
Abstract
Background
Activating mutation of KIT or PDGFRA is the primary molecular mechanism for gastrointestinal stromal tumors (GISTs). Although imatinib has a revolutionary effect on GIST therapeutics, the benefits are not durable. Increasing reports have demonstrated that cell cycle checkpoint plays critical roles in GIST. Here, we explore the role of WEE1 kinase in GIST progression.
Methods
Oncomine public database, western blotting, and immunohistochemistry were used to analyze WEE1 expression in GISTs. Using MTT assays, colony formation analysis, and flow cytometry, we examined the role of WEE1 in GIST cells and the antitumor activity of the inhibitor MK1775 alone, or in combination with imatinib. Cycloheximide chase assay and pharmacological inhibition of autophagy and proteasome pathway were performed to analyze KIT expression. Additionally, autophagic markers Beclin1 and LC3B were detected by western blotting.
Results
Upregulated WEE1 expression was observed in GIST tissues and correlated with tumor size, mitotic count, and risk grade. Inhibition of WEE1 significantly suppressed GIST cell proliferation, induced apoptosis and cell cycle arrest. Imatinib and MK1775 co-treatment markedly enhanced the antitumor activity. Targeting WEE1 decreased the expression of KIT expression. Moreover, WEE1 stabilized KIT protein and KIT reduction observed upon WEE1 inhibition could be reversed by pharmacological inhibition of autophagy, but not proteasome pathway. WEE1 inhibition also increased Beclin1 expression and LC3B II/I ratio in GIST cells.
Conclusions
Our data suggest that WEE1 plays a pivotal role in GIST proliferation. WEE1 inhibition could promote KIT autophagic degradation and, therefore, targeting WEE1 might represent a novel strategy for GIST therapies.
Similar content being viewed by others


Introduction
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumors of the gastrointestinal tract and originates from interstitial cells of Cajal (ICCs) [1, 2]. Activating mutations of the receptor tyrosine kinase KIT (75–80% of GISTs) or platelet-derived growth factor receptor alpha (PDGFRA) (5–8% of GISTs) are often the initial drivers for GIST progression. However, approximately 10% of GISTs lack KIT or PDGFRA mutations [3]. Activated KIT and PDGFRA in turn activate various downstream signaling pathways to regulate GIST cell proliferation [4].
Imatinib, a specific tyrosine kinase inhibitor (TKI), was approved as a first-line treatment for unresectable or metastatic GISTs, and a number of clinical studies demonstrated that imatinib significantly increased overall survival and progression-free survival in patients with GIST [5, 6]. Despite the clinical success, 50% of patients develop resistance to imatinib within 2 years of treatment [7]. Secondary KIT or PDGFRA mutations and the activation of alternative oncogenic signaling pathways have been identified in the majority of resistant GISTs [8,9,10]. To overcome this treatment bottleneck, multi-targeting TKIs sunitinib and regorafenib have been approved as second- and third-line treatments for primary or secondary resistant GISTs. However, the effect of sunitinib and regorafenib are limited [11,12,13]. Thus, it is crucial to better understand the molecular mechanisms involved in GIST malignancy and develop novel therapeutic strategies.
Recently, a number of studies have demonstrated that dysregulated cell cycle-related proteins are involved in tumor progression [14]. Using cDNA microarray analysis of 30 human GIST tissues, Hur et al. found that six cell cycle-related pathways were upregulated in GIST tissues from high-risk patients [15]. G2–M checkpoint regulators, including cyclin B1 and CDK1, were reported to predict the biological behavior of GISTs. Moreover, decreased levels of checkpoint with forkhead and ring finger (CHFR) may contribute to the proliferative activity of higher grade GISTs [16]. Notably, the tyrosine kinase WEE1 is a crucial component of the G2–M cell cycle checkpoint. WEE1 selectively phosphorylates the Tyr15 residue of CDK1 and prevents complex formation with cyclin B, which inhibits cell entry into mitosis at the G2–M transition [17]. Previous studies showed the WEE1 expression is upregulated in various cancers, such as gastric cancer, melanoma and breast cancer [18,19,20]. Inhibition of WEE1 has been shown to impair the G2–M checkpoint, which causes cancer cells with DNA damage to enter successive replication cycles, leading to cell death from mitotic catastrophe [21]. Notably, MK1775, a selective small molecule WEE1 inhibitor, showed potent antitumor effects when treated alone or in combination with conventional chemotherapy or radiotherapy in p53-deficient cancer cells [22, 23]. Thus, targeting of WEE1 appears to be a potential therapeutic strategy although the potential role of WEE1 in GIST remains largely unknown.
Here, we investigated the expression of WEE1 in GIST tissues and the role of the kinase in GIST progression, and reveal that WEE1 inhibition may serve as a novel target for GIST therapy.
Methods
Cell lines, antibodies, and reagents
GIST-T1 and GIST882 cells were kindly provided by Dr. Jonathan Fletcher (Dana-Farber Cancer Institute, Boston, MA). Cells were cultured in RPMI-1640 medium (Gibco, USA) supplemented with 15% fetal bovine serum (FBS; ScienCell, USA), and 1% l-glutamine (#25030081, Gibco, USA) in a humidified atmosphere with 5% CO2 at 37 °C.
