other acidic ecosystems of different types (Kühl et al. 1998; Koschorreck et al. 2003; 2007a; Wendt-Potthoff et al. 2012). Net SO42− reduction rates, calculated either by modelling the pore water profiles of SO42− or \({\text{S}}^{ 2- }_{\text{tot}}\) were similar, about 60 and 220 µmol SO42− m−2 h−1 in HI and OX, respectively (Fig. 6a). These net SO42− reduction rates in the sediment of the Sancho reservoir were in the lower part of the wide range of values available for acid lakes (Holmer and Storkholm 2001; Koschorreck 2008; Wendt-Potthoff et al. 2012).
Counterintuitively, net SO42− reduction rates were higher during OX than HI or HF (Figs. 4b, 5), due to higher SO42− availability as consequence of an increase of oxidation of S-reduced compounds, such as \({\text{S}}^{ 2- }_{\text{tot}}\), FexS, other metal sulfides, and possibly organic S, in the presence of higher O2 concentration and a deeper zox (Kling et al. 1991). In addition to an increase of sulfate reduction, the higher net production of SO42− in the sediment resulted in a net transfer of S to the water column in the form of regenerated SO42− (Table 2, Fig. 1b) (Kling et al. 1991; Holmer and Storkholm 2001; Küsel 2003). Part of this SO42− flux to the water column (about 30%) might be due to the dissolution of hydrobasaluminite as well (Table 1, R13; Sánchez-España et al. 2011). However, once released to the water column, only about 28% of the SO42− remained in the water column during OX according to calculations based on total S and SO42− budgets in the sediment and the water column, respectively. The rest precipitated most likely mainly as schwertmannite (Table 2; R 11), a phenomenon that was most intensely observed in the first 10 days in OX; when schwertmannite covered all solid surfaces within the mesocosm (e.g. tank walls, tank bottom, cores) (Torres et al. 2014).
Net SO42− production rates calculated from the \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation or consumption rates in the oxic and the anoxic part of the sediment increased along OX (Fig. 4b). Oxic \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation (R6, Table 1) occurred in a narrow layer above the SO42− reduction zone (up oxidation) where O2 and H2S profiles generally overlapped as previously found in other systems (Jørgensen and Revsbech 1983). Turnover times in the SO42− reducing layer and the oxic and anoxic \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation layers, assuming constant average concentrations and rates, were about 10, 17, and 86 days, respectively. These values are higher than those reported for biofilms and microbial mats (seconds to hours, Kühl et al. 1992; Dillon et al. 2007), but lower than those for acid lakes (80 days, Koschorreck et al. 2003) or the Black Sea anoxic sediments (about 5 years, Jørgensen et al. 2001).
In the absence of O2, \({\text{S}}^{ 2- }_{\text{tot}}\) can (1) be oxidized anaerobically by manganese and iron oxides, either abiotically or biotically by bacteria (Jørgensen and Bak 1991; Jost et al. 2010) (2) precipitate with Fe2+ and other divalent metals to form different metal sulfides (R9 and R10) or (3) be adsorbed to organic matter (Table 1; R11). In the sediment of the Sancho Reservoir, anaerobic consumption of \({\text{S}}^{ 2- }_{\text{tot}}\) occurs mainly by oxidation with FeOOH (Table 1; R7), as Mn concentration in the pore water and solid phase were very low (Torres et al. 2014). Precipitation of Fe and other metal sulfides during OX in the anoxic sediment layer was most likely of little important since we observed a decrease in total S within the sediment. In addition to oxic \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation, anaerobic \({\text{S}}^{ 2- }_{\text{tot}}\) oxidation by R8 probably increased as a consequence of the higher FeOOH(s) availability in OX due to the oxidation of Fe2+ by O2 in the water column and sediment (Table 1; R6). Our results clearly show that seasonal redox fluctuations driven by higher availability of O2 affect the permanent burial of S and metals in the sediment (Küsel 2003; Blodau 2006; Wendt-Potthoff et al. 2012), likely reducing the efficiency of natural attenuation or bioremediation treatments.
