
Abstract
Oxyanions are ubiquitous in soils, organisms and the environment. Due to their unique chemical structure, oxyanions can be easily transferred into other systems. Carbonate (CO32−), nitrate (NO3−), phosphate (PO43−), silicate (SiO42−) and sulfate (SO42−) are the major oxyanions in organisms and the soil environment, whereas arsenate (AsO43−), antimonate (SbO43−), borate (BO33−), selenate (SeO42−), and tellurate (TeO42−) are generally reported as toxic chemicals found at trace levels. Excessive oxyanions leached from soils into water have caused severe environmental problems. Here, we review the factors affecting the structural configuration of oxyanions and organic acids adsorbed on iron oxides and hydroxides. The configuration of oxyanions on iron (hydr)oxides is controlled by surface loading, pH, sample phase, competing ions and organic acids. Under conditions of low surface loading and low pH at the interface in the absence of competing ions, oxyanions with high affinity possibly form a complex with higher denticity. But an increase in pH decreases the number of sorption sites; thus, a transition from a tri- or bidentate complex to monodentate and outer-sphere complexes occurs.
Similar content being viewed by others
Introduction
An oxyanion is a negatively charged molecule with the generic formula AxOzy; it is ubiquitous on Earth and is a fundamental constituent of organisms and the environment. Carbonate (CO32−), nitrate (NO3−), phosphate (PO43−), silicate (SiO42−) and sulfate (SO42−) are the major oxyanions in organisms and the soil environment, whereas arsenate (AsO43−), antimonate (SbO43−), borate (BO33−), selenate (SeO42−) and tellurate (TeO42−) are generally reported as toxic chemicals found at trace levels (Johnson 1971; Sparks 2000). Among the oxyanions, the oxyanions with arsenic, nitrogen and phosphorus are the most notorious compounds. The arsenic had known as a poison throughout history, while the nitrogen and phosphorus containing compounds had acted as limiting nutrients for the agriculture (Elser et al. 2007; Vahidnia et al. 2007). Recent anthropogenic activities such as mining, agricultural chemicals and wood preservatives have caused the severe arsenic contamination over 20 countries, and invention of Haber–Bosch process and mining of rock phosphate (phosphorite) have caused the severe environmental problems such as overfertilization in an agricultural land and the eutrophication in water system worldwide (Barberis et al. 1995; Jain and Ali 2000; Leermakers et al. 2006; Elser et al. 2007).
Due to their unique chemical structures, oxyanions can easily transform into other chemical species in a process called species transformation (Templeton and Fujishiro 2017). Environmental fluctuations, such as changes in the pH, redox potential and concentrations of ligands and adsorbents, lead to the species transformation of oxyanions in the soil environment, which have unpredictable fate because of the soil heterogeneity (Masscheleyn et al. 1991; Georgiadis et al. 2006; Han and Ro 2018b). For example, the reduction of arsenate to arsenite based on the redox potential and the protonation of phosphate based on the pH are well-known phenomena in the soil environment, and both reactions are species transformations. As a consequence, the mobility and bioavailability of these species are modified, and their environmental fates are reported to be different depending on the environmental fluctuations. Species transformation affects the affinity of a given compound to an adsorbent; thus, it determines the behavior of oxyanions in organisms and the soil environment (Pantsar-Kallio and Manninen 1997; Mir et al. 2007).
Not only environmental fluctuations but also organisms affect the species transformation of oxyanions in the soil environment. Within the rhizosphere, a plant influences the soil through various mechanisms, most significantly by releasing compounds into the soil (Bais et al. 2006; Huang et al. 2014). This process is referred to as rhizodeposition (Hütsch et al. 2002; Bais et al. 2006), and the major types of rhizodeposited substances include exudates, mucilage, border cells and gases (Haichar et al. 2014). An exudate is a soluble, low-weight molecule that undergoes a passive diffusion (Bertin et al. 2003), and recent studies have revealed that exudates such as root-derived organic acids increase bioavailability by competing with sorbed oxyanions, by dissolving oxyanion-sorbed minerals or by causing secondary exudation from microbes (Eick et al. 1999; Dakora and Phillips 2002; Jones et al. 2009). Citric, glutaric, oxalic and malonic acids are the most common exudates found in the rhizosphere (Wang et al. 2008a; Shi et al. 2010; Baetz and Martinoia 2014) and are known as nutrient sources for the microbial community and as stimulators for nutrient uptake, such as those of Al, Cu, Fe, N and P (Dakora and Phillips 2002; Tu et al. 2004). The organic acids not only affect the sorption of oxyanions directly but also might cause changes in the soil indirectly. The specific mechanisms and effects of the competition between oxyanions and organic acids have not yet been fully addressed because of the heterogeneity and complexity of the soil environment and current limitations on analytical techniques (Dakora and Phillips 2002).
The solid component of soil is a complex of various materials, such as phyllosilicates, metal (hydr)oxides and organic matter. Iron (hydr)oxide is one of the key sorbents in retaining both nutrients and pollutants because of its abundance, reactivity, surface area and surface charge (Panias et al. 1996; Lefevre 2004; Adegoke et al. 2013). As mentioned above, environmental fluctuation causes not only species transformation of oxyanions but also the transformation of iron (hydr)oxides, which changes their crystal structure and morphology, ultimately affecting the sorption characteristics of oxyanions in the soil environment. The transformation of oxyanion sorbate species and iron (hydr)oxide sorbents occurs separately or simultaneously by environmental fluctuations, and the unpredictable simultaneous transformation of sorbates and sorbents determines their behavior and fate in the soil environment. Not only the sorbate species and sorbent characteristics but also the structural configuration of the oxyanions with iron (hydr)oxides is important. There are two main types of interactions: an inner-sphere complex is formed by adsorption through covalent bonds, while an outer-sphere complex is formed by adsorption through electrostatic attraction and dispersion interactions (Stumm and Morgan 1981; Sparks 2003). The inner-sphere complex can be monodentate, bidentate or tridentate, and its structural configuration is essential to understanding the behavior of oxyanions in the soil environment because it determines their leachability and bioavailability (Fendorf et al. 1997; Zhu et al. 2013b; Liu et al. 2015).
With the recent development and production of chemical fertilizers, minor elements are no longer considered limiting factors in cultivation (Riley et al. 2001; López-Arredondo et al. 2014). With the use of overfertilization to maximize agricultural productivity, the attention paid to optimizing bioavailability through soil research and management has decreased (Timmons and Dylla 1981; Andrew et al. 2000; Riley et al. 2001; López-Arredondo et al. 2014). Furthermore, the demands of industrialization have exposed rare elements in the crust, such as arsenic, antimony and tellurium (Gleyzes and Tellier 2001; Babula et al. 2008; Rosen and Liu 2009). As a consequence, excessive nutrients and toxicants have leached from the pedosphere to the hydrosphere, causing severe environmental problems worldwide such as eutrophication and metal poisoning (Jain and Ali 2000; Smith et al. 2006; Smith and Schindler 2009; Han et al. 2014, 2016; Species and Hartley-whitaker 2015). Essential methods to mitigate such problems include maintaining the optimum bioavailability of nutrients by taking advantage of the uptake ability of plants, minimizing the bioaccessibility of toxicants by controlling environmental conditions, and reducing the leachability of nutrients and toxicants by regulating both plants and the environment; however, the heterogeneity and complexity of soil make bioavailability difficult to understand.
Therefore, it is important to understand not only the individual characteristics of oxyanions, organic acids and iron (hydr)oxides but also their complexly interconnected interactions in the rhizosphere to obtain a better comprehension of and insight into bioavailability. Thus, herein, we have summarized the literature regarding the transformation of iron (hydr)oxides and the structural configuration of oxyanions and organic acids on iron (hydr)oxides to understand the complex mechanisms that occur in the soil environment.
Transformation of iron (hydr)oxides
Formation and abundance of iron (hydr)oxides
Iron is the fourth most abundant element in the crust of the Earth, at 5.1% by mass (Cox 1989). Biotite [K(Fe2+)3Si3(AlO10(OH)2], pyroxene [(Fe2+)2(Si,Al)2O6], olivine [Fe22+SiO4], magnetite (Fe3O4) and pyrite (FeS2) are the main iron-containing minerals (Cornell and Schwertmann 2003), and weathering by hydrolysis and oxidation forms iron (hydr)oxide in the soil (Cornell and Schwertmann 2003; Schwertmann and Cornell 2008). During the weathering process, once the primary rock containing iron is exposed to oxic conditions, Fe2+ is readily oxidized to Fe3+ by providing an electron to oxygen under soil pH conditions, and then, soluble Fe3+ oxides are easily formed by hydrolysis. Based on the parent materials, climate, time, redox conditions and organisms involved, various iron (hydr)oxides can be formed, such as amorphous iron (hydr)oxide (AIO), goethite (α-FeOOH), lepidocrocite (γ-FeOOH), ferrihydrite (Fe2O30.5H2O), hematite (α-Fe2O3) and maghemite (γ-Fe2O3) (Cornell and Schwertmann 2003; Schwertmann and Cornell 2008).
AIO is the term for an iron and oxygen complex without crystallization, and recent studies have reported that AIO has a large surface area, superior catalytic activity and superparamagnetic behavior (Machala et al. 2007; Yusuf et al. 2010). The abundance of AIO in the soil environment has not yet been fully investigated, but is likely to be low because of its high reactivity and low stability (Srivastava et al. 2002). Goethite is a common iron hydroxide because of its thermodynamic stability and is a yellow–brown mineral (Schwertmann 1993). It is found in all regions with oxic and anoxic soils, often in the low part of a toposequence (Fig. 1) (Schwertmann 1985). Like goethite, hematite also has high thermodynamic stability, but it is mostly found in warm and oxic climates (Schwertmann 1985). The red color of topsoil at low latitudes is mainly caused by the presence of hematite (Schwertmann 1993). Goethite and hematite sometimes occur together, and the color indirectly indicates their ratio (Shaw et al. 2005). Lepidocrocite is less common in soil than are goethite and hematite but is generally found under redoximorphic conditions in cool and temperate climates (Vodyanitskii 2010). In the presence of a high level of carbonate or bacterial activity, the formation of goethite is preferred, while no lepidocrocite is formed; however, lepidocrocite is frequently associated with goethite in noncalcareous soils (Cornell and Schwertmann 2003). Ferrihydrite is structurally unstable and is found only in less weathered soil in cool and temperate zones under anoxic conditions (Schwertmann 1993). A high oxidation rate of Fe2+ with organic matter or silicate yields ferrihydrite instead of goethite or hematite in the soil environment (Johnson and McBride 1989; Cornell and Schwertmann 2003). Magnetite is a common lithogenic mineral in the heavy mineral fraction of soils with high magnetic properties (Schwertmann 1985; Cornell and Schwertmann 2003). Pedogenic magnetite was also discovered by recent studies, but the pedogenic process is still debatable (Fine et al. 1995; Geiss and Zanner 2006). Maghemite is widespread in the topsoil in tropical regions (Cornell and Schwertmann 2003), and its known formation pathways are the aerial oxidation of lithogenic magnetite and the heating of lepidocrocite, goethite and ferrihydrite.

Abundance of iron (hydr)oxides in different climate zones and the common pathways of formation and transformation in the environment. The rectangular and hexagonal shapes in the pedosphere indicate cubic and hexagonal phases, and the colors of the shapes resemble the colors of each iron (hydr)oxide. The size of the polygon shows the distribution with respect to the climate zone. Round-edged rectangular and round shapes indicate the amorphous solid phase and ions in the aqueous phase, respectively. Colored and overlapping arrows show the chemical or physical reactions involved in transformation and simultaneous reactions involved in transformation
Physiochemical transformation of iron (hydr)oxides
The transformation of iron (hydr)oxides can be categorized by two factors: changes in chemical composition and changes in crystal structure (Cornell and Schwertmann 2003). Dehydration, dehydroxylation and oxidation/reduction are examples of chemical transformation that result in changes in composition and structure, while topotactic and reconstructive transformations involve direct and indirect changes in the structure alone (Cornell and Schwertmann 2003; Vodyanitskii 2010; Adegoke et al. 2013). The weathering of primary minerals in the lithosphere releases Fe2+ ions, which are readily oxidized to form Fe3+ ions; the Fe3+ ions then hydrolyze the groundwater at typical soil pH to form Fe(III) oxides and (hydr)oxides. This process is referred to as reconstructive transformation employing dissolution and reprecipitation (Mackay 1960) and is the most common pathway for the iron cycle, while topotactic transformation is a structural change in the solid state that results from the modification of chemical characteristics (Cornell and Schwertmann 2003). Intensive weathering accompanied by large amounts of O2 and H2O increases the iron concentration in the pedosphere by drawing it from the lithosphere (Schwertmann 1985). The aqueous iron ions speciate into Fe2+ and Fe3+ ions depending on the redox potential, and the concentration of hydroxide ion (pOH) determines the formation of iron (hydr)oxides. Cornell and Schwertmann (2003) summarized the reconstructive transformation as follows: (1) the crystal growth of iron (hydr)oxides on the primary mineral, (2) pseudomorphosis, (3) formation of an amorphous coating on the primary mineral, and (4) the diffusion interval between the release and oxidation of Fe2+ ions. Fe3+ oxide is prevalent in the soil environment, and amorphous Fe(III) (hydr)oxide mainly forms goethite, which is a thermodynamically stable hexagonal structure (Machala et al. 2007; Yusuf et al. 2010).
Figure 1 summarizes the transformation of iron (hydr)oxide in the lithosphere, hydrosphere and pedosphere. The dehydration and dehydroxylation of goethite yield a topotactic transformation to hematite without organic matter and yield maghemite in the presence of organic matter (Cudennec and Lecerf 2005). The dissolution of goethite leads to the formation of amorphous iron3+ (hydr)oxide again; spatiotemporal variability leads to crystallization into the various iron (hydr)oxides; and repeated circulation through the iron cycle distributes iron throughout the soil profile (Schwertmann 1985, 1993). Except for goethite, which is distributed worldwide, iron (hydr)oxides can be divided into two groups: iron oxides (maghemite and hematite), which form under oxic and tropical conditions, and iron hydroxides (lepidocrocite and ferrihydrite), which form in cool and temperate climates under anoxic conditions (Cornell and Schwertmann 2003; Cudennec and Lecerf 2005). Interestingly, maghemite and lepidocrocite both show a cubic structure, while hematite and ferrihydrite are hexagonal (Cornell and Schwertmann 2003). No topotactic transformation among goethite, lepidocrocite and ferrihydrite has been reported, but the reconstructive transformation has been observed (Schwertmann and Taylor 1972; Cornell and Schwertmann 2003; Cudennec and Lecerf 2005). In contrast to the iron hydroxides, among the iron oxides, we observed direct transformation, such as the topotactic transformation from maghemite to hematite caused by thermal oxidation and the opposite transformation caused by dehydroxylation with organic matter (Swaddle and Oltmann 1980; Petrovsk et al. 1996; MacHala et al. 2011). The combination of dehydration and thermal events led to transformation from ferrihydrite to hematite (Cudennec and Lecerf 2006), while dehydration alone caused transformation from lepidocrocite to maghemite (Cornell and Schwertmann 2003; Cudennec and Lecerf 2005).
Topotactic transformation is observed only within iron hydroxides or iron oxides, while reconstructive transformation is more widely occurring. Spatiotemporal variability in thermal events, redox potential, pH, background ions and organic matter governs the interconversion of iron (hydr)oxides (Cornell and Schwertmann 2003; MacHala et al. 2011) and leads to perturbation in the behavior of oxyanions in soil because the transformation of iron (hydr)oxides determines their fundamental characteristics (Cornell and Schwertmann 2003; Schwertmann and Cornell 2008; MacHala et al. 2011; Weng et al. 2012). The observation of reconstructive transformation is a challenging task because it occurs with nanoparticles, which are difficult to separate from ions, the abundance on the surface is minor, and the process could be negatively affected by the phase transition during the preparation process of measurement (Usman et al. 2013; Šutka et al. 2015). For that reason, the identification of intermediates during dissolution/reprecipitation is extremely difficult; thus, it is hard to separate the topotactic and reconstructive processes at the solid–solution interface in the soil environment. Furthermore, the heterogeneity of soil makes it impossible to determine the intermediates among the numerous compounds and precipitates present on the soil surface; however, laboratory-scale experiments with relatively simple compositions have revealed the process of reconstructive transformation to extrapolate the reaction in nature (Usman et al. 2013; Šutka et al. 2015).
Numerous papers observed the laboratory-scale transformation of iron (hydr)oxides in simple systems by confirming a difference in the crystal structure induced by environmental conditions such as the elemental composition of reactants, Fe2+/Fe3+ ratio, reaction or stirring time, annealing temperature, redox potential and surface morphology for growth. The majority of papers observed a difference in the crystal structure before and after the experiment; however, most of them did not identify the intermediate structure during the transformation (Machala et al. 2007; Alibeigi and Vaezi 2008; Iwasaki et al. 2011; MacHala et al. 2011; Ray et al. 2013; Yuan et al. 2013; Šutka et al. 2015). This lack of information is mainly caused by the current limitations of analytical techniques because laboratory-scale X-ray diffractometry, X-ray photoemission spectroscopy and electron microscopy involve solid-phase measurement while controlling the atmosphere under purging or vacuum conditions. A synchrotron-based technique, such as X-ray absorption, X-ray scattering and X-ray crystallography, might detect the intermediate during the reconstructive transformation without phase transition of the samples at the solid–solution interface, but it is still a challenging problem because of the low abundance, rapid reaction rate and mostly amorphous structure of the intermediates.
