
Abstract
Rapid and reliable pathogen identification is compulsory to confirm ventilator-associated pneumonia (VAP) in order to initiate appropriate antibiotic treatment. In the present proof of concept, the effectiveness of rapid microorganism identification with a targeted bottom-up proteomics approach was investigated in endotracheal aspirate (ETA) samples of VAP patients. To do so, a prototype selected-reaction monitoring (SRM)-based assay was developed on a triple quadrupole mass spectrometer tracking proteotypic peptide surrogates of bacterial proteomes. Through the concurrent monitoring of 97 species-specific peptides, this preliminary assay was dimensioned to characterize the occurrence of six most frequent bacterial species responsible for over more than 65% of VAP. Assay performance was subsequently evaluated by analyzing early and regular 37 ETA samples collected from 15 patients. Twenty-five samples were above the significant threshold of 105 CFU/mL and five samples showed mixed infections (both pathogens ≥ 105 CFU/mL). The targeted proteomics assay showed 100% specificity for Acinetobacter baumannii, Escherichia coli, Haemophilus influenzae, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniae. No false bacterial identification was reported and no interference was detected arising from the commensal flora. The overall species identification sensitivity was 19/25 (76%) and was higher at the patient level (84.6%). This successful proof of concept provides a rational to broaden the panel of bacteria for further clinical evaluation.
Similar content being viewed by others

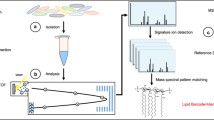

Introduction
Ventilator-associated pneumonia (VAP) is the most frequent health care-associated infection in critically ill patients and is associated with a significant mortality and morbidity [1]. So far, clinicians empirically treat the patients based on guidelines [2] and on the epidemiology of the intensive care unit (ICU). Without early microbiological identification, the treatment includes usually large broad-spectrum antibiotic, increasing the risk of multidrug-resistant bacteria (MDR) selection [3]. The early identification of the causative pathogen is thus crucial for the management of the patient. Indeed, an early targeted and appropriate antibiotic therapy is correlated with the reduction of mechanical ventilation duration, ICU length stay, and hospitalization costs [4].
Besides rapid molecular methods developed recently, semi-quantitative cultures of lower respiratory tract samples (bronchoalveolar lavage (BAL), protected specimen brush (PSB), or endotracheal aspirates (ETA)) remain routinely used for therapeutic decision and less costly. However, semi-quantitative cultures are difficult to standardize and delay the microbial identification for at least 18–24 h. Although controverted [5,6,7], ETA, which are non-invasive specimens, have been proposed for both pre-infection monitoring and VAP diagnosis [8].
Various etiologic pathogens are involved in VAP, including Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus influenzae, Streptococcus pneumoniae, Acinetobacter baumannii, and Enterobacteriaceae covering almost 80% of the cases with little epidemiological modification overtime [2, 9].
With regards to pathogen identification, matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MS) undoubtedly represents a real revolution in that it has drastically shortened the delay of the identification and resistance characterization step, even if a prior culture if usually still required, for MS sensitivity issues. Furthermore, MALDI-TOF-MS-based identification fails when considering a complex, faintly multicontaminated biological matrix [10] and, as a result, is not compatible with direct bacterial identification in ETA samples. As an alternative, recent studies illustrated the value of targeted bottom-up proteomics approaches for bacteria characterization after trypsin digestion of the extracted whole proteome [11]. Developed on low- or high-resolution mass spectrometers, they relied on the monitoring of well-selected peptide panels for, e.g., the detection of four bacteria involved in respiratory tract infections [12], the concomitant identification, resistance and virulence characterization of S. aureus in positive blood cultures [13], and the deciphering of resistance mechanisms [11,12,13,14,15,16,17].
In this proof of concept, a targeted multiplex assay was developed and implemented for assessing the effectiveness of targeted MS in identifying a panel of six microorganisms predominantly associated with VAP directly from ETA samples without culture.
Materials and methods
Reagents and chemicals
LC-MS grade acetonitrile and water, formic acid (FA), dithiothreitol (DTT), iodoacetamide (IAA), ammonium bicarbonate, and porcine trypsin were purchased from Sigma-Aldrich-Fluka (Lyon, France). Blood culture bottles and agar plates for bacterial culture were obtained from bioMérieux (Marcy L’Etoile, France).