Antibodies to WEE1 (13084), CDK1 (77055), phospho-CDK1T14 (4539), KIT (3074), phospho-KITT703 (3073), AKT (4685), phospho-AKTS473 (4060), mTOR (2893), phospho-mTORS2448 (5536), ERK1/2 (4695), phospho-ERK1/2 (4370), and the protein synthesis inhibitor cycloheximide (2112) were purchased from Cell Signaling Technology (MA, USA). Anti-cleaved PARP antibody (ab32064) was purchased from Abcam (MA, USA). The cyclin B1 antibody (A5016) and LC3B antibody (A5202) were obtained from Bimake (TX, USA). γH2AX antibody (2716681) was purchased from Millipore (MA, USA). Anti-GAPDH (60004–1-Ig), anti-Beclin1 (11306–1-AP), and HRP-conjugated secondary antibodies (SA00001-1 and SA00001-2) were obtained from Proteintech (Wuhan, China). CY3-conjugated anti-mouse IgG (GB21301) and DAPI (G1012) were purchased from Servicebio (Wuhan, China). Imatinib mesylate (S1026), MG132 (S2619), and MK1775 (S1525) were purchased from SELLECK (TX, USA). Chloroquine (C6628) was obtained from Sigma (MO, USA).
Immunohistochemistry
GIST specimens were obtained from the Department of Gastrointestinal Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology after obtaining informed consent from each patient. This study was approved by the Huazhong University of Science and Technology Ethics Committee. According to the manufacturer’s instruction from the immunohistochemistry kit (SA1020, Boster, Wuhan, China), immunohistochemistry was performed using anti-WEE1 (1:100). For statistical analysis, the staining intensity assessment and staining area calculations were used. The following staining intensity scores were used: negative (0), weak (1), moderate (2), and strong (3). The staining area scores were as follows: none (0), 1–25% (1), 26–50% (2), 51–75% (3), and 76–100% (4). Next, the staining intensity score and staining area score were multiplied to generate the final score.
RNA interference
Specific WEE1 siRNAs were synthetized by RiboBio (Guangzhou, China). The sequences were as follows: siRNA#1, 5′-AAUAUGAAGUCCCGGUAUA-3′; siRNA#2, 5′-GGUAGCAGCUGAUUUCGUU-3′ and non-targeting siRNA, 5′-UUCUCCGAACGUGUCACGU-3′. According to the manufacturer’s instructions for Lipofectamine 3000 (Thermo, USA), siRNAs were transfected into cells. After culturing for 48 h, the knockdown efficiency of the siRNAs was determined by western blotting.
Cell viability assay
The methylthiazolyldiphenyl-tetrazolium bromide assay kit (MTT; C0009, Beyotime, Shanghai, China) was used to determine cell viability. GIST-T1 and GIST-882 cells (5 × 103 cells/well) were transfected with WEE1 siRNA, seeded in a 96-well plate and treated with 10 μL of 5 mg/mL MTT solution. After incubation with MTT for 4 h, dimethylsulfoxide (DMSO; ST038, Beyotime, Shanghai, China) was used to dissolve the crystal and the absorbance value was measured at 490 nm. The half inhibitory concentration (IC50) of MK1775 and combination index (CI) with imatinib and MK1775 were assessed after the 48-h treatment. The CI was calculated using CompuSyn software, based on the Chou–Talalay equation CI < 1 indicates synergistic effects, CI = 1 indicates additive effects, and CI > 1 indicates antagonistic effects.
Colony formation assay
GIST-T1 and GIST-882 cells (5 × 102 cells/well) were transfected with WEE1 siRNAs, seeded in a six-well plate and cultured for 14 days. For MK1775 treatment in colony formation assay, cells were treated with MK1775 for 48 h, the medium was removed, and the cells were then supplied with fresh medium and cultured for 14 days. To examine colony formation, cells were fixed with 4% paraformaldehyde for 20 min. Next, crystal violet (0.5%) was used to stain the cells for 20 min. Colonies were photographed and then counted using Image J software.
Western blotting
The total protein was extracted with RIPA buffer (V900854, Sigma, MO, USA) supplemented with protease inhibitor (B14001, Bimake, TX, USA) and phosphatase inhibitor (G2007, Servicebio, Wuhan, China). Total proteins (20 μg) were separated via SDS-PAGE and transferred to PVDF membranes. Membranes were then blocked in 5% skim milk for 1 h and incubated with primary antibodies overnight at 4 °C. The blots were then washed with TBST and incubated with secondary antibodies (1:3000). Bands were detected after incubation with ECL solution and imaged using a Bio-Rad Imaging system detector. GAPDH was used as a control.
Apoptosis and cell cycle analysis
Cells (5 × 105 cells/well) were harvested by trypsinization and washed twice with PBS, according to the manufacturer’s instructions (640930, BioLegend, CA, USA). The cells then were suspended in 100 µL Annexin V binding buffer, followed by 5 µL APC–Annexin V and 5 µL 7-AAD, the cells were next gently vortexed and incubated for 15 min at room temperature. Finally, 400 µL of Annexin V binding buffer was added and the cells were analyzed by flow cytometry.