Protons, iron cycling and acidity exchange at the sediment–water interface
The pH profiles at the sediment–water interface in the Sancho Reservoir were similar to those found in other acid lakes (Koschorreck et al. 2007a, b; Geller et al. 2009). Sediment pH was always higher than in the water column, being the maximum pH gradient, this is the maximum net H+ consumption layer, at or below the zox (Figs. 2a, c, 3g, h). In the pH range measured in the Sancho sediment, H+ consumption rates by iron reduction coupled to either organic matter or H2S oxidation (Table 1; R4 and R7, respectively) were the main consuming processes, probably an order of magnitude higher than sulfate reduction and denitrification (Table 1; R2 and R3) (Jourabchi et al. 2005; Soetaert et al. 2007). The switch to OX changed the H+ consumption profile to a production profile, where H+ production occurred at sediment surface and extended downward during OX (Figs. 2d, 3g–i). This was apparently due to the increase of zox and consequently of aerobic oxidation of H2S and Fe2+ followed by the precipitation of Fe(III) oxyhydroxides (Table 1; R5, R6 and R12).
Part of the H+ produced within the sediment during OX was exported to the water column, changing the role of the sediment from net sink during HI to net source, thus contributing to the observed acidification of the water column (Fig. 1). Two clear successive phases were observed in OX. During the first phase, the increase of H+ in the water column was 5.5 times higher than the release from the sediment up to day 10 (Table 3). The main contributor to this increase was probably the precipitation of schwertmannite (Table 1; R12) (Torres et al. 2014). During the second phase (days 20–50), the release of H+ from the sediment was neutralized (Table 3), likely due to the dissolution of Al hydroxides and hydroxysulfates (Table 1; R13) at the sediment surface (Totsche et al. 2003; Sánchez-España et al. 2011).
The efflux of acidity during OX was in the range of those measured in other acid lakes (Peine et al. 2000) and likely occurred via an efflux of Fe2+ from the sediment to the water column. Reduced iron in water column was immediately oxidized by O2 over the sediment surface (Table 1; R6), where Fe3+ precipitated mainly as schwertmannite releasing H+ (Table 1; R12) (Peine et al. 2000; Küsel 2003; Meier et al. 2004). The drop in pH increased the solubility of Al hydroxides, with Al being released to the water column. This, on the one hand, contributed to an increase of acidity and, on the other, to buffer further decreases of pH (Totsche et al. 2003; Sánchez-España et al. 2011). Another very important consequence of the interaction between Fe and Al cycling at the sediment surface is the transfer of e− acceptors in the form of Fe(III) minerals from the oxic water column to the sediment, where Fe(III) minerals can support high rates of anoxic oxidation of reduced sulfide minerals and organic matter (Peine et al. 2000; Wendt-Potthoff and Koschorreck 2002; Meier et al. 2004). This mechanism is a drawback for bioremediation of acid pit lakes based on carbon-driven alkalinity generation treatments (Geller et al. 2009; Nixdorf et al. 2010) and should be taken into account in all types of bioremediation strategies in acid mine drainage impacted systems. The dynamics of H+, acidity, and the solid and soluble phases of Fe and Al compounds at the sediment–water interface were therefore strongly interconnected and were highly dependent on O2 availability.
Carbon and nitrogen cycling during redox transitions in acidic environments
Despite the special characteristics of the AMD impacted Sancho Reservoir, the Corg and N contents and C:N ratio in the upper sediment layer were within the wide range observed in lakes (Kemp et al. 1977; Meyers and Ishiwatari 1993). Temporal changes in the Corg and N content of the sediment during the redox transition were only restricted to upper 2 cm. In this reactive layer, given an average net sedimentation rate of 1.6 cm year−1 for this system (Cánovas et al. 2016), OM presumably deposited during the last 1.25 years at most. Redox transitions exerted a strong effect on the exchange of DOC and DON across the sediment–water interface. During HI-OX transition, we observed a transient efflux of DOC and DON from the sediment (0.88 mM DOC m−2 h−1 and 0.08 mM DON m−2 h−1). Most likely, the higher O2 availability stimulated aerobic microbial OM degradation within the sediment (Freeman et al. 2001; Arndt et al. 2013), and soluble compounds diffused to the water column. The high efflux of NH4+ to the water column during OX (see below) supports an increase of OM degradation. In addition, the pH and redox changes in OX could have affected the adsorption–desorption balance of OM to sediment particles releasing OM adsorbed during the previous anoxic conditions (Sinninghe Damste and De Leeuw 1990; Zonneveld et al. 2010). The decrease of DOC and DON during the second half of OX was probably due to (1) their co-precipitation with mineral phases of Fe(III), mostly Fe(III) oxyhydroxysulfates like schwertmannite or jarosite (Küsel 2003; Meier et al. 2004), and (2) to the formation of macromolecular iron-organic carbon complexes, which has been shown to increase the transfer of OM to the sediment and protects it from degradation (Eglinton 2012; Lalonde et al. 2012; Barber et al. 2017). The overall result was the partial return of the DOC and DON released to the water column back to the sediment, which explains the minimal difference observed in Corg between HI and OX (Table 2).