Bacterial strains
In this preliminary proof of concept, a VAP diagnosis assay (VAP-6) was developed to detect the six bacterial species most frequently detected in VAP, i.e., A. baumannii, Escherichia coli, H. influenzae, P. aeruginosa, S. aureus, and S. pneumoniae. In order to assess the specificity of the peptide panel of VAP-6 SRM assay, shotgun proteomics experiments were also carried out on 36 other species used as negative controls. Among these 36 species, 6 species represented less frequent bacteria that could be involved in respiratory tract infection (Enterobacter aerogenes, Enterobacter cloacae, Klebsiella oxytoca, Klebsiella pneumoniae, Proteus mirabilis, and Serratia marcescens), 22 species were from the commensal oro-pharyngeal flora, often identified in respiratory samples (Haemophilus (H. haemolyticus, H. parainfluenzae), Neisseria (N. sicca, N. subflava), Staphylococcus (S. epidermidis, S. hominis, S. haemolyticus, S. saprophyticus, S. simulans, S. warneri), Streptococcus (S. agalactiae, S. anginosus, S. constellatus, S. infantis, S. intermedius, S. mitis, S. mutans, S. oralis, S. pseudopneumoniae, S. pyogenes, S. salivarius, S. sanguinis), and the remaining eight species were phylogenetically closely related to the pathogens (Acinetobacter (A. calcoaceticus, A. haemolyticus, A. junii), Pseudomonas (P. alcaligenes, P. putida, P. stutzeri), Morganella morganii, and Serratia fonticola). The bacterial strains used in this study came from the bioMérieux collection (Supplementary data Table 1).
ETA samples and microbiological analysis
A prospective sample collection was conducted in the ICU of Limoges University Hospital (France) to obtain 37 ETA specimens from 15 patients (> 18 years old) who had been under mechanical ventilation for at least 48 h and who were suspected of VAP.
For each ETA, an aliquot was frozen at − 80 °C for further proteomic analysis (see below ETA sample preparation) and one aliquot was used for a cultivation-dependent analysis performed in the microbiology laboratory of the Limoges University hospital according to the recommendations of the European Society for Clinical Microbiology and Infectious Diseases society (ESCMID) [18]. Bacterial identification was performed with the VITEK® MS (bioMérieux) and antibiotic susceptibility testing was performed for bacteria considered as pathogens with the VITEK® 2 system (bioMérieux).
Informed consent for inclusion in the study was obtained from an authority member of the family. A non-opposition certificate was transmitted to the patient. The data conservation and the experimental protocol were conducted according to national regulations and approved by national ethical committee CCTIRS (French Consultative Committee of Information Treatment concerning health Research) (File no 12.001) and the CNIL (National commission of Informatics and freedom) (file no EGY/VCS/AR125816).
Strain preparation
One milliliter of bacterial inoculum at 4 McF was centrifuged at 6000g during 10 min. The pellet was suspended into 150 μL of 5 mM DTT, 50 mM ammonium bicarbonate buffer, pH 8.0. Bacterial lysis was accomplished in 5 min using ultrasound (Microlab ID STARlet, Hamilton Robotics, Reno, NV, USA) with beads from Biospec Product (Bartlesville, OK, USA), followed by alkylation in 12.5 mM IAA during 5 min in the dark. Protein digestion was then carried out using trypsin (50 μL at 1 mg/mL) during 15 min at 50 °C. The enzyme activity was stopped by the addition of 0.05% FA.
ETA sample preparation
ETA samples were fluidized by the addition of 20 U/mL DNase (Promega, Madison, WI, USA) followed by incubation for 15 min at room temperature. For a complete fluidification, samples were further half-diluted in Digester (Eurobio, les Ulis, France) for 15 min at room temperature. Saponin was used for differential lysis between human cells and bacteria [19]; then, the bacterial cells were sedimented by centrifugation at 12,000g during 10 min.
The pellet was suspended in 700 μL of 5 mM DTT, 50 mM ammonium bicarbonate buffer, pH 8.0. Bacterial lysis and digestion were performed as described above for the reference strains. Lysed and digested samples were desalted using a solid-phase extraction (SPE) OASIS® HLB cartridge (Waters, Milford, MA, USA) into the RapidTrace SPE workstation (Biotage, Uppsala, Sweden) according to manufacturer instructions. The eluted sample was dried using a TurboVap (Biotage) and suspended in 200 μL of 0.05% FA containing water. The sample preparation time was 2 h per sample batch (16 samples).