For cell cycle analysis, harvested cells were fixed in 75% ethanol overnight at 4 °C. Cells were then washed twice with cold PBS buffer, resuspended in propidium iodide/RNaseA-mixed staining solution (KGA511, KeyGEN, Nanjing, China) for 30 min and analyzed by flow cytometry. The results were analyzed by ModFit 5.0 software.
RNA extraction and quantitative PCR (qPCR)
Total RNA was extracted by RNAiso Plus (9109, TaKaRa, Dalian, China) and reverse transcribed into cDNA using the PrimeScript RT Master Mix (RR036A, TaKaRa) according to the manufacturer’s instructions. Gene expression level was determined using SYBR® Premix Ex TaqTM (RR820A, TaKaRa) on a StepOnePlus™ Real-Time PCR System and calculated using the 2−ΔΔCt method. Data were normalized by GAPDH. Experiments were carried out in triplicate. The primer sequences were as follows: GAPDH, former primer: 5′-ACAACTTTGGTATCGTGGAAGG-3′, reverse primer: 5′-GCCATCACGCCACAGTTTC-3′; KIT, former primer: 5′-ACTTGAGGTTTATTCCTGACCCC-3′, reverse primer: 5′-GCAGACAGAGCCGATGGTAG-3′.
Immunofluorescence staining
Cells were treated with imatinib, MK1775, or combination of both and fixed with 4% formaldehyde for 20 min, permeabilized with 0.2% Triton X-100 for 20 min, blocked with 2% BSA for 60 min, and incubated with anti-γH2AX (1:500) overnight at 4 °C successively. The slides were then washed three times by PBST, and incubated with CY3-conjugated secondary antibody (1:500) for 60 min in the dark and DAPI (1:100,000) for 5 min. After mounting with anti-fade mounting medium, cells were examined using a microscope and imaged.
Cycloheximide (CHX) chase assay
GIST-T1 and GIST-882 cells were treated with WEE1 siRNA or 1 μM MK1775 and collected after the addition of 50 μg/mL CHX at the indicated time points (0, 2, 4, and 6 h). The relative protein expression of the cell lysates was determined by western blotting and quantified by Image J software. The KIT degradation rate at each time point is presented as the percentage of KIT/GAPDH expression.
Statistical analysis
Statistical analysis was performed using SPSS 20.0 software. A two-tailed t test was used to analyze statistical differences between the two groups and one-way ANOVA was utilized among multiple groups. Data are presented as the mean ± S.D. P < 0.05 was considered to indicate statistically significant differences between groups.
Results
WEE1 is highly expressed in GIST tissues
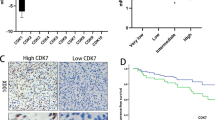
To study the expression of WEE1 in GISTs, we first analyzed the data from the Oncomine database (https://www.oncomine.org/). We found that WEE1 levels were upregulated in 6 GISTs compared to those in 19 normal tissues (Supplementary Fig. 1). To validate this finding, we further assessed WEE1 expression in our clinical GIST specimens, including 41 GIST tissues and 16 adjacent non-tumor tissues. Western blotting analysis demonstrated that WEE1 protein expression was elevated in GIST tissues compared to adjacent non-tumor tissues (Fig. 1a, b). Immunohistochemistry analysis showed that WEE1 mainly localized in the nucleus (Fig. 1c). According to the IHC scores, negative staining (score 0–5) and positive staining (score 6–12) were distinguished. As shown in Fig. 1d, 75% (12/16) of specimens showed negative staining and 25% (4/16) showed positive staining in adjacent non-tumor tissues. In contrast, for GIST tissues, 43.9% (18/41) and 56.1% (23/41) of samples showed negative staining and positive staining, respectively.

WEE1 is overexpressed in GISTs. a WEE1 protein expression in adjacent non-tumor and GIST tissues was examined by western blotting. b WEE1 protein expression was quantified by Image J software (t test). c Immunohistochemical staining showed the expression of WEE1 in adjacent non-tumor and GIST tissues. d Frequency of WEE1-negative staining and WEE1-positive staining in adjacent non-tumor and GIST tissues. **P < 0.01. N adjacent non-tumor tissue, T GIST tissue
Next, we investigated the relationship between WEE1 expression and the clinicopathological features of patients with GIST. As shown in Table 1, WEE1-positive staining was significantly correlated with tumor size (P = 0.031), mitotic count (P = 0.026), and risk grade (P = 0.003), but not with gender, age or tumor site. Thus, these results suggest that WEE1 kinase may be involved in GIST progression.
WEE1 ablation inhibits proliferation of GIST cells
To investigate the function of WEE1 in GIST progression, we used two independent siRNAs to silence WEE1 expression in GIST-T1 and GIST-882 cells. As shown in Fig. 2a, WEE1 protein expression was efficiently inhibited in siRNA-treated groups, followed by inhibition of phosphorylated CDK1 (p-CDK1). The MTT assay revealed that WEE1 knockdown inhibited the proliferative ability of GIST-T1 and GIST-882 cells (Fig. 2b). We further found that silencing WEE1 markedly decreased the number and size of the cell colonies (Fig. 2c).