The mechanisms of reconstructive transformation could be explained step by step. The dissolved ferrous and ferric ions from the Fe-containing minerals form a hexacoordinated aquo complex, and hydroxylation occurs at different rates depending on the pH (Jolivet et al. 2006). The hydroxylated Fe complex is unstable in solution; thus, condensation occurs through two mechanisms, namely olation and oxolation (Combes et al. 1990). The olation mechanism is a condensation process between two hydroxo ligands in which a water molecule is eliminated to form a hydroxo bridge, while the oxolation mechanism is a two-step condensation process between a hydroxo ligand and partially positively charged Fe center of a hydroxylated Fe complex (Jolivet et al. 2006). As a result, the excess hydrogen and hydroxyl ligand form a water molecule. Due to the high reactivity of the hydroxylated Fe complex, condensation as a ferric complex is rapid and hard to observe. The sequential condensation via olation and oxolation leads to the growth of amorphous iron hydroxides, and the elemental composition of reactants and the Fe2+/Fe3+ ratio are the key factors determining the reconstructive transformation (Alibeigi and Vaezi 2008). In the Fe2+-abundant condition, the brucite [Fe(OH)2] structure is mainly yielded, and green rust and magnetite are the major structures in the presence of mixtures of Fe2+ and Fe3+ (Usman et al. 2018). In the oxic condition, ferrihydrite is formed by the reconstructive transformation of ferric ions, but it has an unstable structure; thus, the transformation occurs (Jolivet et al. 2006; MacHala et al. 2011). The pH is the key for determining the product of reconstructive transformation from the ferrihydrite; hematite and goethite could be formed under circumneutral and extreme pH conditions, respectively (Combes et al. 1990; Jolivet et al. 2006). During the transformation, topotactic or reconstructive processes could occur together, and the presence of complexing ligands such as oxyanions could delay the transformation (Masion et al. 1997; Rose et al. 1997; Han and Ro 2018b). Drying or thermolysis yields the topotactic transformation from iron hydroxides to iron oxides (Vodyanitskii 2010; MacHala et al. 2011). In the soil environment, the phase changes by drying and wetting or freezing and thawing and the elemental concentration changes due to pH, redox potential, addition and loss always occur simultaneously, and varied iron (hydr)oxides are present. In this review, we focus on the effect of oxyanions as the complexing ligand modifying the rate of transformation. Iron (hydr)oxides are key sorbents for oxyanions, but the formation of iron (hydr)oxides is also determined by the presence of oxyanions. As a consequence, the abundant oxyanions bind with the small aggregates of hydroxylated Fe complexes, and oxyanion-hydroxylated Fe complexes are more stable than hydroxylated Fe complexes alone; thus, the former complexes are easily mobilized or leached out.
It was found that most studies on sorption experiments thoroughly characterized the iron (hydr)oxide properties, such as the pH, EC, surface area, point of net zero charge, morphology and crystal structure, before the adsorption experiment, but only a few studies observed the stability or integrity of the iron (hydr)oxides during the adsorption experiment. According to Bolanz et al. (2013a, b), the presence of oxyanions, concentration and pH controlled the transformation of iron (hydr)oxides via various processes, and much attention was dedicated to determining the stability of the iron (hydr)oxides in the adsorption experiments. Based on their studies, a high concentration (6 mM) of arsenate and phosphate blocked the transformation from ferrihydrite to other species, but a high concentration (6 mM) of antimonate led to transformation to feroxyhyte (Bolanz et al. 2013b; Michael Bolanz et al. 2013). The authors also examined the effect of pH and observed transformation from ferrihydrite to hematite at pH 4 and to goethite at pH 12, while transformation to both hematite and goethite was observed at pH 7. The transformation of goethite to bernalite was also observed at pH 10 with arsenate concentrations of 1–10 mM in an adsorption experiment (Han and Ro 2018a), and the bernalite showed higher arsenate adsorption capacity than did the goethite without aggregation, which would facilitate massive colloidal transport. However, numerous previous sorption experiments did not pay adequate attention to confirming structural stability and integrity, which could be one of the reasons for the conflicting results of sorption experiments.
Possible biological effects on the transformation of iron (hydr)oxides
Over 1.2 million living species have been cataloged on Earth, and the number of species has been predicted to be approximately 8.7 million (Mora et al. 2011). Only 25% of predicted species are oceanic organisms, while most of the others interact with the soil (Mora et al. 2011). Among the various organisms, microbes and plants are the major mediators that influence the chemical and physical characteristics of soil (Bertin et al. 2003; Bais et al. 2006; Baetz and Martinoia 2014). Uncountable interactions take place under the ground, but chemoautotrophic bacteria are known as redox controllers of metal (hydr)oxides and oxygen-containing molecules (Ilbert and Bonnefoy 2013). Bacteria utilize metal (hydr)oxides as electron acceptors to obtain energy. During the redox process, metal (hydr)oxide transformations occur, and oxyanions at the surface migrate to other sorption sites or precipitate with ions (Fakour and Lin 2014; Borer and Hug 2014). Not only the redox potential but also other environmental factors influence redox reactions; for example, pH and ion concentration have been shown to affect weathering and dissolution (Dowling 2002; Shi et al. 2011).
Reactions involving bacteria directly transform the crystal structure of iron (hydr)oxide in the soil, while plants influence the material belowground by inputting a massive amount of energy in the form of organic compounds (Bais et al. 2006; Baetz and Martinoia 2014). Rhizodeposition is one of the ways by which plants survive and shape their surrounding environment. Various molecules have been identified as root exudates, such as organic acids, amino acids, sugars, proteins, inorganic ions, gases and phenolic compounds, and their functions are not yet fully understood; however, they act as nutrient sources, chemoattractants, chelators, acidifiers, detoxifiers, catalysts, growth promotors, growth inhibitors and defenders against pathogens (Hütsch et al. 2002; Bais et al. 2006; Baetz and Martinoia 2014). The organic molecules involved in rhizodeposition vary significantly depending on the plant cultivar, growth stage and environmental conditions; thus, determining a fundamental mechanism is very difficult (Aulakh et al. 2001; Bais et al. 2006). A few studies have been conducted to reveal the effects of root exudates on the oxyanion sorption of iron (hydr)oxides, which include the following processes: 1) reductive dissolution by oxalate and ascorbate, 2) chelation, 3) competition at sorption sites, and 4) surface precipitation caused by hindering or facilitating sorption (Panias et al. 1996; Dakora and Phillips 2002; Erbs et al. 2010; Reza et al. 2010; Zhu et al. 2011).
The reconstructive transformation by reductive dissolution of oxalate on the goethite surface is an appropriate example to understand the biological transformation of iron (hydr)oxides. Kubicki et al. (2017) simulated the reconstructive transformation of goethite by oxalate and Fe2+ ions, and they found that the adsorbed Fe2+ complexes may have a catalytic effect on the dissolution of goethite by electron transfer to surface defect sites, while the oxalate was shown to cause ligand-enhanced dissolution on the goethite surface and the formation of Fe3+–oxalate complexes in the aqueous phase (Kubicki et al. 2017). The electron transfer from the Fe2+ complex to the mineral structure and the high affinity of oxalate to Fe3+ would enhance the dissolution of goethite mineral, and the structure would be transformed into other structures upon environmental fluctuations. The enhanced dissolution would weaken the neighboring oxyanion adsorbed on the goethite surface; then, the oxyanion–Fe3+ complex would be dissolved. In addition, the organic acid would work as a coating to inhibit the crystallized growth or the bridge to aggregate the nanoparticles of iron (hydr)oxides. Li et al. (2018) confirmed that the concentration ratio between oxalate and ferrihydrite determines the structural configuration from the bidentate binuclear complex to the bidentate mononuclear complex. As a result, the stability and morphology of ferrihydrite aggregates were changed, and the bidentate mononuclear complex led to more dissolution of Fe ions.
Reactivity with ascorbate represents another example for understanding the reduction of iron (hydr)oxides. Ascorbate is abundantly found in the exudates of plant roots, and it functions as a reductant of Fe(III) (hydr)oxides. Larsen and Postma (2001) studied the reductive dissolution rate of lepidocrocite, ferrihydrite and goethite in the presence of ascorbate, and they found that this rate was mainly controlled by the crystal structures and that the surface area determined the dissolution rate in the same crystal structure. A recent study by Huang et al. (2017) confirmed the importance of the structural configuration of ascorbate on the hematite surface, and the monodentate mononuclear complex on the (012) surface led to a higher reductive dissolution rate than that of the bidentate monodentate complex on the (001) surface. The specific mechanism of ascorbate in the Fe2+/Fe3+ cycle was suggested, and the reduced Fe2+ caused molecular oxygen activation (Hou et al. 2016). As a result, reactive oxygen species were generated, possibly causing the transformation of iron (hydr)oxides. In addition to the examples of oxalate and ascorbate, other various organic compounds are delivered from the plant root. The complex mechanism caused by various organic compounds and inorganic substances promotes or depresses the aggregation, adsorption and dissolution of iron (hydr)oxides, but there have not been any sufficient studies to understand such a mechanism comprehensively.
Example of transformation in upland versus paddy fields
The redox condition is the key factor that governs iron (hydr)oxide transformation, but it is hard to observe this transformation in nature (Zhang et al. 2012; Fan et al. 2014). For that reason, an example of redox fluctuating conditions is presented in this section to describe the possible natural reactions. Cycling between upland and paddy fields represents the typical land-use regime for crop cultivation in which the fields have distinctive redox potentials. There are few studies dealing with iron (hydr)oxide transformation in relation to the land-use regime (Takahashil et al. 1999; Kögel-Knabner et al. 2010; Fan et al. 2014). In the soil profile, the redox potential decreases with increasing depth (Mansfeldt 2004; Reza et al. 2010), and the redox gradient yields complex and varied oxidation/reduction reactions in heterogeneous soil (Fakour and Lin 2014; Fan et al. 2014). We schematically illustrated the redox potential change and iron (hydr)oxide transformation with interconversion from a paddy to an upland field (Fig. 2). In the upland field, iron2+ oxide was mainly found in the subsurface, while iron3+ oxide typically crystallized at the soil surface (Cornell and Schwertmann 2003). With increasing depth, the reduction potential governs chemical reactions by reducing the oxygen-containing compounds or minerals sequentially. Conversion to a paddy field is achieved by flooding the upland field over enough time. In the paddy field, the redox potential dramatically decreases with depth and time (Kögel-Knabner et al. 2010), and iron3+ oxide is hardly found because of its reduction by chemotrophic bacteria. An interesting phenomenon observed in rice cultivation in paddy fields is the formation of so-called iron plaque (Chen et al. 2006). Previous studies identified the iron plaque as precipitates oxidized by the exudation of O2 from rice roots, and it contributes to nutrient uptake (Chen et al. 2006; Huang et al. 2015). Iron3+ oxide is transformed into another phase via reconstructive transformation, the Fe2+ ions from the reduction and dissolution of iron3+ oxide precipitate with various anions to form minerals such as siderite (with CO32−), troilite (with S2−), pyrite (with S22−) and vivianite (with PO43−). The reduction along the soil profile is easily observable because of the color change of iron (hydr)oxide from orange brown to gray black.

Iron (hydr)oxide transformation in the soil environment based on the land-use regime for crop cultivation. The hexagons and circles indicate iron (hydr)oxides in the solid phase and iron ions in the aqueous phase, respectively. The red dotted graph illustrates the redox potential change with soil depth
After the vegetative and reproductive stages of development, the ripening stage requires the soil to dry; thus, the oxidation potential governs the ensuing chemical reactions (Aulakh et al. 2001). Thermodynamically unstable reduced compounds and minerals are rapidly oxidized upon contacting the atmosphere, and iron3+ oxide is formed (Takahashil et al. 1999; Cornell and Schwertmann 2003). However, a very distinctive phenomenon observed between the converted field and the upland field is the size of iron3+ oxide particles in the soil profile (Schwertmann 1993; Anschutz and Penn 2005). The precipitation during the reconstructive transformation determines the size of iron (hydr)oxides, and newly precipitated iron3+ oxide is found to be nanosized. Based on environmental factors, this nanosized iron3+ oxide can be grown to macrosized particles or can remain nanosized (Williams and Scherer 2004; Iwasaki et al. 2011; Hinkle et al. 2015). It is extremely important to describe the oxyanion dynamics in the soil because the nanosized particles cause colloidal transport (Zhang and Selim 2007; Borer and Hug 2014). As mentioned above, iron (hydr)oxide has a positively charged surface complexed with organic matter and anions to result in a neutral surface charge, which causes high mobility by decreasing interactions with the soil component. This colloid transport phenomenon is one of the possible reasons for the massive leaching of nutrients from the soil to the water system (Van Riemsdijk et al. 2007; Zhang and Selim 2007; Borer and Hug 2014) and is thus a severe environmental problem worldwide (Smith et al. 2006; Smith and Schindler 2009; Zamparas and Zacharias 2014; Han et al. 2016).
In addition to the particle size, the morphology of iron (hydr)oxide is critical for the availability of oxyanions. There is no concrete and direct evidence on the effect of morphology on oxyanion sequestration, but a previous study by Han and Ro (2019) suggested the possible effect of morphology. During a phosphate adsorption study on binary goethite and maghemite using ATR-FTIR spectroscopy, shrinking and swelling properties were found in goethite and maghemite, respectively. However, the shrinking properties were diminished over repeated wetting and drying processes with phosphate solution. They also reported significantly different spectra between goethite and maghemite, which might be caused by complexes bridging between the nanorods. If the phenomenon is valid, it would explain the low availability of phosphate in the cultivation because the goethite is known as the most abundant structure among the iron (hydr)oxides in the soil, and only the rod shape was reported in previous studies. After the formation of goethite as nanorods, the oxyanions and organic acids act as the glue between the nanorods, and the sequential attachment leads to nanorod aggregation. The tight bridging structure between the nanorods over the matrix potential of water to separate the nanorods and the oxyanions is sequestrated until the safe box is destroyed. Not only the goethite but also several iron (hydr)oxides form the rod shape; thus, they also work to help sequester the oxyanions. However, more concrete evidence should be provided to understand the morphology effect.
Structural configuration
Several studies have attempted to reveal the structural configuration of oxyanions and organic acids on an iron (hydr)oxide surface. There are few experimental and theoretical methods for identifying this structural configuration. The surface complexation model is the most versatile method and is based on theoretical approximation combined with sorption data from batch experiments (Sharif et al. 2011; Goldberg 2013). Theoretical calculation based on computational chemistry is a promising approach to reveal the structural configuration (Kubicki et al. 2007; Johnston and Chrysochoou 2012; Acelas et al. 2013), but it is still unsuitable for simulating a multicomponent system because it requires excessive computational resources, and the structure must be confirmed by comparison with experimental data (Luengo et al. 2015; Yang et al. 2016). Spectroscopy is a promising experimental technique, and X-ray and infrared are common light sources for such experiments (Ferrari et al. 2004; Lefevre 2004; Ona-Nguema et al. 2005; Mudunkotuwa et al. 2014; Liu et al. 2015). X-ray absorption spectroscopy (XAS) includes extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES), both techniques that enable us to see the local structure and oxidation number of the target element (Scott et al. 1992; Waychunas et al. 1993). Fourier transform infrared (FTIR) spectroscopy equipped with attenuated total reflection (ATR) or diffuse reflectance (DRIFT) accessories is frequently used to reveal the structural configuration of materials after adsorption (Madejová 2003; Mudunkotuwa et al. 2014).
Structural configuration of oxyanions
Oxyanion adsorption on an iron (hydr)oxide surface can be classified into the formation of two types of complexes: an outer-sphere complex formed by electrostatic attraction and hydrogen bonding with its configuration stabilized by interfacial water (Catalano et al. 2008) and an inner-sphere complex formed by covalent bonds (Sparks 2003). The charge difference between the positively charged iron (hydr)oxide surface and the negatively charged oxyanion keeps the oxyanion near the surface at the interface (Adegoke et al. 2013; Sparks 2003). The outer-sphere complex is a weak and exchangeable complex, whereas the inner-sphere complex is a strong and nonreversible complex (Sparks 2003). The inner-sphere complex can be specified based on the number of covalent bonds and the number of ligands. As the number of covalent bonds to the metal center increases, the ligand is called monodentate, bidentate and tridentate, while as the number of ligands coordinated to the complex increases, it is called mononuclear, binuclear and trinuclear. For example, the oxyanion formed by two covalent bonds with two polyhedra is called a bidentate binuclear complex (BB).
Table 1 summarizes the dominant structural configurations of 9 oxyanions and 8 organic acids on 6 types of iron (hydr)oxides. Only 60 articles are cited in the table. We reviewed over 400 articles and selected articles that employed pure iron (hydr)oxides with descriptions of the crystal structure and complexation. For the oxyanions and iron (hydr)oxide, 7 elements (As, C, Cr, P, S, Se and Si) and 6 iron (hydr)oxides (goethite, lepidocrocite, ferrihydrite, hematite, maghemite and magnetite) were chosen based on abundance in the soil, deleterious effects on the environment and number of published articles. The reason for focusing only on iron (hydr)oxide is to discuss the effect of crystal structure on the structural configuration of oxyanions and organic acids. Taking arsenate on goethite as an example, the keywords arsenate, goethite and complex were searched on Scopus within the article title, abstract and keywords; 79 articles were found, and the abstracts were carefully reviewed to determine the descriptions of the complexes. Numerous organic acids are present in the soil, and 19 organic acids and 24 amino acids were chosen from the previous literature summarizing root exudates (Dakora and Phillips 2002); however, only a few studies have addressed the structural configuration of organic acids and amino acids on pure iron (hydr)oxide.
The subscripted number in Table 1 indicates the pH range in each experiment, the first subscripted letter denotes the experimental method, and the last subscripted letter denotes the sample preparation method. Further description is included in the table caption. Arsenate and goethite are the most common sorbate and sorbent, respectively, and arsenate sorption on a goethite surface is the most frequently studied. The experimental method is quite different for each oxyanion because of the differing utility of XAS and other methods. As, Cr and Se are mainly analyzed via XAS measurement within a hard X-ray regime (> 2 keV), whereas lighter elements such as C, P and S are frequently measured with infrared spectroscopy (Newville 2014). XAS can theoretically measure the soft X-ray region (< 2 keV), where the auger processes predominate over fluorescence (Josefsson et al. 2012; Newville 2014). The EXAFS and XANES measurements provide us with direct information on the local structure, coordination and oxidation state, but species transformation by protonation and deprotonation cannot be easily distinguished (Kubicki et al. 2007; Waychunas et al. 1993). In contrast, infrared spectroscopy offers only indirect evidence through molecular vibrations, but it enables us to interpret the species transformation from protonation and deprotonation because those processes significantly alter the vibration mode of the complex, while the local structure shows no significant difference (Brechbühl and Christl 2012; Yang et al. 2016).
Toxicants
Arsenic is one of the most prevalent elements causing severe health threats to humans (Vahidnia et al. 2007; Zhu et al. 2013a), and arsenite and arsenate are arsenic-based oxyanions that form depending on the redox potential. Arsenite is more abundant at low redox potential, while arsenate is the opposite, and it has been reported that arsenite has higher toxicity and mobility than that of arsenate (Welch and Stollenwerk 2003; Babula et al. 2008; Vamerali et al. 2010). The natural abundance of arsenic in bedrock and anthropogenic activity from gold mining and arsenical pesticides are the major reason for the pollution. Populations from over 20 countries have suffered due to groundwater contamination by arsenic originating from both natural and anthropogenic sources (Smedley and Kinniburgh 2002), and the median concentration of arsenic was reported to 6 mg kg−1 in soils worldwide (Bowen 1979). Biomethylation in soil transforms the arsenic elements into organoarsenic (Kim et al. 2014), and reductive conditions at low pH cause the transformation into arsane (AsH3), but arsenite and arsenate are the prevalent species based on the previous literature (Bose and Sharma 2002; Singh et al. 2015).