Interestingly, we observed larger and statistically significant differences in Corg and N sediment contents after 10 days in HF. The decrease of 5.3 mg Corg cm−3 in HF was likely due to its release again to the water phase as DOC (0.42 mM DOC m−2 h−1) due to re-dissolution of ferric hydroxides and a subsequent desorption of bound organic compounds (Gu et al. 1995; Skoog et al. 2009; Riedel et al. 2013). The DOC:Fe stoichiometric ratio of the released DOC and Fe after dissolution of Fe hydroxides–Corg complex was about 0.46 probably due to the relatively long exposure to oxygen during the OX (Lalonde et al. 2012; Barber et al. 2017). Similarly, nitrogen in the sediment decreased significantly, suggesting the release of DON to the water phase (0.11 mM DON m−2 h−1) also due to the dissolution of ferric hydroxides.
Both DOC and DON were released right after each redox transition; however, the DOC:DON ratio was very different in each case. The DOC:DON ratio (10.8) was similar to that of sediment OM at the beginning of OX, but this ratio was considerably lower (3.7) after the transition to HF, suggesting that dissolved OM released in HF was considerably enriched in N. This observation deserves further study and it might be due to a higher co-precipitation of DON and NH4+ with Fe(III) hydroxides compared to DOC during OX.
Inorganic nitrogen was relatively high in our system. Probably, P low concentration due to sorption to ferric hydroxides as in other acid lakes (Beulker et al. 2003; Moser and Weisse 2011) lead to a low N demand overall. The most abundant species of inorganic nitrogen in our experiment was NH4+, probably due to the inhibition of nitrification in acid lakes (Jeschke et al. 2013). The transition to oxic conditions almost doubled sediment ammonification rates within the first 6 days, resulting in the release of NH4+ to the water column (0.5 mmol NH4+ m−2 h−1). This increase in NH4+ coincided with that of DOC. However, contrary to DOC and DON, NH4+ kept increasing during OX (0.1 mmol NH4+ m−2 h−1). This NH4+ release rate is in the range of those found in neutral fresh water environments, including eutrophic and hypereutrophic lakes (Burger et al. 2007; Kuwabara et al. 2009). In our experimental conditions, the release of NH4+ in OX might be due to an increase in the mineralization of Corg (Supplementary material, Table S1) and its release from the exchangeable NH4+ fraction of the sediment (Mackin and Aller 1984; Morse and Morin 2005).
The sudden decrease in NH4+ and the parallel release of NO2− (result not shown) and NO3− from the sediment in HF was a rather unexpected result (Fig. 1d). Recently, the anaerobic oxidation of NH4+ to N2, NO2−or NO3− coupled to Fe(III) reduction (Feammox), has been suggested in different iron reducing environments (Clément et al. 2005; Yang et al. 2012; Huang and Jaffé 2015). This process is thermodynamically favorable at the low pH (≤ 4, Thamdrup 2012) found in AMD contaminated systems, and could be the responsible for the production and release of NO2− and NO3− from the sediment in HF. Since nitrification is inhibited in acid lakes, Feammox might play a very important role in cycling of inorganic N in acids lakes. Further research, beyond the scope of this work, is needed to demonstrate the role of Feammox in the cycling of N and Fe in acidic environments such as the Sancho Reservoir.