Silencing of WEE1 inhibits GIST cell proliferation. a Western blotting was used to examine the expression of WEE1 and downstream signaling proteins after transfection with small interfering RNAs for 48 h. b, c The viability of GIST-T1 and GIST-882 cells transfected with a negative control or WEE1 siRNAs was determined by the MTT and colony formation assays (t test). d Expression of apoptosis-related proteins were evaluated by western blotting after transfection with small interfering RNAs for 48 h. e The cell cycle distribution was assessed by flow cytometry (t test). *P < 0.05, **P < 0.01. NC negative control
Upon WEE1 ablation, we observed a slight change in cellular morphology (data not shown). We examined the expression of apoptosis-related proteins and found that the expression of cleaved PARP was increased compared to that of the negative control group (Fig. 2d). Therefore, WEE1 ablation was shown to induce GIST cell apoptosis. To further clarify the potential cellular mechanism involved, we analyzed the cell cycle distribution. Flow cytometry demonstrated that the percentage of G0/G1 phase cells was significantly reduced in WEE1-knockdown groups and was accompanied by an increased number of S phase cells (Fig. 2e). Taken together, our results suggest that silencing WEE1 appears to inhibit GIST cells proliferation by inducing cell apoptosis and cell cycle arrest.
MK1775, a specific WEE1 inhibitor, can inhibit GIST cell growth and induce DNA damage
Pharmacological inhibition of WEE1 by a small molecule inhibitor MK1775 exerts anti-tumorigenic effects in various cancers. As shown in Fig. 3a, GIST-T1 and GIST-882 cells were treated with different doses of MK1775 for 24 h, and the expression of p-CDK1 was reduced in a dose-dependent manner. Then the cytotoxicity of MK1775 was assessed using the MTT and colony formation assays. MK1775 significantly reduced the viability of GIST cells in a dose-dependent manner. The IC50 value was 0.650 ± 1.539 μM and 1.532 ± 1.111 μM for GIST-T1 and GIST-882, respectively (Fig. 3b). With respect to colony formation, MK1775 treatment led to fewer and smaller colonies than in the control group (Fig. 3c). We hypothesized that inhibition of WEE1 would result in DNA double-strand breaks during DNA replication. Western blotting was performed to analyze γH2AX expression, a surrogate marker for DNA damage. As expected, increased γH2AX expression was observed in GIST-T1 and GIST-882 cells following MK1775 treatment (Fig. 3d). For immunofluorescence, we also detected γH2AX expression in the cell nucleus, which was strikingly increased after MK1775 treatment (Fig. 3e). These data suggest that disruption of WEE1 kinase activity by MK1775 reduced GIST cell viability, increasing the occurrence of DNA double-strand breaks.

Treatment with MK1775 inhibits GIST cell proliferation in a dose-independent manner. a The expression of WEE1 and downstream signaling proteins was analyzed by western blotting after treatment with 0.5, 1, and 2 μM MK1775 for 24 h. b The IC50 of MK1775 in GIST-T1 and GIST-882 cells was determined by the MTT assay after a 48-h treatment with MK1775. c Colony formation assay was used to determine the antitumor activity of MK1775 in GIST-T1 and GIST-882 cells (one-way ANOVA). d Western blot analysis was used to examine the expression of γH2AX after treatment with 0.5 and 1 μM MK1775 for 24 h. e Immunofluorescence was performed to detect the expression of γH2AX in MK1775-treated (1 μM) GIST cells. *P < 0.05, **P < 0.01
MK1775 induces apoptosis and G2–M cell cycle arrest in GIST cells
To assess the extent of apoptosis induced in GIST cells after MK1775 treatment, we detected the proportion of apoptotic cells using Annexin V staining by flow cytometry analysis. The percentage of apoptotic cells was increased in a dose-dependent manner (Fig. 4a). GIST cells treated with DMSO exerted low staining with Annexin V (GIST-T1: 2.45 ± 0.55%; GIST-882: 2.85 ± 0.85%). In contrast, treatment with 0.5 μM MK1775 resulted in an increase in the percentage of Annexin V-positive cells (GIST-T1: 9.15 ± 0.95%; GIST-882: 12.4 ± 0.70%). Additionally, there were more apoptotic cells on the treatment with 1 μM MK1775 (GIST-T1: 31.25 ± 0.55%; GIST-882: 30.85 ± 0.05%). Next, the expression of apoptosis-related proteins was measured to elucidate the specific apoptotic pathways underlying the cytotoxic effect of MK1775. As shown in Fig. 4b, MK1755 treatment markedly increased the expression of cleaved PARP in GIST-T1 and GIST-882 cells. Furthermore, we monitored the cell cycle distribution after treatment with 0.5 μM MK1775 in GIST-T1 and GIST-882 cells. Treatment with MK1775 induced cell G2–M phase arrest, accompanied by a decrease in the proportion of cells in G0/G1 phase, but did not change the proportion of cells in S phase (Fig. 4c). Together, these data suggest that MK1775 treatment induced GIST cell apoptosis and G2–M phase arrest.