The sorption of arsenite on 6 iron (hydr)oxides was studied in 7 papers at pH values ranging from 3 to 10. XAS was used in 6 papers, and FTIR spectroscopy and a surface complexation model (SCM) were employed together in one paper. As mentioned above, XAS is an appropriate technique for detecting arsenic, while ATR-FTIR spectroscopy is a less employed technique because the arsenite bands appear at 850–650 cm−1, where the long-wavelength cutoff occurs. The BB, bidentate mononuclear (BM) and monodentate mononuclear (MM) complexes similar to those of arsenate were identified, but tridentate hexanuclear (TH) complexes were proposed to describe arsenite on maghemite and magnetite surfaces (Auffan et al. 2008; Liu et al. 2015; Wang et al. b). The nano-effect, which occurs in nanosized maghemite smaller than 20 nm, resulted in arsenite sorption on a six-membered iron octahedral ring in a TH configuration (Auffan et al. 2008). Wang et al. (2008a, b) also identified a TH configuration of arsenite on the magnetite surface; they found that a decrease in surface loading determined the structural transformation of arsenite from BM to TH and that the TH complexes on the maghemite significantly decreased its solubility. Liu et al. (2015) confirmed the TH configuration and proposed that the interconversion between arsenate and arsenite occurs by redox fluctuation.
The sorption of arsenate was reported in 12 papers, and the pH of the sorption experiments ranged from 3 to 10. FTIR, SCM, computational chemistry and XAS analyses were used. Based on the experimental results, 9 papers suggested a BB complex as the structural configuration of arsenate sorption on 5 iron (hydr)oxides but not on goethite (Randall et al. 2001; Farquhar et al. 2002; Sherman and Randall 2003; Antelo et al. 2005; Morin et al. 2008; Brechbühl and Christl 2012; Neupane et al. 2014; Liu et al. 2015; Han and Ro 2018a). The interatomic distances calculated with EXAFS were 1.68 Å for As–O and 2.85–3.65 Å for As–Fe. Most papers obtained an interatomic distance for the BB configuration between 3.24 and 3.35 Å, whereas 4 papers (Farquhar et al. 2002; Fendorf et al. 1997; Loring et al. 2009; Waychunas et al. 1993) obtained an interatomic distance ranging from 2.85–2.93 or 3.6–3.65 Å and a different coordination number. Neupane et al. (2014) and Waychunas et al. (1993) identified an As–Fe shell with a distance of 2.85–2.93 Å as a BM complex, whereas Fendorf et al. (1997) and Loring et al. (2009) described the As–Fe shell with a distance of 3.60–3.65 Å as an MM complex. Loring et al. (2009) also determined the As–Fe distance to be 3.29 Å at pH 3 and 10, but they reported the structure as a monodentate trinuclear (MT) complex based on their EXAFS and FTIR results. In that paper, the As–Co distance of pentaamminecobalt(III) arsenate was consistent with a monodentate type (3.25 Å) by EXAFS, and unprotonated, singly protonated and doubly protonated As–O stretching bands were observed in D2O by FTIR analysis. Liu et al. (2015) conducted an experiment to test the difference between a dried and paste sample on magnetite, but no difference was observed; however, the experiment confirmed that the MM complexes were identified only in the paste sample. Interestingly, Han and Ro (2018a, b) identified the transition of structural configuration by varying the pH and surface loading, and they revealed that a higher available sorption site per sorbate (low surface loading or pH) would yield tridentate complexes, while a less available sorption site (high surface loading or pH) was dominated by the formation of monodentate complexes.
Chromium is the seventh most abundant element on Earth, but the average concentration in soil is 84 mg kg−1 (Ure and Berrow 1982). Atmospheric deposition from metallurgical industries and natural high abundance are the reason for the chromium present in the soil. Chromium exists in a number of oxidation states, but chromite (Cr3+) and chromate (Cr6+) are the most stable species in the soil environment (Alloway 1995). Unlike the situation with arsenic oxyanions, chromite is less mobile and is adsorbed more strongly than chromate; thus, chromite is completely precipitated at the neutral pH condition. Chromate exists as monochromate (H2CrO4) and dichromate (H2Cr2O7), and both are toxic to human health. Compared with that of arsenic, the average concentration of chromium in water is 14 times higher, and the WHO guideline for drinking water quality is 10 and 50 mg L−1 for arsenic and chromium, respectively. In the surface soil, chromate is more prevalent, which has higher toxicity than chromite, but the studies on chromium have not appropriately addressed this subject, unlike those on arsenic. Based on its toxicity, distribution and abundance in the soil environment, a comprehensive study on the fate of chromium in the soil environment should be performed to diminish the hazards of this element.
Five studies at pH values of 3–9.5 reported chromate adsorption structures. Fendorf et al. (1997) reported MM, BB and BM complexes similar to those of arsenate. The interatomic distance was 1.89–1.69 Å for Cr–O and 2.91–3.63 Å for Cr–Fe. Johnston and Chrysochoou (2014) reported interatomic distances of Cr–O and Cr–Fe as 1.64 and 3.22–3.51 Å, respectively. Fendorf et al. (1997) determined the interatomic distances of the BB, BM and MM complexes to be 2.91, 3.29 and 3.63 Å, respectively, whereas Johnston and Chrysochoou (2014) determined the interatomic distances of the BB and BM complexes to be 3.32 and 3.51 Å, respectively. The latter authors also identified the BB and BM complexes on the basis of theoretical calculations and IR spectroscopy. Aqueous chromate showed bands at 880 and 848 cm−1 for CrO42− (Td), bands at 950 and 898 cm−1 for HCrO4− (C3v) and bands at 950, 882 and 772 cm−1 for Cr2O72− (C3v). The chromate complex on ferrihydrite had bands at 950, 930, 885, 830 and 750 cm−1 for pH 3.5 and at 910, 873 and 820 for pH 7.0. Kubicki et al. (2018) measured chromate adsorption on the ferrihydrite surface using EXAFS and ATR-FTIR spectroscopy, and density functional theory (DFT) calculations were also conducted. The interatomic distances of Cr–O and Cr–Fe were 1.67 and 3.35–3.58 Å by EXAFS, respectively, and DFT calculations of the Cr–Fe distance yielded 3.40–3.44 Å for monodentate and 3.23–3.48 Å for bidentate complexes by using B3LYP. The peaks of the ATR-FTIR spectra assigned to the monodentate complex were 910, 875, 825 and 800 cm−1, and those assigned to the bidentate complex were 955, 930, 880, 830 and 765 cm−1. Both EXAFS and ATR-FTIR studies were conducted under circumneutral pH conditions, and the surface loading effect was also assessed. These experiments confirmed the presence and transition of monodentate and bidentate chromate on ferrihydrite. Gao et al. (2017) studied chromate and citrate adsorption on hematite by changing the pH. The authors found abundant peaks at 936–942 cm−1, and no peak shift was observed over time. In that study, no detailed information was provided in the manuscript, and no supporting information was provided on how the surface complex of chromate was identified; however, it would be presumed by their previous studies and the hypothesis in this study that both chromate and citrate form the monodentate complex due to electrostatic attraction. An interesting description was suggested because the monodentate complex was more favorable at low pH conditions due to electrostatic attraction. They also observed electron transfer from citrate to reduce chromate. Based on the theoretical results, an unprotonated MM complex was identified under acidic conditions, whereas protonated BB or BM complexes were dominant under basic conditions. We conclude that both MM and BB complexes are the dominant forms with chromate and that an increase in the pH changes the complex from MM to BB with protonation.
The abundance of selenium in the crust is reported to be 0.05–0.09 mg kg−1, but it is an essential element at low concentrations for organisms. Selenium is mainly used as a pest control and dietary supplement for livestock in agricultural soils (Peak and Sparks 2002; Kim et al. 2012; Sharma et al. 2014). Selenium commonly comprises 4 species under environmental conditions, where selenate and selenite are the abundant species, and organic ions of selenium-containing molecules are also reported (Sharma et al. 2014). A high level of selenium is caused by volcanic emission and metallic sulfides associated with igneous activity, and it functions as a toxicant (Alloway 1995; Sharma et al. 2014).
Nine studies have been performed to examine selenate and selenite sorption on iron (hydr)oxide at pH values ranging from 3 to 12. Three studies identified an MM complex of selenate and selenite (Wijnja and Schulthess 2000; Peak and Sparks 2002; Kim et al. 2012), whereas the other five studies identified BB and BM complexes of selenate and selenite (Catalano et al. 2006; Das et al. 2013; Hiemstra et al. 2007; Jordan et al. 2014; Su and Suarez 2000a). Martínez et al. (2006) revealed an outer-sphere complex of selenate, whereas an MM complex of selenite was calculated using SCM. Kim et al. (2012) also identified an MM complex of selenite using SCM. Hiemstra et al. (2007) employed SCM and theoretical calculations to study selenite sorption on goethite, and a BB complex was identified. Catalano et al. (2006) performed X-ray standing wave measurements on a BB complex at pH 4. Manceau and Charlet (1994) showed BB and BM complexes of selenite on ferrihydrite, while only a BB complex of selenite and selenate was identified on the goethite surface. The interatomic distances of selenate and selenite were calculated to be 3.25–3.29 and 3.38 Å for the BB complex and 2.80–2.83 and 2.85 Å for the BM complex, respectively. Peak and Sparks (2002) identified an MM complex on goethite, ferrihydrite and hematite, and the interatomic distance was 1.64–1.65 Å for Se–O and 3.3–3.31 Å for Se–Fe. They noted that the distance of 3.3–3.31 Å was intermediate between the distances associated with an MM and BB complex and that hydrogen bonding led to a contraction of the Se–Fe distance. Das et al. (2013) identified a BB complex on ferrihydrite, goethite and lepidocrocite even though the interatomic distance resembled that found by Peak and Sparks (2002). Jordan et al. (2013) found a BB complex with an interatomic distance of 3.38 Å, but the outer-sphere complex was abundant, while the IS complex made only 15% of the contribution to sorption. Jordan et al. (2014) reported BB and BM complexes of selenite on a maghemite surface, and the interatomic distance was 1.71 Å for Se–O, 2.88–2.91 Å for BM and 3.36–3.38 Å for BB.
From IR spectroscopy, aqueous selenite (pKa1 = 2.46; pKa2 = 7.3) bands were observed at 849 and 825 cm−1 for HSeO3− and at 851, 822 and 731 cm−1 for SeO32− (Duc et al. 2006). The selenate bands (pKa1 = − 3; pKa2 = 1.9) were observed at 867–873 cm−1 (Das et al. 2013). Su and Suarez (2000b) identified a BB complex of selenate with bands at 910, 880 and 820 cm−1. Jordan et al. (2013) also reported a BB complex on maghemite, but the bands were at 904, 879, 859 and 829 cm−1. Peak and Sparks (2002) revealed an MM complex on hematite, and the bands were at 880, 850 and 820 cm−1. For selenium oxyanions, the MM, BB and BM complexes existed together, but the structural configuration changed with the environmental conditions.
Nutrients
Phosphorus is known as an essential nutrient for organisms, and phosphate is its most prevalent oxyanion species. Phosphate is a limiting factor for cultivation, but recent overfertilization has caused severe environmental problems worldwide, such as eutrophication and algae blooms in water systems (Young et al. 1989). Many studies have confirmed that iron (hydr)oxide is the main adsorbent for phosphorus, and the comobilization of phosphorus and iron (hydr)oxide colloids has been reported (Szilas et al. 1998; Makris et al. 2006; Warrinnier et al. 2019).
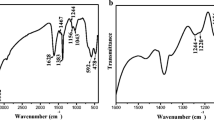
Nine studies revealed the structural configuration of phosphate on iron (hydr)oxides at pH values of 3–13. Two IR studies identified the MM complex at high surface loading and pH (Elzinga and Sparks 2007; Kubicki et al. 2012). Otherwise, all studies reported a BB or BM complex for phosphate sorption. The BB complex was reported using SCM (Antelo et al. 2005; Arai and Sparks 2001), while IR spectroscopy identified the BB or BM complex (Arai and Sparks 2001; Daou et al. 2007; Luengo et al. 2006; Persson et al. 1996; Tejedor-Tejedor and Anderson 1990). X-ray spectroscopy also revealed the BB complex (Daou et al. 2007; Khare et al. 2007). The aqueous band positions of phosphate were reported to be approximately 1172, 1005 and 889 cm−1 for H3PO4 (C3v), 1159, 1077, 940 and 875 cm−1 for H2PO4− (C2v), 1078, 990 and 850 cm−1 for HPO42− (C3v) and 1011 cm−1 for PO43− (Td) (Tejedor-Tejedor and Anderson 1990; Persson et al. 1996; Elzinga and Sparks 2007). Tejedor-Tejedor and Anderson (1990) reported deprotonated BB bands at 1123, 1006 and 986 cm−1 for v(P=O), va(P–OFe) and vs(P–OFe) or v(P–OH), respectively, protonated BB bands at 1094 and 1044 cm−1 for v(P–O), and deprotonated MM bands at 1025 and 1001 for v(P–O). Arai and Sparks (2001) identified bands at 1100, 1040 and 1004 from pH 4.5 to 8.4, and they concluded that the BB complex is formed on the goethite surface. Luengo et al. (2006) assigned bands at 1092, 1047 and 952 cm−1 to the unprotonated BB complex and 1122, 1012 and 933 cm−1 to the protonated BB complex. Persson et al. (1996) studied a dried sample using DRIFT-FTIR but observed a band position that was significantly different from the ATR-FTIR results. Daou et al. (2007) measured dried magnetite at pH 3 using transmission mode and identified BB bands at 1110, 1032, 962 and 816 cm−1 by peak deconvolution. Elzinga and Sparks (2007) reported the bands of an unprotonated BB complex at 1117, 1007 and 964 cm−1, the bands of a monoprotonated BB complex at 1075, 1030 and 936 cm−1 and the bands of an MM complex at 1085, 1040 and 960 cm−1. Kubicki et al. (2012) investigated two types of goethite, micro- and nanosized samples, from pH 4.2 to 8.0. Peak deconvolution identified bands at 1117, 1084, 1044, 1005 and 957 cm−1 in nanosized goethite at pH 4.2 and bands at 1082, 1036, 1006, 966 and 935 cm−1 at pH 8.0. In microsized goethite, the band positions were 1157, 1122, 1091, 1044 and 1009 cm−1 at pH 4.2 and 1095, 1076, 1039 and 1005 cm−1 at pH 8.0. Kubicki et al. (2012) also studied the effect of drying on complexation and found band positions that were completely different from those obtained by interfacial measurement. The theoretical results indicated that the surface structure led to alteration of the complex. Daou et al. (2007) employed X-ray photoemission spectroscopy (XPS) and Mossbauer spectroscopy for complex characterization, and Khare et al. (2007) used a fingerprinting method with molecular orbital calculations; both studies confirmed a BB complex. The IR spectra results showed significant variation in the bands and their interpretation; thus, these results are still debatable for use in interpreting the complex. However, we inferred that phosphate showed dramatic changes with pH and surface loading, and the BB complex was identified as the most abundant phosphate complex on an iron (hydr)oxide surface.
Sulfur is an essential nutrient for constituting amino acids, vitamins and enzymes and exists as sulfate in the oxic soil environment. The mean concentration of sulfur in the crust is 260 mg kg−1, and its cycle in the soil resembles the nitrogen cycle. Under anoxic conditions, sulfate loses oxygen atoms and binds to deoxidized elements such as iron, manganese and zinc. Pyrite (FeS2) is the most common mineral of iron and sulfur. The acid rain caused by fossil fuel combustion and fertilization increases the level of sulfate in the soil environment, eventually leading to competitive adsorption with other oxyanions in the soil.
Eight studies have examined the complexation of sulfate on iron (hydr)oxide. Four studies identified an MM complex of sulfate (Fukushi et al. 2013; Hug 1997; Rahnemaie et al. 2006; Wijnja and Schulthess 2000), whereas two studies concluded that a BB complex is formed (Mansour et al. 2009; Zhu et al. 2013a, b). Lefèvre and Fédoroff (2006) and Eggleston et al. (1998) detected both MM and BB complexes on hematite using ATR-FTIR analysis. Rahnemaie et al. (2006) and Fukushi et al. (2013) used SCM to predict the complexation of sulfate and concluded that an MM complex is formed by comparing the theoretical (BP86) and IR results. Mansour et al. (2009) identified a BB complex on magnetite at pH 3–5, whereas only an outer-sphere complex remained above pH 5. Zhu et al. (2013a, b) employed EXAFS spectroscopy, the differential atomic pair distribution function (d-PDF) and ATR-FTIR measurements. Interatomic distances of 1.46–1.47 Å for S–O and 3.18–3.19 Å for S-Fe were obtained with EXAFS, whereas interatomic distances of 1.46–1.47 Å for S–O and 3.24–3.27 Å for S-Fe were obtained with d-PDF. The IR bands at 1175, 1121, and 1043 cm−1 were assigned to v3 splitting, whereas the bands at 1102 and 977 cm−1 were assigned to degenerate v3 and v1 modes, respectively. As a result, the BB complex was assigned under air-dried conditions. Previous studies observed the effect of drying on complexation; thus, the sulfate possibly forms a complex of the MM type at the aqueous and solid interface. Eggleston et al. (1998) also confirmed a BB complex of sulfate on dried hematite. From the ATR-FTIR results, aqueous sulfate (pKa1 = − 3; pKa2 = 1.99) bands were observed at 1194, 1051 and 891 cm−1 for HSO4− and 1102 cm−1 for SO42− (Td), whereas bands of adsorbed sulfate were found at 1126, 1042 and 980 cm−1 for the wet sample measurement. Hug (1997) and Wijnja and Schulthess (2000) also reported bands at 1128–1130, 1055–1060 and 975–976 cm−1 as belonging to an MM complex. The studies confirmed that the dramatic increase in band intensity at 1200 cm−1 was evidence of a BB complex. Gu et al. (2016) quantified sulfate adsorption on a ferrihydrite surface using DFT calculations, SCM and EXAFS measurements by changing the pH and ionic strength under aqueous and dried sample phases. The S–O and S-Fe interatomic distances by EXAFS were 1.47–1.49 and 3.22–3.25 Å, respectively, while DFT calculations determined the S-Fe interatomic distance of the BB complex under dried conditions to be 3.21 and 3.24 Å, that of the MM complex under dried conditions to be 3.41 and 3.61 Å and that of the MM complex under wet conditions to be 3.28 and 3.58 Å, respectively. The fraction of inner-sphere complex was decreased with increasing pH, and air-drying dramatically converted the outer-sphere complex to the inner-sphere complex. The BB complex confirmed the SCM model. Based on these observations, we concluded that sulfate is primarily present as an MM complex under acidic conditions for all iron (hydr)oxides, and the drying effect and high pH yield a BB complex and outer-sphere complex, respectively.