Conclusions
The results presented here highlight the close and complex coupling between the C, N, O2, H+, S, Fe and Al cycles across the sediment–water interface during redox transitions in acid aquatic environments. In oxic conditions, the release of H+, SO42−, Fe2+ and Al3+ to the water column represents a combined mechanism of acidity and electrons transfer from the sediment to the oxic water column mediated by (1) the efflux of H+ and dissolved Fe(II), with additional smaller contributions of other metals, (2) the reoxidation of Fe(II) to Fe(III) and the high stoichiometric H+ production associated with the precipitation of schwertmannite, and (3) the H+ consumption during the dissolution of Al-hydroxides and hydroxysulfates at the sediment surface, with Al being the main contributor to water column acidity in OX. The precipitation of Fe(III) and SO42− as schwertmannite also represents a mechanism of reintroduction of regenerated e− acceptors to the sediment, which allows further anaerobic oxidation of organic and inorganic reduced compounds. Redox transitions affect the exchange of dissolved C and N between the water column and the sediment by (1) controlling the degradation rate of OM, which depends strongly on the availability electron acceptors, and (2) affecting the physicochemical adsorption–desorption of different organic compounds to sediment particles and their co-precipitation with Fe(III) hydroxides due to redox and pH changes. The unexpected release of NO2− and NO3− in the oxic—hypoxic transition, in parallel with the decrease in NH4+ in the water column, might indicate the presence of Feammox in the sediment of acid lakes for the first time and it might represent an alternative way to nitrification to regenerate NO3−. The prediction of the rates and final outcome of natural attenuation processes and bioremediation treatments requires a better understanding of the multiple biogeochemical effects of redox transitions at the sediment–water interface.
References
Arndt S, Jørgensen BB, LaRowe DE et al (2013) Quantifying the degradation of organic matter in marine sediments: a review and synthesis. Earth Sci Rev 123:53–86. https://doi.org/10.1016/j.earscirev.2013.02.008
Barber A, Brandes J, Leri A et al (2017) Preservation of organic matter in marine sediments by inner-sphere interactions with reactive iron. Sci Rep 7:366. https://doi.org/10.1038/s41598-017-00494-0
Belias C, Dassenakis M, Scoullos M (2007) Study of the N, P and Si fluxes between fish farm sediment and seawater. Results of simulation experiments employing a benthic chamber under various redox conditions. Mar Chem 103:266–275. https://doi.org/10.1016/j.marchem.2006.09.005
Berg P, Risgaard-Petersen N, Rysgaard S (1998) Interpretation of measured concentration profiles in sediment pore water. Limnol Oceanogr 43:1500–1510. https://doi.org/10.4319/lo.1998.43.7.1500
Beulker C, Lessmann D, Nixdorf B (2003) Aspects of phytoplankton succession and spatial distribution in an acidic mining lake (Plessa 117, Germany). Acta Oecol 24:25–31. https://doi.org/10.1016/S1146-609X(03)00002-X
Blodau C (2006) A review of acidity generation and consumption in acidic coal mine lakes and their watersheds. Sci Total Environ 369:307–332. https://doi.org/10.1016/j.scitotenv.2006.05.004
Borch T, Kretzschmar R, Skappler A et al (2010) Biogeochemical redox processes and their impact on contaminant dynamics. Environ Sci Technol 44:15–23. https://doi.org/10.1021/es9026248
Bower CE, Holm-Hansen T (1980) A salicylate-hypochlorite method for determining ammonia in seawater. J Fish Aquat Sci 37:794–798
Burger DF, Hamilton DP, Pilditch CA, Gibbs MM (2007) Benthic nutrient fluxes in a eutrophic, polymictic lake. Hydrobiologia 584:13–25. https://doi.org/10.1007/s10750-007-0582-0
Cai WJ, Reimers CE (1995) Benthic oxygen flux, bottom water oxygen concentration and core top organic carbon content in the deep northeast Pacific Ocean. Deep Res Part I 42:1681–1699. https://doi.org/10.1016/0967-0637(95)00073-F