MK1775 induces apoptosis and cell cycle arrest in GIST cells. a APC Annexin V with 7-AAD apoptosis staining was used to detect the percentage of apoptotic cells by flow cytometry after treatment with MK1775 for 48 h (one-way ANOVA). b Apoptosis-related proteins were evaluated by western blotting after treatment with MK1775 for 48 h. c The cell cycle distribution was assessed by flow cytometry after treatment with MK1775 for 24 h (t test). *P < 0.05, **P < 0.01
Targeting WEE1 facilitates autophagic degradation of KIT protein in GIST cells
To investigate the potential mechanism underlying the effect of WEE1 kinase on GIST cells, we analyzed whether targeting WEE1 could reduce KIT expression. The expression of KIT was suppressed in WEE1-knockdown GIST-T1 and GIST-882 cells and in response to MK1775 treatment in a dose-dependent manner, along with decreased p-KITT703 expression (Fig. 5a, b). Consistently, the downstream signaling molecules, such as AKT, mTOR, and ERK1/2 signal pathway, were inactivated by the inhibition of WEE1. With the reduced expression of KIT and p-KITT703, the levels of p-AKTS473, p-mTORS2448, and p-ERK1/2 were also reduced following WEE1 knockdown and MK1775 treatment (Fig. 5a, b). To clarify the precise mechanism in the downregulation of KIT by WEE1 inhibition, we also analyzed the KIT mRNA levels after WEE1 inhibition. As shown in Supplementary Fig. 2, WEE1 knockdown and MK1775 treatment did not decrease the KIT mRNA expression in GIST-T1 and GIST-882 cells. These results indicated that the decreased KIT expression observed upon targeting WEE1 was independent of the transcription inhibition, and WEE1 may stabilize KIT protein in GIST cells.

Targeting WEE1 promotes autophagic degradation of KIT protein in GIST cells. a GIST-T1 and GIST-882 cells were transfected with siRNAs for 48 h and the total lysates were analyzed by western blotting using antibodies against KIT, p-KITT703, AKT, p-AKTS473, mTOR, p-mTORS2448, ERK1/2, and p-ERK1/2. b GIST-T1 and GIST-882 cells were treated with different dosages of MK1775, and the total lysates were analyzed by western blotting using antibodies against KIT, p-KITT703, AKT, p-AKTS473, mTOR, p-mTORS2448, ERK1/2, and p-ERK1/2. c Western blotting was used to analyze the expression of KIT and p-KITT703 after WEE1 knockdown cells were treated with 50 μg/mL cycloheximide for the indicated time and the half-life of the KIT protein was calculated based on the relative KIT expression normalized by GAPDH levels. d Western blotting was used to determine the expression of KIT and p-KITT703 in GIST-T1 and GIST-882 cells of treated with 50 μg/mL cycloheximide alone or in combination with 1 μM MK1775 for the indicated time and the half-life of the KIT protein was calculated based on the relative KIT expression normalized by GAPDH levels. e Cells were transfected with WEE1 siRNA and pre-incubated with MG-132 (10 μM) or CQ (10 μM) for 4 h. Cell lysates were immunoblotted using an anti-KIT antibody. f Cells were pre-incubated with MG-132 (10 μM) or CQ (10 μM) for 4 h and then treated with 1 μM MK1774 for an additional 8 h. g The expression of Beclin1 and LC3B was evaluated by western blotting after transfection with siRNAs for 48 h. h The expression of Beclin1 and LC3B was analyzed by western blotting after treatment with MK1775 for 24 h. *P < 0.05, **P < 0.01
To directly detect the effect of WEE1 on the endogenous KIT protein stability, the protein synthesis was blocked with CHX in WEE1 knockdown GIST cells. The western blotting results indicated that WEE1 knockdown could enhance the KIT degradation (Fig. 5c). The fold change in KIT protein normalized to GAPDH was used to calculate the half-life of KIT protein. In GIST-T1 cells, the half-life of the control group vs. the WEE1 knockdown group was 4.552 ± 0.655 h vs. 2.195 ± 0.664 h. In GIST-882 cells, the half-life was 4.937 ± 1.881 h vs. 1.194 ± 0.562 h (Fig. 5c). To further examine whether MK1775 would have a similar effect on KIT protein degradation, GIST cells were treated with CHX alone or in combination with 1 μM MK1775. We also found MK1775 promoted KIT degradation and the half-life of the CHX group vs. the CHX plus MK1775 group was 3.836 ± 1.081 h vs. 2.017 ± 1.136 h and 5.112 ± 1.441 h vs. 2.393 ± 1.455 h in GIST-T1 cells and GIST-882 cells, respectively (Fig. 5d). These results highlight that WEE1 prevents KIT protein degradation in GIST cells.