LC-ESI-Q-TOF-MS analysis for the specific peptide discovery phase
Screening for strain-specific peptide identification was performed in Data Dependent Analysis (DDA) mode using a LC Ultimate 3000 chromatography system (Dionex, Sunnyvale, CA, USA) hyphenated to a TripleTOF®5600 MS (Sciex, Framingham, MA, USA). Mobile phase A was water with 0.1% FA, and mobile phase B was acetonitrile with 0.1% FA. The gradient ranged from 2 to 37% of B in 60 min at 300 μL/min. A MS/MS fragmentation spectrum was registered during 75 ms for all precursor ions detected above 200 counts in the survey MS spectrum (250 ms), with a dynamic exclusion time of 3 s. MS control and data acquisitions were performed using Analyst 1.6 software (Sciex).
Data analysis for the specific peptide discovery phase
Spectra analysis and database interrogations were made with ProteinPilot™ software 4.0.8085 (Sciex). Peptides were identified using the rapid identification mode and a confidence score (ProtScore) superior to 95%. Carbamidomethylation was considered as a fixed chemical modification and only one missed cleavage was allowed. The databases used were UniProtKB/Swiss-Prot and UniProtKB/TrEMBL. Databases were restricted at different taxonomic levels depending on the species. The family level was used for Enterobacter spp., Morganella spp., Serratia spp. (Enterobacteriaceae), Neisseria spp. (Neisseriaceae), Haemophilus spp. (Pasteurellaceae), Pseudomonas spp. (Pseudomonadaceae), Staphylococcus spp. (Staphylococcaceae), and Streptococcus spp. (Streptococcaceae); the genus level was used for Acinetobacter spp., Klebsiella spp., and Proteus spp.; the species level was retained for E. coli. Then, each peptide detected in a single species and in at least 70% of the strains of this specie has been validated as specific and inclusive. Moreover, an identity BLAST alignment [20] was made on the two databases previously cited to confirm in silico the species specificity of the peptides.
SRM method construction
The SRM method was developed using a Nexera LC (Shimadzu) system hyphenated to a hybrid quadrupole linear ion trap mass spectrometer (QTRAP® 5500, Sciex) and Analyst 1.5.2 software. The SRM assays were created by selecting a subset of species-specific peptides among the best-flyer candidates identified during the discovery phase. Peptides containing amino acid residues prone to partial chemical modifications, such as methionine and cysteine, were not retained in the final list. The three most intense transitions were selected to build the SRM method. The peptides were separated on a C18 reversed phase column 2.1 × 100 mm, 3.5 μm, 130 Å (BEH, Waters Milford, MA, USA) in 28 min at 300 μL/min using a 2% to 37% of B gradient. Two columns were used in parallel for increased throughput. The scheduled SRM method used 264 s time windows centered on the peptide retention times measured by spiking ETA samples with pure bacteria. Q1 and Q3 quadrupole resolutions were adjusted to unitary resolution. The SRM method (VAP-6 SRM assay) contained 291 transitions tracking 97 species-specific peptides used as surrogate targets of A. baumannii, E. coli, H. influenzae, P. aeruginosa, S. aureus, and S. pneumoniae (Supplementary Data Table 1).
Data analysis: peptide identification in ETA samples
Scheduled SRM data processing was performed using Multiquant 2.0 software (Sciex) and the integration algorithm Signal Finder. The software allows extracting a chromatogram corresponding to each peptide. Post process result interpretation was based on the chromatographic peak area. Peptide detection was considered positive if the three transitions per peptide were simultaneously detected with a standard deviation of the retention time lower than 0.03 s and a conserved ratio (± 20% deviation) between the three transition as established by analyzing the pure strains. Bacteria quantification in ETA was performed using integration of chromatographic peptide areas.
Results and discussion
Identification of species-specific peptides
The panel of peptides, selected in the VAP-targeted MS assay, was designed by combining shotgun proteomics experiments and a bioinformatics pipeline for selection and validation steps (Fig. 1). Fundamentally, two drastic criteria guided the final peptide choice: proteotypicity and high ionization yield. They were assessed using protein extracts of the six species most frequently associated to VAP (A. baumannii, E. coli, H. influenzae, P. aeruginosa, S. aureus, and S. pneumoniae) and 36 “control” species (Table 1).