Canavan RW, Slomp CP, Jourabchi P et al (2006) Organic matter mineralization in sediment of a coastal freshwater lake and response to salinization. Geochim Cosmochim Acta 70:2836–2855. https://doi.org/10.1016/j.gca.2006.03.012
Cánovas CR, Olías M, Macias F et al (2016) Water acidification trends in a reservoir of the Iberian Pyrite Belt (SW Spain). Sci Total Environ 541:400–411. https://doi.org/10.1016/j.scitotenv.2015.09.070
Carignan R (1985) Quantitative importance of alkalinity flux from the sediments of acid lakes. Nature 314:92–94. https://doi.org/10.1038/314141a0
Clément J-C, Shrestha J, Ehrenfeld JG, Jaffé PR (2005) Ammonium oxidation coupled to dissimilatory reduction of iron under anaerobic conditions in wetland soils. Soil Biol Biochem 37:2323–2328. https://doi.org/10.1016/j.soilbio.2005.03.027
Corzo A, Luzon A, Mayayo M et al (2005) Carbonate mineralogy along a biogeochemical gradient in recent lacustrine sediments of Gallocanta Lake (Spain). Geomicrobiol J 22:283–298. https://doi.org/10.1080/01490450500183654
Dillon JG, Fishbain S, Miller SR, Bebout BM, Habicht KS, Webb SM, Stahl DA (2007) High rates of sulfate reduction in a low-sulfate hot spring microbial mat are driven by a low level of diversity of sulfate-respiring microorganisms. Appl Environ Micorbiol. https://doi.org/10.1128/AEM.00357-07
Eglinton TI (2012) Geochemistry: a rusty carbon sink. Nature 483:165–166. https://doi.org/10.1038/483165a
Elberling B, Damgaard LR (2001) Microscale measurements of oxygen diffusion and consumption in subaqueous sulfide tailings. Geochim Cosmochim Acta 65:1897–1905. https://doi.org/10.1016/S0016-7037(01)00574-9
Freeman C, Ostle N, Kang H (2001) An enzymic “latch” on a global carbon store. Nature 409:149. https://doi.org/10.1038/35051650
Frindte K, Eckert W, Attermeyer K, Grossart HP (2013) Internal wave-induced redox shifts affect biogeochemistry and microbial activity in sediments: a simulation experiment. Biogeochemistry 113:423–434. https://doi.org/10.1007/s10533-012-9769-1
Frindte K, Allgaier M, Grossart HP, Eckert W (2016) Redox stability regulates community structure of active microbes at the sediment-water interface. Environ Microbiol Rep 8:798–804. https://doi.org/10.1111/1758-2229.12441
Froelich PN, Klinkhammer GP, Bender ML et al (1978) Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim Cosmochim Acta 43:1075–1090. https://doi.org/10.1016/0016-7037(79)90095-4
García-Robledo E, Corzo A, Papaspyrou S (2014) A fast and direct spectrophotometric method for the sequential determination of nitrate and nitrite at low concentrations in small volumes. Mar Chem 162:30–36. https://doi.org/10.1016/j.marchem.2014.03.002
Geller W, Koschorreck M, Wendt-Potthoff K et al (2009) A pilot-scale field experiment for the microbial neutralization of a holomictic acidic pit lake. J Geochem Explor 100:153–159. https://doi.org/10.1016/j.gexplo.2008.04.003
Glud RN (2008) Oxygen dynamics of marine sediments. Mar Biol Res 4:243–289. https://doi.org/10.1080/17451000801888726
Grasshoff K, Kremling K, Ehrhardt M (1999) Methods of seawater analysis, 3rd edn. Wiley, Weinheim
Gu B, Schmitt J, Chen Z et al (1995) Adsorption and desorption of different organic matter fractions on iron oxide. Geochim Cosmochim Acta 59:219–229. https://doi.org/10.1016/0016-7037(94)00282-Q
Gunnars A, Blomqvist S (1997) Phosphate exchange across the sediment-water interface when shifting from anoxic to oxic conditions—an experimental comparison of freshwater and brackish-marine systems. Biogeochemistry 37:203–226. https://doi.org/10.1023/A:1005744610602
Holmer M, Storkholm P (2001) Sulphate reduction and sulphur cycling in lake sediments: a review. Freshw Biol 46:431–451. https://doi.org/10.1046/j.1365-2427.2001.00687.x