There are three major systems, including proteasome, lysosome, and autolysosome pathways, for controlling protein degradation in eukaryotic cells [24]. We next determined which system contributed to the WEE1-induced KIT stability. We found that WEE1 knockdown-mediated degradation of KIT was completely inhibited by the autophagy inhibitor chloroquine (CQ). However, the proteasome inhibitor MG132 had no inhibited effect on GIST-T1 cells and had limited effect on GIST-882 cells (Fig. 5e). Moreover, the downregulation of KIT protein by MK1775 could be rescued by CQ in both GIST-T1 and GIST-882 cells, but not by MG132 (Fig. 5f). These data suggest that WEE1 mostly inhibited autophagic degradation of KIT protein in GIST cells. To further confirm the involvement of autophagy in the KIT downregulation induced by WEE1 inhibition, we examined the expression of the autophagic biomarkers, Beclin1 and LC3B. The Beclin1 expression and LC3BII/I ratio were increased in WEE1 knockdown GIST cells (Fig. 5g). Coincidently, treatment with MK1775 resulted in the similar results in a concentration-dependent manner (Fig. 5h). Together, these finding indicate that targeting WEE1 promoted autophagic degradation of KIT protein in GIST cells.
MK1775 enhances the antitumor activity of imatinib via reducing KIT expression
Based on the previous results that MK1775 accelerated KIT degradation, we speculated that MK1775 could affect the sensitivity of imatinib for GIST. To validate the hypothesis, a cell proliferation assay was performed in GIST-T1 and GIST-882 cells. After incubation with 50 nM imatinib and 1 μM MK1775 alone or in combination for 48 h, we observed that imatinib plus MK1775 had a greater antitumor activity than single treatment in GIST cells (Fig. 6a). As determined using CompuSyn software, the combination index (CI) of imatinib and MK1775 was 0.6238 in GIST-T1 cells and 0.5687 in GIST-882 cells, which indicated synergistic effects. To further analyze the effect of MK1775 on imatinib-treated GIST cells, we examined the levels of apoptosis-related proteins and Annexin V-positive cells. The expression of cleaved PARP was slightly increased in the imatinib- or MK1775-treatment groups compared to that of the control group, whereas it was significantly increased in the combination group (Fig. 6b). Consistent with the percentage of Annexin V-positive cells, a greater induction of apoptosis was observed in the co-treatment group compared to treatment with imatinib or MK1775 alone (Fig. 6c).

MK1775 augments the effect of imatinib in suppressing GIST cell proliferation. a MTT assays were performed to detect cell viability with 50 nM imatinib, 1 μM MK1775, or a combination of both for 48 h (one-way ANOVA). b Western blotting was used to analyze the expression of cleaved PARP and γH2AX after treatment with 50 nM imatinib, 1 μM MK1775, or a combination of both for 48 h. c Cellular apoptosis was detected by flow cytometry (one-way ANOVA). d Immunofluorescence was performed to detect the expression of γH2AX in GIST cells. e The expression levels of KIT, p-KITT703, AKT, p-AKTS473, ERK1/2, and p-ERK1/2 were examined by western blotting after treatment with 50 nM imatinib, 1 μM MK1775, or a combination of both for 48 h. *P < 0.05, **P < 0.01
Notably, we revealed that MK1775 treatment induced DNA damage in GIST cells. As shown in Fig. 6b, d, imatinib treatment did not lead to γH2AX expression and DNA double-strand breaks. However, treatment with a combination of imatinib and MK1775 profoundly increased γH2AX expression. To address the molecular mechanism underlying the enhanced antitumor activity in GIST cells treated with a combination of imatinib and MK1775, we assessed the activated states of KIT and related downstream signaling pathways. Both imatinib and MK1775 could inhibit p-KITT703 expression, accompanied with decreased p-AKTS473 and p-ERK1/2 levels. More importantly, combination treatment strongly downregulated KIT and p-KITT703 expression and reduced the activity of downstream signaling pathways, including the AKT and ERK1/2 pathway (Fig. 6e). Together, these results provide a rationale for the enhanced antitumor activity induced by combined treatment with imatinib and MK1775.
Discussion
DNA damage commonly occurs in cells from endogenous and exogenous lesions. There are two major DNA damage checkpoints, G1–S and G2–M, to regulate whether a cell continues in the cell cycle or instead undergoes DNA repair or apoptosis [25, 26]. p53 plays a crucial role in the maintenance of genomic integrity and in regulating cell cycle arrest in response to various stress signals. It is clear that wild-type p53-harboring cells arrest at the G1 checkpoint to repair damaged DNA before DNA replication. In contrast, p53-mutant cells rely mainly on DNA repair at the G2 checkpoint [27]. Studied by Henze et al. suggested that a p53 deletion mutation was present in GIST-T1 and GIST-882 cells [28]. Therefore, abrogation of the G2–M checkpoint by inhibition of WEE1 kinase activity is an attractive strategy to drive mutant-p53-harboring GIST cells to enter into unscheduled mitosis and ultimately undergo cell death via mitotic catastrophe.