Others
Carbonate is a ubiquitous oxyanion in the soil environment. Both inorganic and organic carbons are the main source of carbonate in the soil environment; the weathering and dissolution of primary carbon-containing minerals is the inorganic source of carbonate, while microbial respiration is the organic source (Bargar et al. 2005). The carbonate concentration is proportional to the CO2 concentration in soil pores, and it has no significant harmful effect on terrestrial plants. However, depending on the soil profile and microbial activity, a high level of CO2 is frequently measured, and it can compete with other oxyanions to increase the mobility or precipitate with multivalent cations, creating an additional surface for adsorption (Rahnemaie et al. 2007; Brechbühl and Christl 2012).
For carbonate, infrared spectroscopy, theoretical calculations and SCM were mainly used in 5 papers, and the pH range investigated was 3–12. All experiments employed aqueous or paste samples for the measurements. Wijnja and Schulthess (2001) identified bands at 1510 (O-C-O sym. str.), 1315 (O-C-O asym. str.) and 1068 cm−1 (C-O str.) with an H2O background and an additional band at 1462 cm−1 (shift of O-C-O asym. str.) appeared with a D2O background; they assigned the carbonate complex on goethite as an MM complex under high surface loading conditions (10.5 μm m−2). Brechbühl and Christl (2012) identified a band at 1522 cm−1 as that of an MM complex and a band at 1463 cm−1 as that of a BB complex, whereas bands at 1357 and 1063 cm−1 were assigned for both MM and BB complexes; however, the SCM result showed that only the BB complex is abundant at 0.39–100 hPa of PCO2. Both sets of ATR-FTIR results showed more absorbance at neutral pH than at low pH conditions. Hiemstra et al. (2004) interpreted the structural configuration by considering the relationship between the degree of splitting (Δv3) and the field strength of coordination. A large difference (300–340 cm−1) in Δv3 indicated the presence of the BM complex, whereas a relatively small difference (80–120 cm−1) revealed the MM complex. Bargar et al. (2005) reported the bands associated with aqueous carbonate species transformation and a carbonate complex at pH 3.6–8.2. Aqueous HCO3− showed bands at 1355, 1300 and 1000 cm−1, whereas CO32− had only one band at 1393 cm−1. The bands at 1522, 1480, 1355 and 1320 cm−1 were assigned to the BB complex on hematite. DFT (B3LYP/6-31G(d)) calculations identified bands at 1529 and 1339 cm−1 for the BB complex and at 1605 and 1271 cm−1 for the MB complex. Roonasi and Holmgren (2010) found BB complex bands at 1545, 1485, 1335 and 1070 cm−1. All studies confirmed that various complexes, such as MM, MB, BM and BB, were reported, and it was hard to find patterns from the small amount of literature. In addition, the comparison of surface loading in carbonate was not applicable because different methods were applied to input the carbonate in the experiments (that is, adding sodium carbonate vs. maintaining a certain CO2 concentration in the atmosphere).
Except for element oxygen, silicon is the most abundant element in the soil environment, and it plays critical roles in the nutrition of plants and the stability of soil. Higher plants require more silicon compounds to reinforce their structural stability. The chemical weathering of phyllosilicate is the source of silicate, which is the oxyanion species of silicon, and dissolved silicate constitutes the secondary minerals of soils and combines with other rich elements (Alloway 1995). The formation and type of secondary mineral mainly govern the characteristics of soils, and many studies have confirmed the surface precipitation of silicate on the iron (hydr)oxide surface, which would cause a change in the bioavailability of oxyanions on the surface of iron (hydr)oxides (Luxton et al. 2008; Swedlund et al. 2009; Yang et al. 2009).
Five studies were reported to examine the sorption of silicate on iron (hydr)oxide, all of which reported a BB, BM and MM complex. Kersten and Vlasova (2009) performed batch adsorption experiments at various temperatures (10–75 °C). The percentage of adsorbed silicate decreased with time, and the maximum sorption occurred at pH 9. SCM identified two complexes, an unprotonated BB complex and a protonated BB complex, and increasing the temperature yielded a decrease in the amount of unprotonated BB complex at acidic and neutral pH. Swedlund et al. (2009) observed the polymerization of silicate on a ferrihydrite surface. Aqueous H4SiO4 (pKa1 = 9.84; pKa2 = 13.2) showed bands at 1090 and 939 cm−1, which were assigned to Si–O–H deformation and asymmetric Si–O stretching vibrations, respectively. Adsorbed silicate showed broad bands between 1200 and 750 cm−1, and band shifts were observed upon changes in time and silicate concentration. At low silicate concentrations, the maximum intensity appeared at 943 cm−1, and it shifted to 1001 cm−1 and finally to 1110 cm−1 with increasing silicate concentration. The band shift resulted from the polymerization of monomeric silicate to oligomeric silicate and finally to polymeric silicate. Yang et al. (2008) also observed a band shift upon the polymerization of silicate at pH 8.5 and 10.8, which are above and below the pKa1 of silicate. However, the polymerization rate was significantly lower at pH 10.8. Jolsterå et al. (2010) studied the SCM of silicate on maghemite at pH 3–11, and they also found a BB complex or polynuclear silicate complex. Using EXAFS and ATR-FTIR measurements and DFT calculations, Wang et al. (2018) studied silicate adsorption on ferrihydrite when changing the pH from 4 to 10. The interatomic distances of Si–O and Si–Fe were 1.62 and 3.25 Å, respectively, while a Si–Si distance of 3.10 Å was also identified as the result of polymerization. The multivariate curve resolution of the ATR-FTIR spectra confirmed two dominant signals with peaks at 1011 and 935 cm−1, and the peaks were assigned to the monomer of the polymeric bidentate binuclear complex and the MM complex, respectively. The MM complex was abundant under low surface loading conditions, but increased surface loading and pH led to the polymerization of the BB complex on ferrihydrite.
In contrast to the situation for other oxyanions, polymerization is the key mechanism for understanding the dynamics of silicate in the environment, especially at high silicate concentrations and pH values. The band shift in the IR spectrum clearly confirmed the polymerization. Silicate is primarily sorbed as a BB complex and is strongly retained at the iron (hydr)oxide surface, forming an additional surface over the iron (hydr)oxides. In the soil environment, silicate is an abundant oxyanion due to the weathering of primary and secondary minerals; thus, understanding silicate dynamics is important.
Structural configuration of organic acids
It is well known that organic molecules mainly interact with the hydrophobic surface of soil organic matter and that ionizable organic molecules interact with the hydrophilic surface of soil minerals (Hyun and Lee 2008; Zhu et al. 2019). The sources of these organic molecules in the soil environment range from the decomposition of organic matter to rhizodeposition by plants, both of which introduce abundant and varied organic molecules into the soil (Bais et al. 2006; Bertin et al. 2003). Previous studies found that 5–21% of photosynthetic carbon is discharged into the rhizosphere through root exudates (Hütsch et al. 2002; Jones et al. 2009). For example, citrate, glutarate, oxalate, malonate, malate, succinate, acetate, lactate, glycolate and formate have been reported as nutrient sources, chelators, acidifiers and detoxifiers (Bais et al. 2006; Dakora and Phillips 2002; Haichar et al. 2014), but their interaction with soil minerals is not fully understood (Dakora and Phillips 2002). This section discusses previous studies regarding the complexation of organic acids on an iron (hydr)oxide surface to reveal the competition effect on oxyanions.
A literature review found only 11 papers that describe the structural configuration of organic acids on an iron (hydr)oxide surface. A few studies have focused on the structural configuration of organic acids on pure iron (hydr)oxide using a molecular-scale approach. IR spectroscopy was the main tool for describing the complexation. Norén and Persson (2007) studied the adsorption of acetate, benzoate and cyclohexanecarboxylate by varying the ionic strength and pH. They concluded that outer-sphere complexes dominated the sorption of three monocarboxylates on goethite and that two types of outer-sphere complexes are involved in the sorption: hydrated and nonhydrated outer-sphere complex. Persson and Axe (2005) identified the structural configuration of oxalate and malonate on goethite using soft X-ray EXAFS, theoretical calculations (B3LYP, 6-31+G*) and IR spectroscopy. They found that oxalate and malonate interacted with goethite via an outer-sphere complex and an inner-sphere complex and that the inner-sphere complex was favored at low pH. A ring structure–bidentate mononuclear (RS-BM) complex was identified for oxalate and malonate. Hug and Bahnemann (2006) studied oxalate and malonate sorption on lepidocrocite via IR spectroscopy by varying the pH from 3 to 9. RSs of the BM and BB complexes were assumed for oxalate, whereas no specific complex was identified for malonate, but an inner-sphere complex of malonate was assumed because of the shifts in the asymmetric and symmetric carboxylate vibrations. Duckworth and Martin (2001) examined the sorption of oxalate, malonate, glutarate, succinate and adipate on hematite at pH 5. The oxalate, malonate and glutarate showed the RS of a BM or BB complex, whereas succinate and adipate formed an MM complex. The authors also found that the dissolution effect of organic acids on hematite, oxalate, glutarate, malonate and adipate increased the dissolution of the iron (hydr)oxide, whereas the dissolution of adipate and succinate resulted in no difference from the control. Kubicki et al. (2017) studied the adsorption of oxalate on goethite and the mechanism of reductive dissolution using DFT calculations, and they showed that oxalate is most stable as a bidentate mononuclear complex. Borowski et al. (2018) studied oxalate complexation on lepidocrocite using ATR-FTIR spectroscopy and DFT calculations by changing the pH and surface loading, and MM and BM complexes were identified. The increase in pH leads to a decrease in the content of the BM complex, and UV light causes photolysis of the BM complex compared to the MM or outer-sphere complex. Lindegren et al. (2009) showed the molecular structure of citrate and tricarballylate on goethite and confirmed that only an inner-sphere complex was formed for both citrate and tricarballylate; however, they concluded that the inner-sphere complex was prevalent at high pH via a combination of hydroxyl and carboxylic groups. This result contradicted the findings of previous studies, which implied that the IS complex formed mainly at low pH, whereas the outer-sphere complex was dominant at high pH. Gao et al. (2017) tried to evaluate the interaction between citrate and chromate on hematite, and they observed the reduction of chromate to chromite by electron transfer from citrate. The structural configuration of citrate was presumed to be a monodentate or bidentate ring structure, but no detailed description was provided. Hwang and Lenhart (2008) examined the sorption of phthalate, malate, fumarate and succinate on hematite using ATR-FTIR analysis. Phthalate, succinate and malate showed an inner-sphere complex at low pH and an outer-sphere complex at high pH, whereas fumarate showed only an outer-sphere complex. Yang et al. (2016) used ATR-FTIR measurements and theoretical calculations to interpret aspartate sorption on goethite, and formation of the RS-BB structure was the dominant sorption mechanism at pH 3. The outer-sphere complex prevailed above neutral pH via electrostatic attraction and hydrogen bonding. Bürger et al. (2013) also identified aspartate sorption on the magnetite surface through theoretical calculations (force field simulations), and the results confirmed the RS-BM complex of aspartate, the tetradentate trinuclear complex of glutamine and the RS-BB complex of glycine and leucine.
It has been recognized that the inner-sphere complex of an organic acid in various configurations occurs primarily under acidic conditions, whereas the outer-sphere complex dominates under basic conditions. Therefore, organic acids not only compete with oxyanions for sorption sites but also change the sorption sites by the dissolution of iron from the surface. However, the specific mechanism has not been fully verified; thus, these interactions must be examined to elucidate the effect of organic acids on the bioavailability of oxyanions in the soil environment.
Factors affecting the structural configuration
pH effect
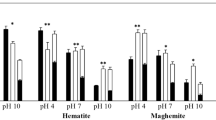
The pH is known as one of the most responsible factors governing the sorption of oxyanions on iron (hydr)oxides and their structural configuration (Elzinga and Sparks 2007; Abdala et al. 2015; Han and Ro 2018a). The pH determines the level of protonation and deprotonation in the surface functional groups of iron (hydr)oxides, and it shifts the surface charge of variably charged surfaces. The relationship between pH and interatomic distance measured by EXAFS is plotted in Fig. 3a to address the pH effect on the structural configuration, while the relationship among the pH, surface loading and surface density is plotted in Fig. 3b to explain the pattern of arsenate complexation. The graph was constructed from 13 studies using EXAFS results of arsenate on six iron (hydr)oxides under various experimental conditions. The interatomic distance of As–O was constant and ranged from 1.62 to 1.71 Å, while that of As–Fe ranged from 2.59 to 3.60 Å. Based on the interpretation of each study, the most abundant structural configuration was plotted, and the As–Fe interatomic distances of TB, BM, BB and MM ranged from 2.59, 2.83–2.93, 3.24–3.38 and 3.57–3.60 Å, respectively. Based on Fig. 3a, it was hard to identify the pH effect except that the TB complex was only observed under acidic and neutral pH conditions.

Scatter plot of arsenate on iron (hydr)oxides between pH and As–Fe distance measured by extended X-ray absorption fine structure studies (a). BB, BM and MM indicate bidentate binuclear, bidentate mononuclear and monodentate mononuclear complexes, respectively. 3D plot among pH, surface density (molecule nm−2) and surface loading (molecule nm−2) of arsenate on iron (hydr)oxide surfaces (b). B, M and T denote the bidentate, monodentate and tridentate complex, respectively. This figure was illustrated with data from the previous studies by Waychunas et al. (1993), Fendorf et al. (1997), Randall et al. (2001), Farquhar et al. (2002), Sherman and Randall (2003), Morin et al. (2008), Neupane et al. (2014); Liu et al. (2015) and Han and Ro (2018a)
For further interpretation, a 3D plot comparing the pH, surface loading and surface density was established, and it showed better results in explaining the transition of the structural configuration. The surface loading could be separated into two groups, namely low surface loading (0–5 molecule nm−2) and high surface loading (5–20 molecule nm−2). The major structural configuration at low surface loading was the bidentate complex, while both bidentate and monodentate complexes were abundant under high surface loading conditions. However, the pH effect was not identifiable when compared with the surface loading, which is because of the difference in experimental conditions among the 9 studies employed as shown in Fig. 3. Many studies have already identified the transition of structural configuration based on the pH (Elzinga and Sparks 2007; Kubicki et al. 2012; Han and Ro 2018a), but comparing the results under various experimental conditions would not lead to a meaningful effect of pH on the structural configuration compared with the effect of surface loading. This fact could be explained by the ratio between the adsorption site and sorbate. The pH change from acidic conditions to basic conditions decreases the number of adsorption sites; for example, a pH change from 4 to 10 caused the maximum surface density (Γmax) to be approximately 20–92% depending on the crystal structure of the iron (hydr)oxides (Han and Ro 2018b), while the minimum and maximum surface loading values were 0.01 and 15.4 molecules nm−2, respectively. The increased pH decreases the number of available adsorption sites to 20% of the initial coverage, while the increased surface loading would lead to a dramatic increase in sorbate concentration of 154,000% from the lowest surface loading condition to the highest surface loading condition. For that reason, the surface loading is as much of a significant factor as the pH to describe the structural configuration, the pH effect should be carefully examined under constant experimental conditions, and the comparison of numerous studies was not effective in describing the pH effect on the structural configuration under various surface loading conditions.
Surface loading effect
As mentioned above in “pH effect” section, the surface loading showed a significant effect on the structural configuration because various surface loading conditions were applied to the experiments. For a detailed interpretation, a scatter plot of the surface density (μmol m2) versus surface loading (mmol m−2) for arsenate sorption on six iron (hydr)oxides is presented in Fig. 4a. We used 19 data points from 8 papers using EXAFS results except one paper that described a tridentate complex, and the minimum and maximum values were employed only when no significant difference was found in the midrange data. The scatter plot clearly showed that the MM complex was observed only in experiments with a relatively high surface loading and low surface density, whereas no pH effect was significantly observed (Fig. 3b), but no monodentate complex was identified at low pH conditions. Fendorf et al. (1997) reported that the addition of surface loading yielded a change in the structural configuration of the complex and that an MM complex gradually became dominant at low surface loading and high pH. One possible explanation for the MM complex formation is the competition with background anions such as NO3− and Cl− or with atmospheric HCO3− and CO32− at high pH, as previous studies confirmed the competitive effects of such anions; thus, a similar effect on the complexation could result from the increase in surface loading (Brechbühl and Christl 2012; Frau et al. 2010). In addition, both experiments used 0.1 M NaNO3 to maintain the background solution, and the atmospheric conditions were not regulated. Another explanation for formation of the MM complex is the ratio among the sorbate, sorbent and solution. Many researchers have reported a sorbent concentration effect, which enhanced the sorption per unit mass or area by increasing the surface loading (Guo et al. 2014; Zhao and Hou 2012). The increased concentration of sorbate per unit area could promote complex formation at the surface, and the competition for limited sorption sites would be increased (Das et al. 2013; Fendorf et al. 1997). Not only the competition and sorbent concentration effect but also the decrease in available sorption sites might yield the MM complex. Environmental changes in pH, aggregation, particulation, organic matter, drying and freezing could alter the number of available sorption sites and the activity coefficient of the surface component (Dixit and Hering 2003; Jordan et al. 2009; Zhang and Selim 2007), and the decreased activity coefficient of the surface component would increase the amount of complex because of the sorbent concentration effect caused by adsorbent particle–particle interactions (Guo et al. 2014; Zhao and Hou 2012).