Huang S, Jaffé PR (2015) Characterization of incubation experiments and development of an enrichment culture capable of ammonium oxidation under iron reducing conditions. Biogeosciences 12:769–779. https://doi.org/10.5194/bgd-11-12295-2014
Hupfer M, Lewandowski J (2008) Oxygen controls the phosphorus release from lake sediments—a long-lasting paradigm in limnology. Int Rev Hydrobiol 93:415–432. https://doi.org/10.1002/iroh.200711054
Jeschke C, Falagán C, Knöller K et al (2013) No nitrification in lakes below pH 3. Environ Sci Technol 47:14018–14023. https://doi.org/10.1021/es402179v
Jørgensen BB, Bak FB (1991) Pathways and microbiology of thiosulphate transformations and sulfate reduction in a marine sediment (Kattegat, Denmark). Appl Environ Microbiol 57:847–856
Jørgensen BB, Revsbech NP (1983) Colorless sulfur bacteria, Beggiatoa spp. and Thiovulum spp., in O2 and H2S microgradients. Appl Environ Microbiol 45:1261–1270. https://doi.org/10.1007/978-3-642-30141-4_78
Jørgensen BB, Weber A, Zopfi J (2001) Sulfate reduction and anaerobic methane oxidation in Black Sea sediments. Deep Sea Res Part I 48:2097–2120. https://doi.org/10.1016/S0967-0637(01)00007-3
Jost G, Martens-Habbena W, Pollehne F et al (2010) Anaerobic sulfur oxidation in the absence of nitrate dominates microbial chemoautotrophy beneath the pelagic chemocline of the eastern Gotland Basin, Baltic Sea. FEMS Microbiol Ecol 71:226–236. https://doi.org/10.1111/j.1574-6941.2009.00798.x
Jourabchi P, Van Cappellen P, Regnier P (2005) Quantitative interpretation of pH distributions in aquatic sediments: a reaction-transport modeling approach. Am J Sci 305:919–956. https://doi.org/10.2475/ajs.305.9.919
Karimian N, Johnston SG, Burton ED (2017) Effect of cyclic redox oscillations on water quality in freshwater acid sulfate soil wetlands. Sci Total Environ 581–582(2017):314–327. https://doi.org/10.1016/j.scitotenv.2016.12.131
Kemp ALW, Thomas RL, Wong HKT, Johnston LM (1977) Nitrogen and C/N ratios in the sediments of Lakes Superior, Huron, St. Clair, Erie, and Ontario. Can J Earth Sci 14:2402–2413. https://doi.org/10.1139/e77-205
Kirby CS, Cravotta CA III (2005) Net alkalinity and net acidity 1: theoretical considerations. Appl Geochem 20:1920–1940. https://doi.org/10.1016/j.apgeochem.2005.07.003
Kling GW, Giblin AE, Fry B, Peterson BJ (1991) The role of seasonal turnover in lake alkalinity dynamics. Limnol Oceanogr 36:106–122. https://doi.org/10.4319/lo.1991.36.1.0106
Koschorreck M (2008) Microbial sulphate reduction at a low pH. FEMS Microbiol Ecol 64:329–342. https://doi.org/10.1111/j.1574-6941.2008.00482.x
Koschorreck M, Wendt-Potthoff K, Geller W (2003) Microbial sulfate reduction at low pH in sediments of an acidic lake in Argentina. Environ Sci Technol 37:1159–1162. https://doi.org/10.1021/es0259584
Koschorreck M, Bozau E, Frömmichen R et al (2007a) Processes at the sediment water interface after addition of organic matter and lime to an Acid Mine Pit Lake mesocosm. Environ Sci Technol 41:1608–1614. https://doi.org/10.1021/es0614823
Koschorreck M, Kleeberg A, Herzsprung P, Wendt-Potthoff K (2007b) Effects of benthic filamentous algae on the sediment-water interface in an acidic mining lake. Hydrobiologia 592:387–397. https://doi.org/10.1007/s10750-007-0776-5
Kühl M, Jørgensen BB, Kuhl M, Jrgensen BOB (1992) Reduction and sulfide oxidation in compact microsensor measurements of sulfate reduction and sulfide oxidation in compact microbial communities of aerobic biofilms. Appl Environ Microbiol 58:1164–1174
Kühl M, Steuckart C, Eickert G, Jeroschewski P (1998) A H2S microsensor for profiling biofilms and sediments: application in an acidic lake sediment. Aquat Microb Ecol 15:201–209. https://doi.org/10.3354/ame015201
Küsel K (2003) Mining lake sediments. Water Air Soil Pollut 3:67–90
Kuwabara JS, Topping BR, Lynch DD et al (2009) Benthic nutrient sources to hypereutrophic upper Klamath Lake, Oregon, USA. Environ Toxicol Chem 28:516. https://doi.org/10.1897/08-207.1