In this study, we determined WEE1 expression in GISTs using the Oncomine public database and found that WEE1 is highly expressed in GISTs relative to normal tissues. Considering that only 6 GISTs were available in the database, we further demonstrated that WEE1 protein expression was elevated in GIST specimens and showed that upregulated expression of WEE1 is correlated with tumor size, mitotic count, and risk grade. These data suggested that WEE1 or related cell cycle pathways may play important roles in GIST progression. Consistent with previous studies, cyclin B1 and CDK1 expression may be useful markers for predicting aggressive GISTs [29]. Therefore, we investigated whether targeting of WEE1 could be a potential therapeutic strategy for GIST.
Silencing of WEE1, which was accompanied by a decrease in p-CDK1 levels, significantly inhibited GIST-T1 and GIST-882 cell proliferation and induced cell cycle arrest. Similarly, suppressed kinase activity of WEE1 by MK1775 decreased cell viability and colony formation in a dose-dependent manner. Therefore, targeting of WEE1 might offer an effective method to control the progression of GISTs. It has been reported that MK1775 causes DNA damage in both p53-mutant and -wild-type cells [30]. However, MK1775 increased the sub-G1 population through G1 arrest in wild-type p53-harboring gastric cells and the sub-G1 population through G2–M arrest in mutant p53-harboring gastric cells [18]. Here, we examined the role of MK1775 in GIST cell cycle distribution. Treatment of GIST-T1 and GIST-882 cells with MK1775 induced G2–M arrest. The γH2AX levels also increased following MK1775 treatment. Our data further indicate that MK1775 has powerful antitumor effects via induction of cycle arrest and cell apoptosis in GIST cells.
In previous studies, many KIT/PDGFRA expression regulators have been examined in GIST. Our study showed that inhibition of WEE1 could reduce KIT and p-KITT703 expression, but had no effect of KIT mRNA, which indicated decreased KIT expression by targeting WEE1 was independent of the transcription inhibition. It is reported that heat-shock protein 90 (HSP90) protects KIT from ubiquitin-dependent proteasome-mediated degradation or autophagy pathway and inhibition of its function in GIST was postulated to be an effective target [31, 32]. Bortezomib, a proteasome inhibitor, plays a role in downregulating KIT expression in GIST cells [33]. To understand the molecular mechanisms by which WEE1 prevented KIT degradation in GIST cells, we show WEE1 inhibition-induced KIT degradation could be reversed by pharmacological inhibition of autophagy, but not proteasome pathway. In addition, WEE1 inhibition increased Beclin1 expression and LC3BII/I ratio in GIST cells.
Although imatinib has provided a revolutionary treatment option for GIST, the benefits are not durable. To overcome these limitations, new targets and therapies for GIST have been developed [34]. It was shown that ETV1 was highly expressed in GIST and regulated cell growth in vitro and in vivo by modulating the expression of KIT [35]. Epigenetic alterations also play important roles in GIST progression. The elevated expression of KDM4D may serve as a biomarker, as KDM4D regulates GIST cell proliferation and migration by directly binding to the HIF1β gene promoter [36]. As with other cancers, many studies have focused on combination strategies for GIST treatment. Combined approaches include targeting the KIT downstream pathway and compensatory active pathways. The PI3K/mTOR signaling is a crucial downstream pathway in GIST. Co-treatment with the PI3K inhibitor GDC-0941 and imatinib in GIST xenografts results in more substantial tumor regression, apoptosis, and durable effects than single treatment with imatinib [37]. A phase I/II study of everolimus (an oral mTOR inhibitor) in combination with imatinib showed partial benefit for PFS 3.5 months in imatinib-resistant GIST [38]. In our study, we found that imatinib and MK1775 co-treatment could increase the antitumor activity compared to single treatment. In contrast to a previous study [39], treatment with imatinib induced mild DNA damage, which was confirmed by γH2AX expression, and it is reasonable that approximately the IC50 dose of MK1775 was used in our study. Unexpectedly, γH2AX expression was more rapidly induced upon co-treatment than with MK1775 treatment alone. Our data suggest that a combined strategy involving imatinib and MK1775 could be a potential therapy for GISTs.
In conclusion, we demonstrated that WEE1 is highly expressed in GISTs and upregulated expression of WEE1 is correlated with tumor size, mitotic count, and risk grade. Moreover, WEE1 kinase is a key molecule in controlling GIST malignancy, and targeting of WEE1 could markedly induce DNA damage and promote cell apoptosis. Importantly, co-treatment with imatinib and MK1775 augmented the antitumor activity via promoting autophagic degradation of KIT protein. Taken together, WEE1 may be a potential target for GIST treatment.
References
Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet. 2013;382:973–83.
Niinuma T, Suzuki H, Sugai T. Molecular characterization and pathogenesis of gastrointestinal stromal tumor. Transl Gastroenterol Hepatol. 2018;3:2.
Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 2008;53:245–66.
Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23:3999–4006.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80.
Joensuu H, Wardelmann E, Sihto H, Eriksson M, Sundby Hall K, Reichardt A, et al. Effect of KIT and PDGFRA mutations on survival in patients with gastrointestinal stromal tumors treated with adjuvant imatinib: an exploratory analysis of a randomized clinical trial. JAMA Oncol. 2017;3:602–9.
Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5.
Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–90.