Discovery process for selecting species-specific peptide targets. Protein identifications were performed with a LC-ESI-Q-TOF. *Peptide specificity was verified experimentally by comparison using 194 bacterial strains including 115 different strains from 36 species considered as control species. Sensitivity was ensured by retaining only peptides present in more than 70% of the strains belonging to a species. **Peptide specificity was verified in silico using identity BLAST and UniProtKB database. The peptide selection was based on the intensity of the chromatographic peak of the peptide
In total, 49,062 tryptic peptides were identified during the proteomics discovery phase after having analyzed strains of reference and representative of the diversity of each of the VAP-6 species (Table 1). A shotgun proteomics strategy was similarly deployed on the 115 bacterial strains from 36 species considered as a control panel (Supplementary data Table 1). This discovery step enables to extract a list of peptides specifically detected in the VAP-6 panel and considered as “best flyer” candidates. However, curated public databases are not yet comprehensive for all bacterial species. For instance, H. influenzae displays only 3090 and 51,400 entries in Swiss-Prot and in TrEMBL databases, respectively, while E. coli exhibits 23,139 and 1,671,579 entries (www.uniprot.org, 2020, April the 8th).
Moreover, different strains of the same species can exhibit an important genomic diversity, i.e., E. coli has only 50% of highly conserved genes among strains [21]. Genes can also be transferred between strains among the same or different species. Consequently, the reliable selection of species-specific peptides should not exclusively rely on protein database mining but requires, in contrast, thorough validation across the analysis of multiple species and of different strains of the same species. As a typical example of such a mandatory extensive validation procedure, the IIELAGHLDTYIPEPERK tryptic peptide arising from K. pneumoniae (Elongation factor Tu) was predicted absent in S. pneumoniae proteome using UniProtKB, while it was experimentally detected (Fig. 2). Last but not least, encoded proteins can exhibit significant differential expression level between clinical isolates, which in turn may induce false-negative detection whether the peptide signals fall below the lower limit of detection.

Extracted SRM chromatogram signals of IIELAGHLDTYIPEPERK peptide from K. pneumoniae and S. pneumoniae species (transitions: 698.71/868.49; 698.71/1031.55; 698.71/1132.60). a Chromatogram of a pure K. pneumoniae digest (API 9103130 strain), peptide retention time: 21.47 min. b Chromatogram of a pure S. pneumoniae digest (ATCC 49619 strain), peptide retention time: 21.59 min. IIELAGHLDTYIPEPERK peptide showed a BLAST identity to K. pneumoniae elongation factor protein A6TEX7 (UniProtKB accession number). It also has been described in Erwinia tracheiphila, Leminorella grimontii, and Serratia ficaria. However, the same peptide was not yet described in S. pneumoniae while it was clearly detected experimentally in b
Hence, we have applied a first selection filter consisting in choosing only the peptides identified during the proteomics experiments in at least 70% of the VAP-6 bacteria and not retrieved in the control panel. Then, their specificity was confirmed by a BLAST query against UniProtKB (Swiss-Prot and TrEMBLE) public databases, which led to characterize from 44 up to 520 species-specific peptides depending on the species (Table 1). As anticipated, no peptide was identified as being strictly proteotypic of E. coli due to close genetic background with the intestinal Shigella genus [22]. However, it is not a hurdle since the Shigella genus is not retrieved in a VAP infection context.
The last step of building the VAP-6 SRM method followed three rules so as to warrant the assay as reliable and sensitive as expected: (i) the peptide panel had to be dispersed throughout the chromatogram in order to limit the number of transitions concurrently monitored within the same scheduled window of retention time; (ii) for each species, the peptides were selected in the BLAST-specific list among those harboring the highest signal to noise ratio at the precursor ion level; (iii) with very few exceptions, peptides harboring a proline or a histidine close to the N terminus were excluded of the selection in order to favor the fragmentation pathway leading to y type fragment ions with close intensity. Ninety-seven peptides (11 to 24 signature peptides per species) were thus ultimately retained in the VAP-6 SRM method. The intrinsic sensitivity of the assay was assessed by monitoring all transitions in a diluted series of trypsin digests of pooled individual cultures of VAP-6 panel. All bacteria were successfully identified (Fig. 3) with a limit of detection (LOD) ranging from 105 to 106 CFU/mL depending on the species (Supplementary data Fig. 1). These detection limits were consistent with the specifications required for true clinical sample analysis considering a VAP pathogenicity threshold at 105 CFU/mL for ETA samples in the European and American guidelines [18, 23].