Scatter plot between surface density (μmol m−2) and surface loading (μmol m−2) for arsenate sorption on six iron (hydr)oxides using extended X-ray absorption fine structure spectroscopy results from previous papers (N = 19) (a). Scatter plot between competition effect and surface loading for the competition of arsenate with arsenite (N = 45) (b). The correlation coefficient of bidentate and monodentate complexes and bidentate complex from (a) was 0.756 and 0.736, while it was 0.877 for arsenite versus arsenate in (b). The BB, BM and MM denote the structural configuration of the inner-sphere complex as bidentate binuclear, bidentate mononuclear and monodentate mononuclear, respectively. This figure was illustrated with data from the previous studies by Waychunas et al. (1993), Fendorf et al. (1997), Jain and Loeppert (2000), Randall et al. (2001), Farquhar et al. (2002), Sherman and Randall (2003), Morin et al. (2008), Neupane et al. (2014), Liu et al. (2015)
Sample phase effect
Numerous papers have studied the sample phase effect on the structural configuration of oxyanions on the iron (hydr)oxide surface. For example, Kubicki et al. (2012) conducted an ATR-FTIR study of phosphate with DFT calculations on various surfaces of microgoethite and nanogoethite under aqueous and dried conditions, and Han and Ro (2018a, b) conducted an EXAFS study of arsenate with DFT calculations on nanogoethite under aqueous and dried conditions. Both studies confirmed the significantly different spectra between the aqueous and dried samples, which indicates a transition of the structural configuration, but several studies have identified no transition. Liu et al. (2015) employed EXAFS and XPS to identify the transition of arsenic and the transformation of ferrous and ferric oxides, and they found that drying under oxic conditions causes the transition from arsenate to arsenite by the oxidation of magnetite to ferric oxides. Only a few studies discussed the sample phase effect; thus, there was not enough information to interpret this effect.
In addition, X-ray-based techniques were mainly employed for relatively heavy oxyanions such as arsenate and selenate, but infrared-based measurements were applied for the lighter oxyanions such as carbonate, phosphate and sulfate. ATR-FTIR spectroscopy is a powerful tool to characterize interfacial reactions, while EXAFS analysis frequently employs sample phase change to a paste, dried or frozen phase for measurement. Drying would cause an effect similar to that of increasing surface loading, causing precipitation of oxyanions. For comparison, the structural configuration data of the sample phases were categorized for each major oxyanion as wet (aqueous and paste) and solid (dried and frozen) (Fig. 5). Before explaining the result, it might not be appropriate to deduce a conclusion using insufficient data and various methods, but it is still reasonable to find a general trend using the acquired data. Interestingly, except for the trigonal arsenite, the tetrahedral arsenate, phosphate, and selenate showed that monodentate complexes were more frequently found in the wet phase than in the solid phase, and the tridentate complex was only identified in arsenite and arsenate. Based on this observation, drying or freezing would transform the structural configuration such that the ligands bind with higher denticity; however, it is essential to accumulate more data to confirm the trend, but it is possible that changing the sample phase would cause a transition of the structural configuration.

Percentage bar graph of three complex types (monodentate, bidentate and tridentate) under wet (aqueous and paste) and solid (air-dried and freeze-dried) conditions for arsenite, arsenate, phosphate, selenate and the total. N indicates the number of studies of conditions for certain oxyanions. This figure was illustrated with the data from the previous studies provided in Table 1
Competition and promotive effect
Based on a previous study, the competition effect was calculated and plotted with the surface loading effect in Fig. 4b (Han and Ro 2018b). The competition effect is the ratio between the amount of sorbed sorbate with the competitor and that without the competitor, and it is based on the difference between single- and dual-batch experiments, with positive and negative values indicating the competition and promotive effect, respectively. For example, for two A or two B molecules present on the sorbent in a single batch experiment, if both one A and one B molecule were added to the sorbent, the sorption would differ by preferential or competitive adsorption. In the case of competition effect = 0, no preferential or competitive adsorption occurred because there was no difference in the adsorption using a single- or dual-batch experiment, while if the competition effect was positive or negative, the sorption was decreased or increased by the competitive or preferential interaction between the A and B molecules (Han and Ro 2018b). As shown in Fig. 3b, the competition effect was increased by the surface loading, which indicates that fewer oxyanions were adsorbed when the competitor was also present. In the soil environment, uncountable species of inorganic and organic compounds coexist together; thus, there is always competition for the limited sites. Arsenate and phosphate are known to be strong competitors for adsorption on the metal (hydr)oxide surface, while carbonate, nitrate and sulfate are weak competitors at similar concentrations. On the other hand, carbonate and silicate are frequently reported as adsorbent precursors by precipitation with cations under certain conditions, and competition is diminished by decreasing the number of competitors and increasing the abundance of surface sites available for adsorption. As discussed above, polymerization is a key mechanism for silicate adsorption on iron (hydr)oxide, but it also interrupts the adsorption of other oxyanions by covering the iron (hydr)oxide surface with polymerized silicate, leading to a more competitive environment for the adsorption of other oxyanions by decreasing the limited quantity of adsorption sites.
In the case of organic compounds, an enormous number of compounds have been found in the rhizosphere, but the majority of these compounds are still unknown regarding their roles in the soil environment. Numerous studies confirmed the possibility of competition among root-exuded compounds, but this work is still not sufficient to discuss their roles and mechanisms in the rhizosphere. A few studies have confirmed the competitive adsorption of organic acids, such as acetate, aspartate, citrate, oxalate, malate and tartrate, while Wijnja and Schulthess (2002) identified the promotive effect between acetate and sulfate on goethite. Only a small portion of organic compounds has been studied; thus, it is not appropriate to discuss their adsorption mechanisms and effects on the structural configuration; however, the presence of organic compounds would lead to a transition of the structural configuration of oxyanions both directly and indirectly. Based on current observations, organic acids would not effectively compete with strong oxyanions such as arsenate and phosphate; thus, it was presumed that the indirect way (for example, reductive dissolution) would dominate to allow plants to acquire essential nutrients from the iron (hydr)oxides. Based on our review of the literature, it was hard to find such studies describing the competitive or promotive effect on the structural configuration of oxyanions, and the indirect way of transformation in the iron (hydr)oxides should be fully studied to understand the effect of organic compounds in the rhizosphere.
Rhizodeposition effect
Rhizodeposition is recognized as a plant survival mechanism to ameliorate the rhizosphere. A significant amount of carbon is deposited as root exudates, but the effect of these exudates has not yet been well addressed. Numerous studies indirectly confirmed that root exudates might affect the bioavailability of oxyanions and the transformation of iron (hydr)oxides, but a detailed study of how they influence the structural configuration and how they change upon environmental fluctuation. As mentioned above, two possible mechanisms are introduced: a direct competitive or promotive effect on adsorption sites and the indirect transformation of iron (hydr)oxides through reductive dissolution (Lindegren and Persson 2009; Kubicki et al. 2017). Numerous compounds are exuded from plant roots and microbes at different compositions over growth stage and time (Jones et al. 2009). Modern analytical technology is not sufficient to identify the crucial mechanisms among the various coexisting compounds; thus, we need more time and effort to separate the meaningful mechanisms by a bottom-up approach. Among the numerous compounds, citrate and oxalate are good examples. The indirect evidence regarding the effect of organic acids on oxyanions and the direct observation of citrate and oxalate could allow the basic principles to be established.
The germination of plants does not require exogenous nutrients; only water, sunlight and temperature initiate the growth. After the rapid development of photosynthesis, the water pumping system of the leaf starts to pull up soil water, and the dissolved nutrients in the soil water are readily utilized for growth. Among various oxyanions, nitrate, phosphate and sulfate are essential macronutrients, but the level of nutrients required and their mobility in the soil water are different (Xu et al. 2006; Acelas et al. 2013). In the case of nitrate and sulfate, their affinity at the soil surface is relatively low compared with that of phosphate; thus, most nitrate and sulfate is dissolved in the soil solution or easily desorbed from the soil surface, while phosphate is strongly adsorbed on the metal (hydr)oxide surface in the soil environment. During photosynthesis and respiration of plants, numerous metabolites are synthesized, and the exudation of such metabolites has been observed (Hütsch et al. 2002). Citrate and oxalate are frequently observed metabolites in the exudate. Previous studies identified the indirect reductive dissolution of oxyanion-sorbed metal (hydr)oxides, and direct competitive adsorption with phosphate was also observed. Under Fe and P deficiency conditions, exudation occurs abundantly, which helps to uptake Fe and P for plant growth (López-Arredondo et al. 2014; Zhu et al. 2015).
The exudates not only affect oxyanions and iron (hydr)oxides but also might cause the severe colloidal mobilization of iron (hydr)oxide nanoparticles because they could stabilize the nanoparticles by complexation on the surface. For example, citrate and aspartate are known to be nanoparticle stabilizers that can be dispersed in the medium and prevent dissolution (Frost et al. 2017; Phenrat et al. 2018). However, the current analytical techniques and knowledge regarding nanoparticles in the soil environment are not enough to discuss the combination of various effects; thus, we should develop more advanced analytical techniques that minimize phase transition, and we should characterize the individual interactions from a complex interconnected soil ecosystem.
Transformation of iron (hydr)oxides and their colloidal mobilization
The reason for focusing on iron (hydr)oxides in this review is to identify the effect of crystal structure on the structural configuration of oxyanions, but there are numerous factors governing the structural configuration; thus, it is difficult to isolate the effect of crystal structure only. Furthermore, the structural configuration of a single iron (hydr)oxide remains controversial. For example, goethite and phosphate are the most frequently employed mineral and oxyanion in studies, respectively: four studies confirmed the bidentate binding fashion, two studies identified both bidentate and monodentate together, and only two studies confirmed the monodentate fashion. Among these studies, interesting phenomena were suggested by Han and Ro (2019) including the formation of side-by-side bridging complexes between goethite nanorods, which would be one of reasons for phosphate sequestration and the low bioavailability. Based on these previous studies, it is still not enough to conclude the effects of crystal structure on the structural configuration, and more accumulated data with reference methods and materials would lead to insights into these complex interactions.
It has been observed that the transformation of iron (hydr)oxides occurs upon environmental fluctuation, but the detailed mechanisms have not yet been clearly identified. In addition, there are several studies in which colloidal mobilization would occur by environmental fluctuations; however, it is extremely difficult to measure transformation and colloidal mobilization at the same time. For example, a recent HR-TEM study identified bernalite formation from goethite at high pH conditions in the presence of arsenate, but the EXAFS and XANES data did not show the formation of bernalite at the Fe edge because the concentration was relatively too low to detect even when using a synchrotron (Han and Ro 2018a). In addition, it is possible that bernalite is formed during the drying stage when making a specimen for HR-TEM analysis. The reactivity and stability of nanoparticles are strong and weak, respectively; thus, it is difficult both to observe the transformation or presence of nanoparticles in nature using current analytical technology and to obtain the sample from nature without changing the conditions. The majority of current analytical technologies employ phase transition to conduct a measurement, for example, solid and vacuum conditions for spectroscopy or microscopy using electrons. For that reason, the phase effect would not be diminished. XAFS and ATR-FTIR techniques are appropriate tools to minimize the phase effect, but it is still hard to detect low concentrations of transforming nanoparticles.
Based on studies examining the effect of oxyanions on the transformation of iron (hydr)oxides, the pH, type and concentration of oxyanions controlled the transformation from ferrihydrite to goethite, hematite or feroxyhyte (Michael Bolanz et al. 2013). Unlike the result by Han and Ro (2018a), a high concentration of oxyanions suppressed the transformation of ferrihydrite except the antimony-based oxyanion, which led to feroxyhyte formation at high concentrations only. During the transformation process, not only the crystal structure but also the morphology is transformed, which determines the surface area, reactivity and stability (MacHala et al. 2011). The change in morphology would significantly increase or decrease colloidal mobilization, but there is no study discussing such an effect to the best of our knowledge. As mentioned above, the oxyanions encounter numerous chemical species at different concentrations over time, but our knowledge is only based on the measurement at a certain time with phase transition; thus, there might be several missing links to describe the transformation and colloidal mobilization of iron (hydr)oxides and oxyanions. However, we believe that understanding the individual interactions among numerous chemicals in the soil environment would lead us to better understand their behaviors and fate.
Problems during spectral interpretation and prospects of computational chemistry
EXAFS and ATR-FTIR spectroscopy are the key methods for identifying structural configurations, but there can be subsidiary problems caused by the spectral processing. There are two ways to interpret the measured spectra, namely theoretical calculations for the ideal structure and experimental measurements of a similar structure. For example, scorodite (FeAsO4·2H2O) is a common mineral structure for shell fitting the structural configuration of arsenic using EXAFS spectroscopy, but it only provides two As–Fe single scattering paths. Many studies employed scorodite for shell fitting, which identified two bidentate and monodentate complexes (Waychunas et al. 1993; Sherman and Randall 2003; Morin et al. 2008; Gao et al. 2013). On the other hand, a recent study by Han and Ro (2018b) using EXAFS analysis and DFT calculations confirmed that the transition of the arsenate complex from tridentate to monodentate depends on the environmental conditions, and Farrell (2017) calculated the tridentate complex to be thermodynamically favorable using DFT calculations. A direct comparison between the shell fitting with the mineral structure and DFT calculations is not applicable because various experimental conditions (surface loading, pH or phase) were applied. In addition, the theoretical structure for computational chemistry is still too simple to represent the real environmental conditions. For that reason, it is difficult to conclude which approach is more appropriate to interpret the spectra.
Not only the methods for spectral interpretation but also the stability of iron (hydr)oxide films in ATR-FTIR studies could cause unexpected results regarding the structural configuration of oxyanions and organic acids. Han and Ro (2019) identified shrinking and swelling effects of phosphate on the goethite and maghemite surface, indicating that the concentration of oxyanions and iron (hydr)oxides in the evanescent wave path would fluctuate by the intercolloidal interactions. However, the stability of iron (hydr)oxides in the ATR-FTIR studies has not fully explained the previous kinetic and equilibrium studies, and it is essential to examine the colloidal stability for further study; moreover, unrevealed errors or misinterpretation would lead to problems in identifying the structural configuration on the iron (hydr)oxides.
The observed spectra from natural reactions mostly contain numerous overlapping signals, and the separation of overlapping signals is the key process for understanding these reactions. The soil heterogeneity causes difficulty in the signal processing, but a bottom-up approach by understanding a simple system to extend the understanding of more complex systems has been successfully undertaken. There are several ways to identify specific signals that explain a given change, including by modulating the environmental conditions such as ionic strength, competing ions, pH and redox potentials or by calculating the theoretical properties using computational chemistry. The structural configuration of arsenic represents a good example because papers published in the 1990s used scorodite as the mineral structure for shell fitting of EXAFS studies described above, but most papers published in the 2000s utilized the simple octahedral iron hydroxide structure for shell fitting. Recent studies in the 2010s have employed more complex structures for computational chemistry by using more efficient approaches such as periodic boundary conditions or projector-augmented wave methods.
Recent publications have discussed the adsorption of oxyanions on the facet of nanoparticle structures. The study of phosphate on goethite by Kubicki et al. (2012) is an appropriate example. The nanorod structure of goethite was employed, and two types of phosphate complex (monodentate and bidentate) on (100), (010), (001), (101) and (210) facets were calculated. As a result, each facet showed an energetically favorable structural configuration: the bidentate complex was favorable on the (101) and (100) facets, and the monodentate complex was favorable on the (210) and (001) facets, but adsorption on the (010) facet was predicted to be unfavorable. This study showed valuable results from computational chemistry, but current analytical techniques could not differentiate the spectra from each facet. There is a theoretical possibility to measure the individual spectrum of each facet by employing different percentages of facets with the same phase, but this approach remains challenging. There are still gaps between the experimental data and theoretical calculation, but the advance of computational chemistry has enabled us to estimate the atomic-scale structures of simple systems. We believe that such developments will eventually reveal the atomic-scale structures of heterogeneous soil.
Conclusion
Iron (hydr)oxide is one of the key sorbents for oxyanions in the soil environment. The seasonal and spatial transformation of iron (hydr)oxide determines the bioavailability and leachability of oxyanions and ultimately influences the geocycling of the Earth. The structural configurations of oxyanions were clearly confirmed to be dependent on environmental factors, such as the crystal structure of iron (hydr)oxide, surface area, type of oxyanions, concentration, competing ions, pH and redox potential, and the sorption percentage is also linked to environmental factors. The reciprocal interactions among the environmental factors hamper the estimation of the dynamics of oxyanions, which cause severe environmental problems worldwide such as eutrophication and metal poisoning.
Based on previous studies and our observations, we concluded that a change in environmental conditions leads to the transformation of iron (hydr)oxides and the transition of the structural configuration of oxyanions on these iron (hydr)oxides, and both results contribute to the dynamics of oxyanions in the soil system. Figure 6 shows the complexly interconnected mechanisms of transformation of iron (hydr)oxides among the oxyanions, organic acids and iron (hydr)oxides for better understanding the natural reactions, but these are still far from the real reactions because they are only limited to simple systems in the laboratory that are not real in nature; however, this bottom-up approach has identified numerous mechanisms that could not be identified before, and the complexity of the system has been advanced with the development of analytics.

Competitive adsorption of toxicants and nutrients on the iron (hydr)oxide nanoparticles and the reconstructive and topotactic transformation with organic acids, oxyanions and organic matters. The colloidal mobilization causes the severe effect on aquatic environment and it causes the harmful effects on the ecosystem and human health eventually
Based on observations, we could not find a general pattern on the transition upon environmental perturbations, but it was possible to presume that the transition of structural configuration occurred by environmental fluctuations, as illustrated in Fig. 7 based on the previous studies. Under conditions of low surface loading and low pH at the interface in the absence of competing ions, oxyanions with high affinity such as arsenate, chromate, selenate and phosphate possibly form a complex with higher denticity, but an increase in pH decreases the number of sorption sites; thus, the transition from a tri- or bidentate complex to monodentate and outer-sphere complexes occurs. An increase in surface loading causes competition at the limited sorption sites, and a similar transition occurs with a pH increase. An increase in both pH and surface loading would change the ratio between the sorbate and sorbent. The sample phase has been shown to cause significantly different phenomena. The charge of the monodentate complex at the interface is compensated by both the iron (hydr)oxide surface and cations in the electrical layer, but drying eliminates compensation by those cations. In addition, diffused oxyanions in the water accumulate at the surface during the drying process, and the actual surface loading increases during the drying process. After complete loss of water molecules at the surface, the precipitates from ions and complexes on the surface remain, and the bidentate complex would be dominant because no cation is available to compensate for the charge of the monodentate complex. The introduction of competing ions would differ based on the affinity of the two sorbates on the iron (hydr)oxide surface. If the affinity and sorption mechanisms are similar, the addition of competing ions would be similar to the surface loading effect, but the difference in the affinity would cause a more complicated transition of the structural configuration. As shown in Fig. 7, the competing ions were presumed to be oxyanions with low affinity such as carbonate, nitrate and sulfate, and the competing ions would bind to the sorption site, where those with high affinity would not be able to bind. The competing ions with low affinity slightly exchange with the oxyanions with high affinity.