Lagauzère S, Moreira S, Koschorreck M (2011) Influence of bioturbation on the biogeochemistry of littoral sediments of an acidic post-mining pit lake. Biogeosciences 8:339–352. https://doi.org/10.5194/bg-8-339-2011
Lalonde K, Mucci A, Ouellet A, Gélinas Y (2012) Preservation of organic matter in sediments promoted by iron. Nature 483:198–200. https://doi.org/10.1038/nature10855
Lorke A, Muller B, Maerki M, Wuest A (2003) Breathing sediments: the control of diffusive transport across the sediment-water interface by peroodic boundary-layer turblence. Limnol Oceanogr 48:2077–2085. https://doi.org/10.4319/lo.2003.48.6.2077
Mackin JE, Aller RC (1984) Ammonium adsorption in marine sediments. Limnol Oceanogr 29:250–257
Maerki M, Müller B, Dinkel C, Wehrli B (2009) Mineralization pathways in lake sediments with different oxygen and organic carbon supply. Limnol Oceanogr 54:428–438. https://doi.org/10.4319/lo.2009.54.2.0428
Matzinger A, Müller B, Niederhauser P et al (2010) Hypolimnetic oxygen consumption by sediment-based reduced substances in former eutrophic lakes. Limnol Oceanogr 55:2073–2084. https://doi.org/10.4319/lo.2010.55.5.2073
Meier J, Babenzien H-D, Wendt-Potthoff K (2004) Microbial cycling of iron and sulfur in acidic coal mining lake sediments. Biogeochemistry 67:135–156. https://doi.org/10.1023/A:1022103419928
Meyers PA, Ishiwatari R (1993) Lacustrine organic geochemistry-an overview of indicators of organic matter sources and diagenesis in lake sediments. Org Geochem 20:867–900. https://doi.org/10.1016/0146-6380(93)90100-P
Millero FJ, Plese T, Fernandez M (1988) The dissociation of hydrogen sulfide in seawater. Limnol Oceanogr 33:269–274. https://doi.org/10.4319/lo.1988.33.2.0269
Morse JW, Morin J (2005) Ammonium interaction with coastal marine sediments: influence of redox conditions on K. Mar Chem 95:107–112. https://doi.org/10.1016/j.marchem.2004.08.008
Moser M, Weisse T (2011) The most acidified Austrian lake in comparison to a neutralized mining lake. Limnologica 41:303–315. https://doi.org/10.1016/j.limno.2011.01.002
Nixdorf B, Uhlmann W, Lessmann D (2010) Potential for remediation of acidic mining lakes evaluated by hydrogeochemical modelling: case study Grünewalder Lauch (Plessa 117, Lusatia/Germany). Limnologica 40:167–174. https://doi.org/10.1016/j.limno.2009.12.005
Peine A, Tritschler A, Sel KK, Peiffer S (2000) Electron flow in an iron-rich acidic sediment—evidence for an acidity-driven iron cycle. Limnol Oceanogr 45:1077–1087. https://doi.org/10.4319/lo.2000.45.5.1077
Rabouille C, Gaillard J (1991) A coupled model representing the deep-sea organic carbon mineralization and oxygen consumption in surficial sediments. J Geophys Res 96:2761–2776. https://doi.org/10.1029/90JC02332
Riedel T, Zak D, Biester H, Dittmar T (2013) Iron traps terrestrially derived dissolved organic matter at redox interfaces. Proc Natl Acad Sci USA 110:10101–10105. https://doi.org/10.1073/pnas.1221487110
Sánchez-España J, Yusta I, Diez-Ercilla M (2011) Schwertmannite and hydrobasaluminite: a re-evaluation of their solubility and control on the iron and aluminium concentration in acidic pit lakes. Appl Geochem 26:1752–1774. https://doi.org/10.1016/j.apgeochem.2011.06.020
Sarmiento AM, Olías M, Nieto JM et al (2009) Natural attenuation processes in two water reservoirs receiving acid mine drainage. Sci Total Environ 407:2051–2062. https://doi.org/10.1016/j.scitotenv.2008.11.011
Schwefel R, Steinsberger T, Bouffard D et al (2017) Using small-scale measurements to estimate hypolimnetic oxygen depletion in a deep lake. Limnol Oceanogr. https://doi.org/10.1002/lno.10723
Sinninghe Damste JS, De Leeuw JW (1990) Analysis, structure and geochemical significance of organically-bound sulphur in the geosphere: state of the art and future research. Org Geochem 16:1077–1101. https://doi.org/10.1016/0146-6380(90)90145-P