Wardelmann E, Thomas N, Merkelbach-Bruse S, Pauls K, Speidel N, Buttner R, et al. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol. 2005;6:249–51.
Wang WL, Conley A, Reynoso D, Nolden L, Lazar AJ, George S, et al. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 2011;67(Suppl 1):S15–24.
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38.
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–9.
Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302.
Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93–115.
Hur K, Lee HJ, Woo JH, Kim JH, Yang HK. Gene expression profiling of human gastrointestinal stromal tumors according to its malignant potential. Dig Dis Sci. 2010;55:2561–7.
Fujita A, Yamamoto H, Imamura M, Nakamura N, Maehara Y, Tsuneyoshi M, et al. Expression level of the mitotic checkpoint protein and G2–M cell cycle regulators and prognosis in gastrointestinal stromal tumors in the stomach. Virchows Arch. 2012;460:163–9.
Matheson CJ, Backos DS, Reigan P. Targeting WEE1 kinase in cancer. Trends Pharmacol Sci. 2016;37:872–81.
Kim HY, Cho Y, Kang H, Yim YS, Kim SJ, Song J, et al. Targeting the WEE1 kinase as a molecular targeted therapy for gastric cancer. Oncotarget. 2016;7:49902–16.
Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res Treat. 2010;122:347–57.
Magnussen GI, Holm R, Emilsen E, Rosnes AK, Slipicevic A, Florenes VA. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: potential for targeted therapy. PLoS ONE. 2012;7:e38254.
De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17:4200–7.
Mueller S, Haas-Kogan DA. WEE1 kinase as a target for cancer therapy. J Clin Oncol. 2015;33:3485–7.
Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000.
Liu T, Tang Q, Liu K, Xie W, Liu X, Wang H, et al. TRIM11 suppresses AIM2 Inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep. 2016;16:1988–2002.
Abroudi A, Samarasinghe S, Kulasiri D. A comprehensive complex systems approach to the study and analysis of mammalian cell cycle control system in the presence of DNA damage stress. J Theor Biol. 2017;429:204–28.
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85.
Blandino G, Di Agostino S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J Exp Clin Cancer Res. 2018;37:30.
Henze J, Muhlenberg T, Simon S, Grabellus F, Rubin B, Taeger G, et al. p53 modulation as a therapeutic strategy in gastrointestinal stromal tumors. PLoS ONE. 2012;7:e37776.
Nakamura N, Yamamoto H, Yao T, Oda Y, Nishiyama K, Imamura M, et al. Prognostic significance of expressions of cell-cycle regulatory proteins in gastrointestinal stromal tumor and the relevance of the risk grade. Hum Pathol. 2005;36:828–37.
Aarts M, Linardopoulos S, Turner NC. Tumour selective targeting of cell cycle kinases for cancer treatment. Curr Opin Pharmacol. 2013;13:529–35.
Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66:9153–61.
Hsueh YS, Yen CC, Shih NY, Chiang NJ, Li CF, Chen LT. Autophagy is involved in endogenous and NVP-AUY922-induced KIT degradation in gastrointestinal stromal tumors. Autophagy. 2013;9:220–33.
Bauer S, Parry JA, Muhlenberg T, Brown MF, Seneviratne D, Chatterjee P, et al. Proapoptotic activity of bortezomib in gastrointestinal stromal tumor cells. Cancer Res. 2010;70:150–9.
Wozniak A, Gebreyohannes YK, Debiec-Rychter M, Schoffski P. New targets and therapies for gastrointestinal stromal tumors. Expert Rev Anticancer Ther. 2017;17:1117–29.
Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–53.
Hu F, Li H, Liu L, Xu F, Lai S, Luo X, et al. Histone demethylase KDM4D promotes gastrointestinal stromal tumor progression through HIF1beta/VEGFA signalling. Mol Cancer. 2018;17:107.
Floris G, Wozniak A, Sciot R, Li H, Friedman L, Van Looy T, et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19:620–30.
Schoffski P, Reichardt P, Blay JY, Dumez H, Morgan JA, Ray-Coquard I, et al. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010;21:1990–8.
Liu Y, Tseng M, Perdreau SA, Rossi F, Antonescu C, Besmer P, et al. Histone H2AX is a mediator of gastrointestinal stromal tumor cell apoptosis following treatment with imatinib mesylate. Cancer Res. 2007;67:2685–92.
Acknowledgements
This study was supported by the National Key Basic Research Program of China (2015CB5540007), the National Science and Technology Major Project (2017YFC0113503), the National Natural Science Foundation of China (No. 81572413, 81702386, and 81874184), the Foundation of Independent Innovation Fund of Huazhong University of Science and Technology (2017KFYXJJ230 and 2017KFYXJJ256), and the Natural Science Foundation of Hubei Province (2016CFA100).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, W., Zeng, X., Yin, Y. et al. Targeting the WEE1 kinase strengthens the antitumor activity of imatinib via promoting KIT autophagic degradation in gastrointestinal stromal tumors. Gastric Cancer 23, 39–51 (2020). https://doi.org/10.1007/s10120-019-00977-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-019-00977-1