Transition of structural configuration with the change in pH, surface loading, drying and competing ions. The white tetrahedron indicates an outer-sphere complex, while the blue, gray and red tetrahedra represent monodentate, bidentate and tridentate complexes, respectively. The light-gray triangle represents an outer-sphere complex, while the light-blue and pink triangles represent monodentate and bidentate complexes, respectively. The tetrahedra are presumed to represent sorbates with relatively high affinity such as arsenate, chromate, selenate and phosphate, whereas the triangles represent a sorbate with relatively low affinity such as carbonate, nitrate and sulfate. The OS, TB, TM, BB, BM, MB and MM indicate the outer-sphere, tridentate binuclear, tridentate mononuclear, bidentate binuclear, bidentate mononuclear, monodentate binuclear and monodentate mononuclear complex, respectively
However, the presumptions regarding the transition of structural configuration are not yet valid. There are numerous possibilities of misinterpretation in this paper as well as errors in the experiments and data interpretation; however, after careful review of hundreds of papers, we made conclusions regarding the transition of structural configuration. Developments in analytical and theoretical chemistry have enabled us to see the structural configuration of the complex at the atomic level, but many questions remain for future study. The following questions need to be answered to address environmental problems and to understand oxyanion behavior in the soil environment.
- 1.
Based on our observations, the surface loading is the critical factor for the structural configuration of complexes. However, studies using EXAFS spectroscopy mainly utilize a paste or freeze-dried sample for the examination. The drying process would be similar to subjecting the sample to high surface loading conditions; thus, the sample phase effect should be carefully identified to determine the actual structural configuration of the complex.
- 2.
Several studies identified the MM complex of arsenate and phosphate using various methodologies, but the environmental conditions of the experiments were not sufficiently consistent to obtain a better understanding. Any undiscovered factors controlling the structural configuration of the MM complex should be determined. If the structural configuration could be changed on demand, this ability would be applicable to controlling the environmental problems caused by oxyanions.
- 3.
Recent developments in computational chemistry have led us to interpret experimental data through comparison with theoretical calculations. This approach is applicable only to small molecular clusters for now, but advances in computation technology will enable us to address complex clusters, and we should accumulate data for better computation and interpretation.
- 4.
The characteristics of laboratory-synthesized iron (hydr)oxides differed case by case, making interpretation of the results difficult. In addition, the available data were not sufficient to reach statistical conclusions. More data, a standard method and standard materials are essential for achieving compatible and credible experiments.
- 5.
Most studies overlook the possibility of transformation of metal (hydr)oxides during the experiments and pretreatments for further measurement; however, iron (hydr)oxides easily transform to various nanoparticles under environmental fluctuations. The stability of iron (hydr)oxides should be fully assessed during the experiment and measurement process to better understand the dynamics of oxyanions and iron (hydr)oxides in the soil environment.
In ancient agriculture, limited oxyanions were a weighty matter, but we are now concerned with an excessive abundance of oxyanions. The industrialization of cultivation has brought us prosperity, but this prosperity is accompanied by severe problems. We should be prepared to solve these problems by fully and correctly understanding the dynamics of oxyanions and the interactions among plants, oxyanions and soil components.
References
Abdala DB, Northrup PA, Arai Y, Sparks DL (2015) Surface loading effects on orthophosphate surface complexation at the goethite/water interface as examined by extended X-ray Absorption Fine Structure (EXAFS) spectroscopy. J Colloid Interface Sci 437:297–303. https://doi.org/10.1016/j.jcis.2014.09.057
Acelas NY, Mejia SM, Mondragón F, Flórez E (2013) Density functional theory characterization of phosphate and sulfate adsorption on Fe-(hydr)oxide: reactivity, pH effect, estimation of Gibbs free energies, and topological analysis of hydrogen bonds. Comput Theor Chem 1005:16–24. https://doi.org/10.1016/j.comptc.2012.11.002
Adegoke HI, Adekola FA, Fatoki OS, Ximba BJ (2013) Sorptive interaction of oxyanions with iron oxides: a review. Pol J Environ Stud 22:7–24
Ahmed AA, Gypser S, Leinweber P et al (2019) Infrared spectroscopic characterization of phosphate binding at the goethite-water interface. Phys Chem Chem Phys 21:4421–4434. https://doi.org/10.1039/c8cp07168c
Alibeigi S, Vaezi MR (2008) Phase transformation of iron oxide nanoparticles by varying the molar ratio of Fe2+:Fe3+. Chem Eng Technol 31:1591–1596. https://doi.org/10.1002/ceat.200800093
Alloway B (1995) Heavy metals in soils trace metals and metalloids in soils and their bioavailability. Springer, Berlin
Andrew S, Bob F, Paul W (2000) Practical and innovative measures for the control of agricultural phosphorus losses to water. J Environ Qual 29:1–9
Anschutz AJ, Penn RL (2005) Reduction of crystalline iron(III) oxyhydroxides using hydroquinone: influence of phase and particle size. Geochem Trans 6:60–66. https://doi.org/10.1063/1.2037887
Antelo J, Avena M, Fiol S et al (2005) Effects of pH and ionic strength on the adsorption of phosphate and arsenate at the goethite-water interface. J Colloid Interface Sci 285:476–486. https://doi.org/10.1016/j.jcis.2004.12.032
Arai Y, Sparks DL (2001) ATR–FTIR spectroscopic investigation on phosphate adsorption mechanisms at the ferrihydrite-water interface. J Colloid Interface Sci 241:317–326. https://doi.org/10.1006/jcis.2001.7773
Arroyave JM, Puccia V, Zanini GP, Avena MJ (2018) Surface speciation of phosphate on goethite as seen by InfraRed Surface Titrations (IRST). Spectrochim Acta Part A Mol Biomol Spectrosc 199:57–64. https://doi.org/10.1016/j.saa.2018.03.043
Auffan M, Rose J, Proux O et al (2008) Enhanced adsorption of arsenic onto maghemites nanoparticles: As(III) as a probe of the surface structure and heterogeneity. Langmuir 24:3215–3222. https://doi.org/10.1021/la702998x
Aulakh MS, Wassmann R, Bueno C et al (2001) Characterization of root exudates at different growth stages of ten rice (Oryza sativa L.) cultivars. Plant Biol 3:139–148. https://doi.org/10.1055/s-2001-12905
Babula P, Adam V, Opatrilova R et al (2008) Uncommon heavy metals, metalloids and their plant toxicity: a review. Environ Chem Lett 6:189–213. https://doi.org/10.1007/s10311-008-0159-9
Baetz U, Martinoia E (2014) Root exudates: the hidden part of plant defense. Trends Plant Sci 19:90–98. https://doi.org/10.1016/j.tplants.2013.11.006
Bais HP, Weir TL, Perry LG et al (2006) The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol 57:233–266. https://doi.org/10.1146/annurev.arplant.57.032905.105159
Barberis E, Ajmone Marsan F, Scalenghe R et al (1995) European soils overfertilized with phosphorus: part 1. Basic properties. Fertil Res 45:199–207. https://doi.org/10.1007/BF00748590
Bargar JR, Kubicki JD, Reitmeyer R, Davis JA (2005) ATR-FTIR spectroscopic characterization of coexisting carbonate surface complexes on hematite. Geochim Cosmochim Acta 69:1527–1542. https://doi.org/10.1016/j.gca.2004.08.002
Bertin C, Yang X, Weston LA (2003) The role of root exudates and allelochemicals in the rhizosphere. Plant Soil 256:67–83. https://doi.org/10.1023/A:1026290508166
Bolanz RM, Bläss U, Ackermann S et al (2013a) The effect of antimonate, arsenate, and phosphate on the transformation of ferrihydrite to goethite, hematite, feroxyhyte, and tripuhyite. Clays Clay Miner 61:11–25. https://doi.org/10.1346/CCMN.2013.0610102
Bolanz RM, Wierzbicka-Wieczorek M, Čaplovičová M et al (2013b) Structural incorporation of As5+ into hematite. Environ Sci Technol 47:9140–9147. https://doi.org/10.1021/es305182c
Borer P, Hug SJ (2014) Photo-redox reactions of dicarboxylates and α-hydroxydicarboxylates at the surface of Fe(III)(hydr)oxides followed with in situ ATR-FTIR spectroscopy. J Colloid Interface Sci 416:44–53. https://doi.org/10.1016/j.jcis.2013.10.030
Borowski SC, Biswakarma J, Kang K et al (2018) Structure and reactivity of oxalate surface complexes on lepidocrocite derived from infrared spectroscopy, DFT-calculations, adsorption, dissolution and photochemical experiments. Geochim Cosmochim Acta 226:244–262. https://doi.org/10.1016/j.gca.2018.01.024
Bose P, Sharma A (2002) Role of iron in controlling speciation and mobilization of arsenic in subsurface environment. Water Res 36:4916–4926
Bowen HJM (1979) Environmental chemistry of the elements. Academic Press, London
Brechbühl Y, Christl I (2012) Competitive sorption of carbonate and arsenic to hematite: combined ATR-FTIR and batch experiments. J Colloid Interface Sci 377:313–321. https://doi.org/10.1016/j.jcis.2012.03.025
Bürger A, Magdans U, Gies H (2013) Adsorption of amino acids on the magnetite-(111)-surface: a force field study. J Mol Model 19:851–857. https://doi.org/10.1007/s00894-012-1606-x
Catalano JG, Zhang Z, Fenter P, Bedzyk MJ (2006) Inner-sphere adsorption geometry of Se(IV) at the hematite (100)-water interface. J Colloid Interface Sci 297:665–671. https://doi.org/10.1016/j.jcis.2005.11.026
Catalano JG, Zhang Z, Park C et al (2007) Bridging arsenate surface complexes on the hematite (0 1 2) surface. Geochim Cosmochim Acta 71:1883–1897. https://doi.org/10.1016/j.gca.2007.01.015
Catalano JG, Park C, Fenter P, Zhang Z (2008) Simultaneous inner- and outer-sphere arsenate adsorption on corundum and hematite. Geochim Cosmochim Acta 72:1986–2004. https://doi.org/10.1016/j.gca.2008.02.013
Chen RF, Shen RF, Gu P et al (2006) Response of rice (Oryza sativa) with root surface iron plaque under aluminium stress. Ann Bot 98:389–395. https://doi.org/10.1093/aob/mcl110
Combes JM, Manceau A, Calas G (1990) Formation of ferric oxides from aqueous solutions: a polyhedral approach by X-ray absorption spectroscopy: II. Hematite formation from ferric gels. Geochim Cosmochim Acta 54:1083–1091. https://doi.org/10.1016/0016-7037(90)90440-V
Cornell RM, Schwertmann U (2003) The iron oxides : structure, properties, reactions, occurrences, and uses. Wiley, Amsterdam
Cox PA (1989) The elements: their origin, abundance and distribution. Oxford University Press, Oxford
Cudennec Y, Lecerf A (2005) Topotactic transformations of goethite and lepidocrocite into hematite and maghemite. Solid State Sci 7:520–529. https://doi.org/10.1016/j.solidstatesciences.2005.02.002
Cudennec Y, Lecerf A (2006) The transformation of ferrihydrite into goethite or hematite, revisited. J Solid State Chem 179:716–722. https://doi.org/10.1016/j.jssc.2005.11.030
Dakora FD, Phillips DA (2002) Root exudates as mediators of mineral acquisition in low-nutrient environments. Plant Soil 245:35–47. https://doi.org/10.1023/A:1020809400075
Daou TJ, Grenche JM, Thomas F et al (2007) Phosphate adsorption properties of magnetite-based nanoparticles phosphate adsorption properties of magnetite-based nanoparticles. Chem Mater 19:4494–4505. https://doi.org/10.1021/cm071046v
Das S, Jim Hendry M, Essilfie-Dughan J (2013) Adsorption of selenate onto ferrihydrite, goethite, and lepidocrocite under neutral pH conditions. Appl Geochem 28:185–193. https://doi.org/10.1016/j.apgeochem.2012.10.026
Dixit S, Hering JG (2003) Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility. Environ Sci Technol 37:4182–4189. https://doi.org/10.1021/es030309t
Dowling CB (2002) Geochemical study of arsenic release mechanisms in the Bengal Basin groundwater. Water Resour Res 38:1173–1190
Duc M, Lefèvre G, Fédoroff M (2006) Sorption of selenite ions on hematite. J Colloid Interface Sci 298:556–563. https://doi.org/10.1016/j.jcis.2006.01.029
Duckworth OW, Martin ST (2001) Surface complexation and dissolution of hematite by C1–C6 dicarboxylic acids at pH = 5.0. Geochim Cosmochim Acta 65:4289–4301. https://doi.org/10.1016/S0016-7037(01)00696-2
Dzade NY, De Leeuw NH (2018) Density functional theory characterization of the structures of H3AsO3 and H3AsO4 adsorption complexes on ferrihydrite. Environ Sci Process Impacts 20:977–987. https://doi.org/10.1039/c7em00608j
Eggleston CM, Hug S, Stumm W et al (1998) Surface complexation of sulfate by hematite surfaces: FTIR and STM observations. Geochim Cosmochim Acta 62:585–593. https://doi.org/10.1016/S0016-7037(97)00372-4
Eick MJ, Peak JD, Brady WD (1999) The effect of oxyanions on the oxalate-promoted dissolution of goethite. Soil Sci Soc Am J 63:1133–1141
Elser JJ, Bracken MES, Cleland EE et al (2007) Global analysis of nitrogen and phosphorus limitation of primary producers in freshwater, marine and terrestrial ecosystems. Ecol Lett 10:1135–1142. https://doi.org/10.1111/j.1461-0248.2007.01113.x
Elzinga EJ, Sparks DL (2007) Phosphate adsorption onto hematite: an in situ ATR-FTIR investigation of the effects of pH and loading level on the mode of phosphate surface complexation. J Colloid Interface Sci 308:53–70. https://doi.org/10.1016/j.jcis.2006.12.061
Erbs JJ, Berquó TS, Reinsch BC et al (2010) Reductive dissolution of arsenic-bearing ferrihydrite. Geochim Cosmochim Acta 74:3382–3395. https://doi.org/10.1016/j.gca.2010.01.033
Fakour H, Lin T (2014) Effect of humic acid on As redox transformation and kinetic adsorption onto iron oxide based adsorbent (IBA). Int J Environ Res Public Health 11:10710–10736. https://doi.org/10.3390/ijerph111010710
Fan JX, Wang YJ, Liu C et al (2014) Effect of iron oxide reductive dissolution on the transformation and immobilization of arsenic in soils: new insights from X-ray photoelectron and X-ray absorption spectroscopy. J Hazard Mater 279:212–219. https://doi.org/10.1016/j.jhazmat.2014.06.079
Farquhar ML, Charnock JM, Livens FR, Vaughan DJ (2002) Mechanisms of arsenic uptake from aqueous solution by interaction with goethite, lepidocrocite, mackinawite, and pyrite: an X-ray absorption spectroscopy study. Environ Sci Technol 36:1757–1762. https://doi.org/10.1021/es010216g
Farrell J (2017) Tridentate arsenate complexation with ferric hydroxide and its effect on the kinetics of arsenate adsorption and desorption. Chemosphere 184:1209–1214. https://doi.org/10.1016/j.chemosphere.2017.06.099
Fendorf S, Eick MJ, Grossl P, Sparks DL (1997) Arsenate and chromate retention mechanisms on goethite. 1. Surface structure. Environ Sci Technol 31:315–320. https://doi.org/10.1021/es950653t
Ferrari M, Mottola L, Quaresima V (2004) Principles, techniques, and limitations of near infrared spectroscopy. Can J Appl Physiol 29:463–487. https://doi.org/10.1139/h04-031
Fine P, Verosub KL, Singer MJ (1995) Pedogenic and lithogenic contributions to the magnetic susceptibility record of the Chinese loess/palaeosol sequence. Geophys J Int 122:97–107. https://doi.org/10.1111/j.1365-246X.1995.tb03539.x
Frau F, Addari D, Atzei D, Biddau R (2010) Influence of major anions on As(V) adsorption by synthetic 2-line ferrihydrite. Kinetic investigation and XPS study of the competitive effect of bicarbonate. Water Air Soil Pollut 205:25–41. https://doi.org/10.1007/s11270-009-0054-4
Frost MS, Dempsey MJ, Whitehead DE (2017) The response of citrate functionalised gold and silver nanoparticles to the addition of heavy metal ions. Colloids Surfaces A Physicochem Eng Asp 518:15–24. https://doi.org/10.1016/j.colsurfa.2016.12.036
Fukushi K, Aoyama K, Yang C et al (2013) Surface complexation modeling for sulfate adsorption on ferrihydrite consistent with in situ infrared spectroscopic observations. Appl Geochemistry 36:92–103. https://doi.org/10.1016/j.apgeochem.2013.06.013
Gao X, Root RA, Farrell J et al (2013) Effect of silicic acid on arsenate and arsenite retention mechanisms on 6-L ferrihydrite: a spectroscopic and batch adsorption approach. Appl Geochem 38:110–120. https://doi.org/10.1016/j.apgeochem.2013.09.005
Gao WG, Liu XC, Chen MF (2017) In situ ATR-FTIR investigation and theoretical calculation of the interactions of chromate and citrate on the surface of haematite (α-Fe2O3). RSC Adv 7:41011–41016. https://doi.org/10.1039/c7ra04587e