Skoog AC, Arias-Esquivel VA (2009) The effect of induced anoxia and reoxygenation on benthic fluxes of organic carbon, phosphate, iron, and manganese. Sci Total Environ 407:6085–6092. https://doi.org/10.1016/j.scitotenv.2009.08.030
Smith J, Melville MD (2004) Iron monosulfide formation and oxidation in drain-bottom sediments of an acid sulfate soil environment. Appl Geochem 19:1837–1853. https://doi.org/10.1016/j.apgeochem.2004.04.004
Soetaert K, Hofmann AF, Middelburg JJ et al (2007) The effect of biogeochemical processes on pH. Mar Chem 105:30–51. https://doi.org/10.1016/j.marchem.2007.06.008
Soetaert K, Petzoldt T, Meysman F (2010) Marelac: tools for aquatic sciences. R package. http://CRAN.R-project.org/package=marelac
Sweerts J-PRA, Bär-Gilissen M-J, Cornelese AA, Cappenberg TE (1991) Oxygen-consuming processes at the profundal and littoral sediment-water interface of a small meso-eutrophic lake (Lake Vechten, The Netherlands). Limnol Oceanogr 36:1124–1133. https://doi.org/10.4319/lo.1991.36.6.1124
Thamdrup B (2012) New pathways and processes in the global nitrogen cycle. Annu Rev Ecol Evol Syst 43:407–428. https://doi.org/10.1146/annurev-ecolsys-102710-145048
Torres E, Ayora C, Canovas CR et al (2013) Metal cycling during sediment early diagenesis in a water reservoir affected by acid mine drainage. Sci Total Environ 461–462:416–429. https://doi.org/10.1016/j.scitotenv.2013.05.014
Torres E, Ayora C, Jiménez-Arias JL et al (2014) Benthic metal fluxes and sediment diagenesis in a water reservoir affected by acid mine drainage: a laboratory experiment and reactive transport modeling. Geochim Cosmochim Acta 139:344–361. https://doi.org/10.1016/j.gca.2014.04.013
Torres E, Galván L, Cánovas CR et al (2016) Oxycline formation induced by Fe(II) oxidation in a water reservoir affected by acid mine drainage modeled using a 2D hydrodynamic and water quality model—CE-QUAL-W2. Sci Total Environ 562:1–12. https://doi.org/10.1016/j.scitotenv.2016.03.209
Totsche O, Pöthig R, Uhlmann W et al (2003) Buffering mechanisms in acidic mining lakes—a model-based analysis. Aquat Geochem 9:343–359. https://doi.org/10.1023/B:AQUA.0000029035.88090.eb
Urban NR, Brezonik PL, Baker LA, Sherman LA (1994) Sulfate reduction and diffusion in sediments of Little Rock Lake, Wisconsin. Limnol Oceanogr 39:797–815. https://doi.org/10.4319/lo.1994.39.4.0797
Wendt-Potthoff K, Koschorreck M (2002) Functional groups and activities of bacteria in a highly acidic volcanic mountain stream and lake in Patagonia, Argentina. Microb Ecol 43:92–106. https://doi.org/10.1007/s00248-001-1030-8
Wendt-Potthoff K, Koschorreck M, Diez Ercilla M, Sánchez España J (2012) Microbial activity and biogeochemical cycling in a nutrient-rich meromictic acid pit lake. Limnologica 42:175–188. https://doi.org/10.1016/j.limno.2011.10.004
Yang WH, Weber KA, Silver WL (2012) Nitrogen loss from soil through anaerobic ammonium oxidation coupled to iron reduction. Nat Geosci 5:538–541. https://doi.org/10.1038/ngeo1530
Zonneveld KAF, Versteegh GJM, Kasten S et al (2010) Selective preservation of organic matter in marine environments; processes and impact on the sedimentary record. Biogeosciences 7:483–511. https://doi.org/10.5194/bg-7-483-2010
Acknowledgements
The research was funded by projects P11-RNM-7199 from the Junta de Andalucía and CTM2013-43857-R, CTM2017-82274-R, CGL2013-48460-CO2, CTM2014-61221-JIN and CGL2016-78783-C2-2-R from the Spanish I + D + I Program. J. L. Jiménez Arias was funded by a PhD grant (Ref:2010-063) from the University of Cadiz.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Jennifer Leah Tank.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Corzo, A., Jiménez-Arias, J.L., Torres, E. et al. Biogeochemical changes at the sediment–water interface during redox transitions in an acidic reservoir: exchange of protons, acidity and electron donors and acceptors. Biogeochemistry 139, 241–260 (2018). https://doi.org/10.1007/s10533-018-0465-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-018-0465-7