Geiss CE, Zanner CW (2006) How abundant is pedogenic magnetite? Abundance and grain size estimates for loessic soil based on rock magnetic analyses. J Geophys Res Solid Earth 111:1–19. https://doi.org/10.1029/2006JB004564
Georgiadis M, Cai Y, Solo-Gabriele HM (2006) Extraction of arsenate and arsenite species from soils and sediments. Environ Pollut 141:22–29. https://doi.org/10.1016/j.envpol.2005.08.028
Gleyzes C, Tellier S (2001) Arsenic characterisation in industrial soils by chemical extractions. Environ Technol 22:27–38. https://doi.org/10.1080/09593332208618313
Goldberg S (2013) Modeling selenite adsorption envelopes on oxides, clay minerals, and soils using the triple layer model. Soil Sci Soc Am J 77:64–71. https://doi.org/10.2136/sssaj2012.0205
Gu C, Wang Z, Kubicki JD et al (2016) X-ray absorption spectroscopic quantification and speciation modeling of sulfate adsorption on ferrihydrite surfaces. Environ Sci Technol 50:8067–8076. https://doi.org/10.1021/acs.est.6b00753
Guo Y, Hou W, Liang J, Liu J (2014) Sorbent concentration effect on adsorption of methyl orange on chitosan beads in aqueous solutions. Chem Res Chin Univ 30:837–843. https://doi.org/10.1007/s40242-014-4042-x
Haichar FEZ, Santaella C, Heulin T, Achouak W (2014) Root exudates mediated interactions belowground. Soil Biol Biochem 77:69–80. https://doi.org/10.1016/j.soilbio.2014.06.017
Han J, Ro H (2018a) Identification of bernalite transformation and tridentate arsenate complex at nano-goethite under effects of drying, ph and surface loading. Sci Rep 8:1–10. https://doi.org/10.1038/s41598-018-26808-4
Han J, Ro HM (2018b) Interpreting competitive adsorption of arsenate and phosphate on nanosized iron (hydr)oxides: effects of pH and surface loading. Environ Sci Pollut Res 25:28572–28582. https://doi.org/10.1007/s11356-018-2897-y
Han J, Ro HM (2019) Characterizing preferential adsorption of phosphate on binary sorbents of goethite and maghaemite using in situ ATR-FTIR and 2D correlation spectroscopy. Sci Rep 9:1–11. https://doi.org/10.1038/s41598-019-42575-2
Han J, Kim J, Kim M et al (2014) Chemical extractability of As and Pb from soils across long-term abandoned metallic mine sites in Korea and their phytoavailability assessed by Brassica juncea. Environ Sci Pollut Res Int 22:1270–1278. https://doi.org/10.1007/s11356-014-3441-3
Han J, Ro H-M, Cho KH, Kim K-W (2016) Fluxes of nutrients and trace metals across the sediment-water interface controlled by sediment-capping agents: bentonite and sand. Environ Monit Assess 188:566. https://doi.org/10.1007/s10661-016-5583-x
Hiemstra T, Rahnemaie R, Van Riemsdijk WH (2004) Surface complexation of carbonate on goethite: IR spectroscopy, structure and charge distribution. J Colloid Interface Sci 278:282–290. https://doi.org/10.1016/j.jcis.2004.06.014
Hiemstra T, Rietra RPJJ, Van Riemsdijk WH (2007) Surface complexation of selenite on goethite: MO/DFT geometry and charge distribution. Croat Chem Acta 80:313–324
Hinkle MAG, Wang Z, Giammar DE, Catalano JG (2015) Interaction of Fe(II) with phosphate and sulfate on iron oxide surfaces. Geochim Cosmochim Acta 158:130–146. https://doi.org/10.1016/j.gca.2015.02.030
Hou X, Shen W, Huang X et al (2016) Ascorbic acid enhanced activation of oxygen by ferrous iron: A case of aerobic degradation of rhodamine B. J Hazard Mater 308:67–74. https://doi.org/10.1016/j.jhazmat.2016.01.031
Huang X, Chaparro JM, Reardon KF et al (2014) Rhizosphere interactions: root exudates, microbes, and microbial communities 1. Botany 275:267–275
Huang Q, Yu Y, Wang Q et al (2015) Uptake kinetics and translocation of selenite and selenate as affected by iron plaque on root surfaces of rice seedlings. Planta 241:907–916. https://doi.org/10.1007/s00425-014-2227-7
Huang X, Hou X, Song F et al (2017) Ascorbate induced facet dependent reductive dissolution of hematite nanocrystals. J Phys Chem C. https://doi.org/10.1021/acs.jpcc.6b09281
Hug SJ (1997) In situ Fourier transform infrared measurements of sulfate adsorption on hematite in aqueous solutions. J Colloid Interface Sci 188:415–422. https://doi.org/10.1006/jcis.1996.4755
Hug SJ, Bahnemann D (2006) Infrared spectra of oxalate, malonate and succinate adsorbed on the aqueous surface of rutile, anatase and lepidocrocite measured with in situ ATR-FTIR. J Electron Spectrosc Relat Phenomena 150:208–219. https://doi.org/10.1016/j.elspec.2005.05.006
Hütsch BW, Augustin J, Merbach W (2002) Plant rhizodeposition—an important source for carbon turnover in soils. J Plant Nutr Soil Sci. https://doi.org/10.1002/1522-2624(200208)165
Hwang YS, Lenhart JJ (2008) Adsorption of C4-dicarboxylic acids at the hematite/water interface. Langmuir 24:13934–13943. https://doi.org/10.1021/la801793p
Hyun S, Lee LS (2008) Pentachlorophenol sorption by variable-charge soils in methanol-water mixture: pH effect at the low solvent volume fraction. Chemosphere 70:503–510. https://doi.org/10.1016/j.chemosphere.2007.06.027
Ilbert M, Bonnefoy V (2013) Insight into the evolution of the iron oxidation pathways. Biochim Biophys Acta Bioenergy 1827:161–175. https://doi.org/10.1016/j.bbabio.2012.10.001
Iwasaki T, Sato N, Kosaka K et al (2011) Direct transformation from goethite to magnetite nanoparticles by mechanochemical reduction. J Alloys Compd 509:34–37. https://doi.org/10.1016/j.jallcom.2010.10.029
Jain C, Ali I (2000) Arsenic: occurrence, toxicity and speciation techniques. Water Res 34:4304–4312
Jain A, Loeppert RH (2000) Effect of competing anions on the adsorption of arsenate and arsenite by ferrihydrite. J Environ Qual 29:1422–1430. https://doi.org/10.2134/jeq2000.00472425002900050008x
Johnson DL (1971) Simultaneous determination of arsenate and phosphate in natural waters. Environ Sci Technol 5:411–414. https://doi.org/10.1016/j.apmr.2010.12.011
Johnson MG, McBride MB (1989) Mineralogical and chemical characteristics of adirondack spodosols: evidence for para- and noncrystalline aluminosilicate minerals. Soil Sci Soc Am J 53:482. https://doi.org/10.2136/sssaj1989.03615995005300020030x
Johnston CP, Chrysochoou M (2012) Investigation of chromate coordination on ferrihydrite by in situ ATR-FTIR spectroscopy and theoretical frequency calculations. Environ Sci Technol 46:5851–5858. https://doi.org/10.1021/es300660r
Johnston CP, Chrysochoou M (2014) Mechanisms of chromate adsorption on hematite. Geochim Cosmochim Acta 138:146–157. https://doi.org/10.1016/j.gca.2014.04.030
Jolivet JP, Tronc E, Chanéac C (2006) Iron oxides: from molecular clusters to solid. A nice example of chemical versatility. C R Geosci 338:488–497. https://doi.org/10.1016/j.crte.2006.04.014
Jolsterå R, Gunneriusson L, Forsling W (2010) Adsorption and surface complex modeling of silicates on maghemite in aqueous suspensions. J Colloid Interface Sci 342:493–498. https://doi.org/10.1016/j.jcis.2009.10.080
Jones DL, Nguyen C, Finlay RD (2009) Carbon flow in the rhizosphere: carbon trading at the soil-root interface. Plant Soil 321:5–33. https://doi.org/10.1007/s11104-009-9925-0
Jordan N, Lomenech C, Marmier N et al (2009) Sorption of selenium(IV) onto magnetite in the presence of silicic acid. J Colloid Interface Sci 329:17–23. https://doi.org/10.1016/j.jcis.2008.09.052
Jordan N, Ritter A, Foerstendorf H et al (2013) Adsorption mechanism of selenium(VI) onto maghemite. Geochim Cosmochim Acta 103:63–75. https://doi.org/10.1016/j.gca.2012.09.048
Jordan N, Ritter A, Scheinost AC et al (2014) Selenium(IV) uptake by maghemite (γ-Fe2O3). Environ Sci Technol 48:1665–1674
Josefsson I, Kunnus K, Schreck S et al (2012) Ab initio calculations of X-ray spectra: atomic multiplet and molecular orbital effects in a multiconfigurational scf approach to the L-edge spectra of transition metal complexes. J Phys Chem Lett 3:3565–3570. https://doi.org/10.1021/jz301479j
Kersten M, Vlasova N (2009) Silicate adsorption by goethite at elevated temperatures. Chem Geol 262:372–379. https://doi.org/10.1016/j.chemgeo.2009.02.002
Khare N, Martin JD, Hesterberg D (2007) Phosphate bonding configuration on ferrihydrite based on molecular orbital calculations and XANES fingerprinting. Geochim Cosmochim Acta 71:4405–4415. https://doi.org/10.1016/j.gca.2007.07.008
Kim SS, Min JH, Lee JK et al (2012) Effects of pH and anions on the sorption of selenium ions onto magnetite. J Environ Radioact 104:1–6. https://doi.org/10.1016/j.jenvrad.2011.09.013
Kim EJ, Yoo J-C, Baek K (2014) Arsenic speciation and bioaccessibility in arsenic-contaminated soils: sequential extraction and mineralogical investigation. Environ Pollut 186:29–35. https://doi.org/10.1016/j.envpol.2013.11.032
Kögel-Knabner I, Amelung W, Cao Z et al (2010) Biogeochemistry of paddy soils. Geoderma 157:1–14. https://doi.org/10.1016/j.geoderma.2010.03.009
Kubicki JDJ, Kwon KD, Paul KW, Sparks DL (2007) Surface complex structures modelled with quantum chemical calculations: carbonate, phosphate, sulphate, arsenate and arsenite. Eur J Soil Sci 58:932–944. https://doi.org/10.1111/j.1365-2389.2007.00931.x
Kubicki J, Paul K, Kabalan L, Zhu Q (2012) ATR–FTIR and density functional theory study of the structures, energetics, and vibrational spectra of phosphate adsorbed onto goethite. Langmuir 28:14573–14587
Kubicki JD, Tunega D, Kraemer S (2017) A density functional theory investigation of oxalate and Fe(II) adsorption onto the (010) goethite surface with implications for ligand- and reduction-promoted dissolution. Chem Geol 464:14–22. https://doi.org/10.1016/j.chemgeo.2016.08.010
Kubicki JD, Kabengi N, Chrysochoou M, Bompoti N (2018) Density functional theory modeling of chromate adsorption onto ferrihydrite nanoparticles. Geochem Trans 19:1–12. https://doi.org/10.1186/s12932-018-0053-8
Larsen O, Postma D (2001) Kinetics of reductive bulk dissolution of lepidocrocite, ferrihydrite, and goethite. Geochim Cosmochim Acta 65:1367–1379. https://doi.org/10.1016/S0016-7037(00)00623-2
Leermakers M, Baeyens W, De Gieter M et al (2006) Toxic arsenic compounds in environmental samples: speciation and validation. TrAC Trends Anal Chem 25:1–10. https://doi.org/10.1016/j.trac.2005.06.004
Lefevre G (2004) In situ Fourier-transform infrared spectroscopy studies of inorganic ions adsorption on metal oxides and hydroxides. Adv Colloid Interface Sci 107:109–123. https://doi.org/10.1016/j.cis.2003.11.002
Lefèvre G, Fédoroff M (2006) Sorption of sulfate ions onto hematite studied by attenuated total reflection-infrared spectroscopy: kinetics and competition with other ions. Phys Chem Earth 31:499–504. https://doi.org/10.1016/j.pce.2006.04.001
Li F, Koopal L, Tan W (2018) Roles of different types of oxalate surface complexes in dissolution process of ferrihydrite aggregates. Sci Rep 8:1–13. https://doi.org/10.1038/s41598-018-20401-5
Lindegren M, Persson P (2009) Competitive adsorption between phosphate and carboxylic acids: quantitative effects and molecular mechanisms. Eur J Soil Sci 60:982–993. https://doi.org/10.1111/j.1365-2389.2009.01171.x
Lindegren M, Loring JS, Persson P (2009) Molecular structures of citrate and tricarballylate adsorbed on α-FeOOH particles in aqueous suspensions. Langmuir 25:10639–10647. https://doi.org/10.1021/la900852p
Liu CH, Chuang YH, Chen TY et al (2015) Mechanism of arsenic adsorption on magnetite nanoparticles from water: thermodynamic and spectroscopic studies. Environ Sci Technol 49:7726–7734. https://doi.org/10.1021/acs.est.5b00381
López-Arredondo DL, Leyva-González MA, González-Morales SI et al (2014) Phosphate nutrition: improving low-phosphate tolerance in crops. Annu Rev Plant Biol 65:95–123. https://doi.org/10.1146/annurev-arplant-050213-035949
Loring JS, Sandström MH, Norén K, Persson P (2009) Rethinking arsenate coordination at the surface of goethite. Chem A Eur J 15:5063–5072. https://doi.org/10.1002/chem.200900284
Luengo C, Brigante M, Antelo J, Avena M (2006) Kinetics of phosphate adsorption on goethite: comparing batch adsorption and ATR-IR measurements. J Colloid Interface Sci 300:511–518. https://doi.org/10.1016/j.jcis.2006.04.015
Luengo CV, Castellani NJ, Ferullo RM (2015) Quantum chemical study on surface complex structures of phosphate on gibbsite. Spectrochim Acta Part A Mol Biomol Spectrosc. https://doi.org/10.1016/j.saa.2015.03.013
Luxton TP, Eick MJ, Rimstidt DJ (2008) The role of silicate in the adsorption/desorption of arsenite on goethite. Chem Geol 252:125–135. https://doi.org/10.1016/j.chemgeo.2008.01.022
Machala L, Zboril R, Gedanken A (2007) Amorphous iron(III) oxide—a review. J Phys Chem B 111:4003–4018. https://doi.org/10.1021/jp064992s
MacHala L, Tuček J, Zbořil R (2011) Polymorphous transformations of nanometric iron(III) oxide: a review. Chem Mater 23:3255–3272. https://doi.org/10.1021/cm200397g
Mackay AL (1960) Some aspects of the topochemistry of the iron oxides and hydroxides. In: Proceedings of 4th internbational symposium reactivity of solids, Amsterdam, pp 571–583
Madejová J (2003) FTIR techniques in clay mineral studies. Vib Spectrosc 31:1–10
Makris KC, Grove JH, Matocha CJ (2006) Colloid-mediated vertical phosphorus transport in a waste-amended soil. Geoderma 136:174–183. https://doi.org/10.1016/j.geoderma.2006.03.027
Manceau A, Charlet L (1994) The mechanism of selenate adsorption on goethite and hydrous ferric oxide. J Colloid Interface Sci 168:87–93. https://doi.org/10.1006/jcis.1994.1396
Manning BA, Fendorf SE, Goldberg S, Goldberg S (1998) Surface structures and stability of arsenic (III) on goethite : spectroscopic evidence for inner-sphere complexes. Environ Sci Technol 32:2383–2388. https://doi.org/10.1021/es9802201
Mansfeldt T (2004) Redox potential of bulk soil and soil solution concentration of nitrate, manganese, iron, and sulfate in two Gleysols. J Plant Nutr Soil Sci 167:7–16. https://doi.org/10.1002/jpln.200321204
Mansour C, Lefèvre G, Pavageau EM et al (2009) Sorption of sulfate ions onto magnetite. J Colloid Interface Sci 331:77–82. https://doi.org/10.1016/j.jcis.2008.11.009
Martínez M, Giménez J, De Pablo J et al (2006) Sorption of selenium(IV) and selenium(VI) onto magnetite. Appl Surf Sci 252:3767–3773. https://doi.org/10.1016/j.apsusc.2005.05.067
Masion A, Rose J, Bottero JY et al (1997) Nucleation and growth mechanisms of iron oxyhydroxides in the presence of PO4 ions. 4. Structure of the aggregates. Langmuir 13:3886–3889. https://doi.org/10.1021/la970041h
Masscheleyn PH, Delaune RD, Patrick WH (1991) Effect of redox potential and pH on arsenic speciation and solubility in a contaminated soil. Environ Sci Technol 25:1414–1419. https://doi.org/10.1021/es00020a008
Michael Bolanz R, Bläss U, Ackermann S et al (2013) The effect of antimonate, arsenate, and phosphate on the transformation of ferrihydrite to goethite, hematite, feroxyhyte, and tripuhyite. Clays Clay Miner 61:11–25. https://doi.org/10.1346/CCMN.2013.0610102
Mir KA, Rutter A, Koch I et al (2007) Extraction and speciation of arsenic in plants grown on arsenic contaminated soils. Talanta 72:1507–1518. https://doi.org/10.1016/j.talanta.2007.01.068
Mora C, Tittensor DP, Adl S et al (2011) How many species are there on earth and in the ocean? PLoS Biol 9:1–8. https://doi.org/10.1371/journal.pbio.1001127
Morin G, Ona-Nguema G, Wang Y et al (2008) Extended X-ray absorption fine structure analysis of arsenite and arsenate adsorption on maghemite. Environ Sci Technol 42:2361–2366. https://doi.org/10.1021/es072057s
Mudunkotuwa IA, Al Minshid A, Grassian VH (2014) ATR-FTIR spectroscopy as a tool to probe surface adsorption on nanoparticles at the liquid-solid interface in environmentally and biologically relevant media. Analyst 139:870–881. https://doi.org/10.1039/c3an01684f
Neupane G, Donahoe RJ, Arai Y (2014) Kinetics of competitive adsorption/desorption of arsenate and phosphate at the ferrihydrite-water interface. Chem Geol 368:31–38. https://doi.org/10.1016/j.chemgeo.2013.12.020
Newville M (2014) Fundamentals of XAFS. Rev Miner Geochem 78:33–74. https://doi.org/10.2138/rmg.2014.78.2
Nilsson N, Persson P, Lövgren L, Sjöberg S (1996) Competitive surface complexation of o-phthalate and phosphate on goethite (α-FeOOH) particles. Geochim Cosmochim Acta 60:4385–4395. https://doi.org/10.1016/S0016-7037(96)00258-X
Norén K, Persson P (2007) Adsorption of monocarboxylates at the water/goethite interface: the importance of hydrogen bonding. Geochim Cosmochim Acta 71:5717–5730. https://doi.org/10.1016/j.gca.2007.04.037
Ona-Nguema G, Morin G, Juillot F et al (2005) EXAFS analysis of arsenite adsorption onto two-line ferrihydrite, hematite, goethite, and lepidocrocite. Environ Sci Technol 39:9147–9155. https://doi.org/10.1021/es050889p
Panias D, Taxiarchou M, Paspaliaris I, Kontopoulos A (1996) Mechanisms of dissolution of iron oxides in aqueous oxalic acid solutions. Hydrometallurgy 42:257–265. https://doi.org/10.1016/0304-386X(95)00104-O
Pantsar-Kallio M, Manninen PKG (1997) Speciation of mobile arsenic in soil samples as a function of pH. Sci Total Environ 204:193–200
Peak D, Sparks DL (2002) Mechanisms of selenate adsorption on iron oxides and hydroxides. Environ Sci Technol 36:1460–1466. https://doi.org/10.1021/es0156643
Persson P, Axe K (2005) Adsorption of oxalate and malonate at the water-goethite interface: molecular surface speciation from IR spectroscopy. Geochim Cosmochim Acta 69:541–552. https://doi.org/10.1016/j.gca.2004.07.009
Persson P, Nilsson N, Sjöberg S (1996) Structure and bonding of orthophosphate ions at the iron oxide-aqueous interface. J Colloid Interface Sci 177:263–275. https://doi.org/10.1006/jcis.1996.0030
Petrovsk E, Krop V, Dekkers MJ, Ambafiello A (1996) Transformation of Hematite to maghemite as observed by changed in magentic prameters: effects of mechanical activation? Geophys Res Lett 23:1477–1480
Phenrat T, Schoenfelder D, Kirschling TL et al (2018) Adsorbed poly(aspartate) coating limits the adverse effects of dissolved groundwater solutes on Fe0 nanoparticle reactivity with trichloroethylene. Environ Sci Pollut Res 25:7157–7169. https://doi.org/10.1007/s11356-015-5092-4
Rahnemaie R, Hiemstra T, van Riemsdijk WH (2006) Inner- and outer-sphere complexation of ions at the goethite-solution interface. J Colloid Interface Sci 297:379–388. https://doi.org/10.1016/j.jcis.2005.11.003
Rahnemaie R, Hiemstra T, van Riemsdijk WH (2007) Carbonate adsorption on goethite in competition with phosphate. J Colloid Interface Sci 315:415–425. https://doi.org/10.1016/j.jcis.2007.07.017
Randall SR, Sheerman DM, Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4 3H2O) and lepidocrocite? (–FeOOH): surface complexes from EXAFS spectroscopy. Geochim Cosmochim Acta 65:1015–1023
Ray JR, Wan W, Gilbert B, Jun YS (2013) Effects of formation conditions on the physicochemical properties, aggregation, and phase transformation of iron oxide nanoparticles. Langmuir 29:1069–1076. https://doi.org/10.1021/la3034319
Reza AHMS, Jean JS, Lee MK et al (2010) Implications of organic matter on arsenic mobilization into groundwater: evidence from northwestern (Chapai-Nawabganj), central (Manikganj) and southeastern (Chandpur) Bangladesh. Water Res 44:5556–5574. https://doi.org/10.1016/j.watres.2010.09.004
Riley W, Ortiz-Monasterio I, Matson P (2001) Nitrogen leaching and soil nitrate, nitrite, and ammonium levels under irrigated wheat in Northern Mexico. Nutr Cycl Agroecosyst 61:223–236
Roonasi P, Holmgren A (2010) An ATR-FTIR study of carbonate sorption onto magnetite. Surf Interface Anal 42:1118–1121. https://doi.org/10.1002/sia.3382
Rose J, Flank AM, Masion A et al (1997) Nucleation and growth mechanisms of Fe oxyhydroxide in the presence of PO4 ions. 2. P K-edge EXAFS study. Langmuir 13:1827–1834. https://doi.org/10.1021/la961039d
Rosen BP, Liu Z (2009) Transport pathways for arsenic and selenium: a minireview. Environ Int 35:512–515. https://doi.org/10.1016/j.envint.2008.07.023
Schwertmann U (1985) The effect of pedogenic environments on iron oxide minerals. In: Stewart BA (ed) Advances in soil science. Springer, New York, pp 171–200
Schwertmann U (1993) Relations between iron oxides, soil color, and soil formation. Soil Color. https://doi.org/10.2136/sssaspecpub31.c4
Schwertmann U, Taylor RM (1972) The transformation of lepidocrocite to goethite. Clays Clay Miner 20:151–158. https://doi.org/10.1346/CCMN.1972.0200307
Schwertmann U, Cornell RM (2008) Iron oxides in the laboratory: preparation and characterization, 2nd edn. Wiley, Germany
Scott RA, Yamashita S, Taniguchi K et al (1992) X-ray absorption spectroscopy. X-Ray Spectrom 21:91–97. https://doi.org/10.1002/xrs.1300210209
Sharif MSU, Davis RK, Steele KF et al (2011) Surface complexation modeling for predicting solid phase arsenic concentrations in the sediments of the Mississippi River Valley alluvial aquifer, Arkansas, USA. Appl Geochem 26:496–504. https://doi.org/10.1016/j.apgeochem.2011.01.008
Sharma VK, McDonald TJ, Sohn M et al (2014) Biogeochemistry of selenium. A review. Environ Chem Lett 13:49–58. https://doi.org/10.1007/s10311-014-0487-x
Shaw S, Pepper SE, Bryan ND, Livens FR (2005) The kinetics and mechanisms of goethite and hematite crystallization under alkaline conditions, and in the presence of phosphate. Am Miner 90:1852–1860. https://doi.org/10.2138/am.2005.1757
Sherman DM, Randall SR (2003) Surface complexation of arsenic(V) to iron(III) (hydr)oxides: structural mechanism from ab initio molecular geometries and EXAFS spectroscopy. Geochim Cosmochim Acta 67:4223–4230. https://doi.org/10.1016/S0016-7037(03)00237-0
Shi Z, Li F, Yao S (2010) Effect of small organic acid anions on the adsorption of phosphate anions onto synthetic goethite from aqueous solution. Adsorpt Sci Technol 28:885–893. https://doi.org/10.1260/0263-6174.28.10.885
Shi Z, Krom MD, Bonneville S et al (2011) Influence of chemical weathering and aging of iron oxides on the potential iron solubility of Saharan dust during simulated atmospheric processing. Global Biogeochem Cycles. https://doi.org/10.1029/2010GB003837
Singh R, Singh S, Parihar P et al (2015) Arsenic contamination, consequences and remediation techniques: a review. Ecotoxicol Environ Saf 112:247–270. https://doi.org/10.1016/j.ecoenv.2014.10.009
Smedley PL, Kinniburgh DG (2002) A review of the source, behaviour and distribution of arsenic in natural waters. Appl Geochem 17:517–568. https://doi.org/10.1016/S0883-2927(02)00018-5
Smith VH, Schindler DW (2009) Eutrophication science: where do we go from here? Trends Ecol Evol 24:201–207. https://doi.org/10.1016/j.tree.2008.11.009
Smith VH, Joye SB, Howarth RW (2006) Eutrophication of freshwater and marine ecosystems. Limnol Oceanogr 51:351–355. https://doi.org/10.4319/lo.2006.51.1_part_2.0351
Sparks DL (2000) New frontiers in elucidating the kinetics and mechanisms of metal and oxyanion sorption at the soil mineral/water interface. J Plant Nutr Soil Sci 163:563–570. https://doi.org/10.1002/1522-2624(200012)163:6%3c563:aid-jpln563%3e3.0.co;2-0
Sparks DL (2003) Environmental soil chemistry. Academic Press, Cambridge
Species P, Hartley-whitaker J (2015) review Tansley Arsenic and metabolism in uptake and nonresistant resistant arsenic plant species. New Phytol 154:29–43
Srivastava DN, Perkas N, Gedanken A, Felner I (2002) Sonochemical synthesis of mesoporous iron oxide and accounts of its magnetic and catalytic properties. J Phys Chem B 106:1878–1883. https://doi.org/10.1021/jp015532w
Stumm W, Morgan JJ (1981) Aquatic chemistry : an introduction emphasizing chemical equilibria in natural waters. Wiley, New York
Su C, Suarez DL (2000a) Selenate and selenite sorption on iron oxides. Soil Sci Soc Am J 64:101. https://doi.org/10.2136/sssaj2000.641101x
Su C, Suarez DL (2000b) Selenate and selenite sorption on iron oxides: an infrared and electrophoretic study. Soil Sci Soc Am J 64:101–111
Šutka A, Lagzdina S, Käämbre T et al (2015) Study of the structural phase transformation of iron oxide nanoparticles from an Fe2+ ion source by precipitation under various synthesis parameters and temperatures. Mater Chem Phys 149:473–479. https://doi.org/10.1016/j.matchemphys.2014.10.048
Swaddle TW, Oltmann P (1980) Kinetics of the magnetite-maghemite-hematite transformation, with special reference to hydrothermal. Can J Chem 58:1763–1772
Swedlund PJ, Miskelly GM, McQuillan AJ (2009) An attenuated total reflectance IR study of silicic acid adsorbed onto a ferric oxyhydroxide surface. Geochim Cosmochim Acta 73:4199–4214. https://doi.org/10.1016/j.gca.2009.04.007
Szilas CP, Borggaard OK, Hansen HCB (1998) Potential iron and phosphate mobilization during flooding of soil material re-establishment of wetlands on formerly reclaimed agricultural areas is an ongo- ing activity in Denmark (Møller, 1995). The aims of these projects comprise improvement of biol. Water Air Soil Pollut 106:97–109
Takahashil T, Park C-Y, Nakajima H et al (1999) Ferric iron transformation in soils with rotation of irrigated rice-upland crops and effect on soil tillage properties. Soil Sci Plant Nutr 45:163–173. https://doi.org/10.1080/00380768.1999.10409332
Tejedor-Tejedor M, Anderson M (1990) The protonation of phosphate on the surface of goethite as studied by CIR-FTIR and electrophoretic mobility. Langmuir 6:602–611. https://doi.org/10.1021/la00093a015
Templeton DM, Fujishiro H (2017) Terminology of elemental speciation—an IUPAC perspective. Coord Chem Rev 352:424–431. https://doi.org/10.1016/j.ccr.2017.02.002
Timmons DR, Dylla AS (1981) Nitrogen leaching as influenced by nitrogen management and supplemental irrigation level. J Environ Qual 10:421–426. https://doi.org/10.2134/jeq1981.00472425001000030036x
Tu S, Ma L, Luongo T (2004) Root exudates and arsenic accumulation in arsenic hyperaccumulating Pteris vittata and non-hyperaccumulating Nephrolepis exaltata. Plant Soil 258:9–19
Ure AM, Berrow ML (1982) The elemental constituents of soils. In: Bowen HJM (ed) Environmental chemistry, vol 2. The Royal Society of Chemistry, Cambridge, pp 94–204
Usman M, Abdelmoula M, Faure P et al (2013) Transformation of various kinds of goethite into magnetite: effect of chemical and surface properties. Geoderma 197–198:9–16. https://doi.org/10.1016/j.geoderma.2012.12.015
Usman M, Byrne JM, Chaudhary A et al (2018) Magnetite and green rust: synthesis, properties, and environmental applications of mixed-valent iron minerals. Chem Rev 118:3251–3304. https://doi.org/10.1021/acs.chemrev.7b00224
Vahidnia A, Van Der Voet GB, De Wolff FA (2007) Arsenic neurotoxicity—a review. Hum Exp Toxicol 26:823–832. https://doi.org/10.1177/0960327107084539
Vamerali T, Bandiera M, Mosca G (2010) Field crops for phytoremediation of metal-contaminated land. A review. Environ Chem Lett 8:1–17. https://doi.org/10.1007/s10311-009-0268-0
Van Riemsdijk WH, Weng L, Hiemstra T (2007) Ion–colloid–colloid interactions. In: Frimmel FH, Von Der Kammer F, Flemming HC (eds) Colloidal transport in porous media. Springer, Berlin, pp 205–249
Vodyanitskii YN (2010) Iron hydroxides in soils: a review of publications. Eurasian Soil Sci 43:1244–1254. https://doi.org/10.1134/S1064229310110074
Wang Y, He Y, Zhang H et al (2008a) Phosphate mobilization by citric, tartaric, and oxalic acids in a clay loam ultisol. Soil Sci Soc Am J 72:1263
Wang Y, Morin G, Ona-Nguema G et al (2008b) Arsenite sorption at the magnetite-water interface during aqueous precipitation of magnetite: EXAFS evidence for a new arsenite surface complex. Geochim Cosmochim Acta 72:2573–2586. https://doi.org/10.1016/j.gca.2008.03.011
Wang X, Kubicki JD, Boily JF et al (2018) Binding geometries of silicate species on ferrihydrite surfaces. ACS Earth Space Chem 2:125–134. https://doi.org/10.1021/acsearthspacechem.7b00109
Warrinnier R, Goossens T, Amery F et al (2019) Investigation on the control of phosphate leaching by sorption and colloidal transport: column studies and multi-surface complexation modelling. Appl Geochem 100:371–379. https://doi.org/10.1016/j.apgeochem.2018.12.012
Waychunas GA, Rea BA, Fuller CC, Davis JA (1993) Surface chemistry of ferrihydrite: part 1. EXAFS studies ongeometrie of coprecipitated and adsorbed arsenate. Geochim Cosmochim Acta 57:2251–2269. https://doi.org/10.1016/0016-7037(93)90567-G
Welch AH, Stollenwerk KG (2003) Arsenic in ground water. Kluwer Academic Publishers
Weng L, Van Riemsdijk WH, Hiemstra T (2012) Factors controlling phosphate interaction with iron oxides. J Environ Qual 41:628–635. https://doi.org/10.2134/jeq2011.0250
Wijnja H, Schulthess CPC (2000) Vibrational spectroscopy study of selenate and sulfate adsorption mechanisms on Fe and Al (hydr)oxide surfaces. J Colloid Interface Sci 229:286–297. https://doi.org/10.1006/jcis.2000.6960
Wijnja H, Schulthess CP (2001) Carbonate adsorption mechanism on goethite studied with ATR – FTIR, DRIFT. and proton coadsorption measurements. Soil Sci Soc Am J 65:324–330
Wijnja H, Schulthess CP (2002) Effect of carbonate on the adsorption of selenate and sulfate on goethite. Soil Sci Soc Am J 66:1190–1197. https://doi.org/10.2136/sssaj2002.1190
Williams AGB, Scherer MM (2004) Spectroscopic evidence for Fe(II)-Fe(III) electron transfer at the iron oxide-water interface. Environ Sci Technol 38:4782–4790. https://doi.org/10.1021/es049373g
Xu N, Christodoulatos C, Braida W (2006) Modeling the competitive effect of phosphate, sulfate, silicate, and tungstate anions on the adsorption of molybdate onto goethite. Chemosphere 64:1325–1333. https://doi.org/10.1016/j.chemosphere.2005.12.043
Yang X, Roonasi P, Holmgren A (2008) A study of sodium silicate in aqueous solution and sorbed by synthetic magnetite using in situ ATR-FTIR spectroscopy. J Colloid Interface Sci 328:41–47. https://doi.org/10.1016/j.jcis.2008.08.061
Yang X, Roonasi P, Jolsterå R, Holmgren A (2009) Kinetics of silicate sorption on magnetite and maghemite: an in situ ATR-FTIR study. Colloids Surf A Physicochem Eng Asp 343:24–29. https://doi.org/10.1016/j.colsurfa.2009.01.041
Yang Y, Wang S, Xu Y et al (2016) Molecular-scale study of aspartate adsorption on goethite and competition with phosphate. Environ Sci Technol. https://doi.org/10.1021/acs.est.5b05450
Young RA, Onstad CA, Bosch DD, WPA (1989) A nonpoint-source pollution model for evaluating agricultural watersheds. J Soil Water Conserv 44:168–173
Yuan L, Cai R, Jang JI et al (2013) Morphological transformation of hematite nanostructures during oxidation of iron. Nanoscale 5:7581–7588. https://doi.org/10.1039/c3nr01669b
Yusuf SM, Mukadam MD, De Teresa JM et al (2010) Structural and magnetic properties of amorphous iron oxide. Phys B Condens Matter 405:1202–1206. https://doi.org/10.1016/j.physb.2009.11.040
Zamparas M, Zacharias I (2014) Restoration of eutrophic freshwater by managing internal nutrient loads. A review. Sci Total Environ 496:551–562. https://doi.org/10.1016/j.scitotenv.2014.07.076
Zhang H, Selim HM (2007) Colloid mobilization and arsenite transport in soil columns: effect of ionic strength. J Environ Qual 36:1273–1280. https://doi.org/10.2134/jeq2006.0373
Zhang C, Ge Y, Yao H et al (2012) Iron oxidation-reduction and its impacts on cadmium bioavailability in paddy soils: a review. Front Environ Sci Eng China 6:509–517. https://doi.org/10.1007/s11783-012-0394-y
Zhao LX, Hou WG (2012) The effect of sorbent concentration on the partition coefficient of pollutants between aqueous and particulate phases. Colloids Surf A Physicochem Eng Asp 396:29–34. https://doi.org/10.1016/j.colsurfa.2011.12.026
Zhu J, Pigna M, Cozzolino V et al (2011) Sorption of arsenite and arsenate on ferrihydrite: effect of organic and inorganic ligands. J Hazard Mater 189:564–571. https://doi.org/10.1016/j.jhazmat.2011.02.071
Zhu J, Pigna M, Cozzolino V et al (2013a) Higher sorption of arsenate versus arsenite on amorphous Al-oxide, effect of ligands. Environ Chem Lett 11:289–294. https://doi.org/10.1007/s10311-013-0405-7
Zhu M, Northrup P, Shi C et al (2013b) Structure of sulfate adsorption complexes on ferrihydrite. Environ Sci Technol Lett 1:97–101. https://doi.org/10.1021/ez400052r
Zhu XF, Wang ZW, Wan JX et al (2015) Pectin enhances rice (Oryza sativa) root phosphorus remobilization. J Exp Bot 66:1017–1024. https://doi.org/10.1093/jxb/eru461
Zhu J, Fu Q, Qiu G et al (2019) Influence of low molecular weight anionic ligands on the sorption of heavy metals by soil constituents: a review. Environ Chem Lett 17:1271–1280. https://doi.org/10.1007/s10311-019-00881-1
Acknowledgements
This research was supported not only by the Young Researcher Program (NRF-2019R1C1C1007535) and Global Ph.D. Fellowship Program (NRF-2015H1A2A1034068) through the National Research Foundation of Korea (NRF) Grant funded by the Korean government (MSIP and MOE) but also by the Brain Korea 21 Plus Program funded by the Korean government (MOE).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, J., Kim, M. & Ro, HM. Factors modifying the structural configuration of oxyanions and organic acids adsorbed on iron (hydr)oxides in soils. A review. Environ Chem Lett 18, 631–662 (2020). https://doi.org/10.1007/s10311-020-00964-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10311-020-00964-4