Multiplex capability of the VAP-6-SRM method. 108 CFU of each six species pooled (A. baumannii, E. coli, H. influenzae, P. aeruginosa, S. aureus, S. pneumoniae) were injected in a LC-ESI-QqQ-MS system and followed simultaneously in one unique run (28 min) using the VAP-6-SRM method. This figure shows the reconstituted chromatogram of the extraction of three specific peptides per species. *1A. baumannii SGTTGNIEAATK: (B7GW18 UniProtKB accession number; retention time: 9.8 min); NIGLLAGLPK (A0A059ZM33 UniProtKB accession number; retention time: 20.44 min); GQAINVQNIYGK (D0C807 UniProtKB accession number; retention time: 15.56 min). *2E. coli VVAVGDQVEK: (P75691 UniProtKB accession number; retention time: 11.24 min); DYVEGETAAK (P0A9Q7 UniProtKB accession number; retention time: 9.76 min); WNGVTVTPK (P0ADU5 UniProtKB accession number; retention time: 13.99 min). *3H. influenzae: YAYVTLGNK (P43839 UniProtKB accession number; retention time: 14.71 min); VQFEVLHSDK (A0A0D0IG76 UniProtKB accession number; retention time: 14.9 min); DSSAEFDNSK (P44076 UniProtKB accession number; retention time: 8.73 min). *4P. aeruginosa: TALATAVAAGTR (Q8KQ36 UniProtKB accession number; retention time: 13.7 min); NIAIAAGDSAK (P72151 UniProtKB accession number; retention time: 11.9 min); VSEGLVLAEPAK (Q8KQ36 UniProtKB accession number; retention time: 15.96 min). *5S. aureus: AFAQLVTK (Q2FXQ1 UniProtKB accession number; retention time: 14.99 min); TQVVDTVAK (P80544 UniProtKB accession number; retention time: 10.7 min); VTDADFDSK (Q2FHT6 UniProtKB accession number; retention time: 10.45 min). *6S. pneumoniae: DLEVTTVVR (A0A0D6J7I5 UniProtKB accession number; retention time: 16.11 min); IDELDAEIAK (A0A0D6J7I5 UniProtKB accession number; retention time: 15.3 min); EVDDTIAEEK (A0A062WP99 UniProtKB accession number; retention time: 11.11 min)
Method validation using ETA samples
In order to assess the diagnostic value of the newly developed SRM-based proteomics assay, 37 ETA samples were successively collected from 15 patients suspected of VAP, then submitted in parallel to both the conventional cultivation-dependent approach and the direct VAP-6 SRM assay. Using the cultivation-dependent approach, at least one bacterial species was isolated in all the samples and significant clinical thresholds (≥ 105 CFU/mL) were detected for 24 samples collected from 14 patients (Table 2). Five out of the six species targeted with the VAP-6-SRM assay were isolated (E. coli, H. influenzae, P. aeruginosa, S. aureus, and S. pneumoniae) and seven samples contained both of these species (Table 2). A. baumannii was not isolated in any of the cohort samples. Moreover, 29 out of the 37 samples showed growth of other bacterial species, mainly with a oro-pharyngeal flora origin. Despite of the molecular complexity of ETA matrix sample and univocal contaminations by oro-pharyngeal bacteria, as highlighted by the cultivation-dependent assay, the VAP-6 SRM assay exhibits 100% specificity (no false bacterial identification) for E. coli, H. influenzae, P. aeruginosa, S. aureus, and S. pneumoniae (Table 3). This performance features the relevancy of the strategy deployed for selecting the peptide candidates in the present VAP-6 assay, which could be replicated for other species to expand the SRM-based VAP assay.
Microorganisms present at the significant clinical threshold ≥ 105 CFU/mL were identified by SRM in 21 out of 25 ETA samples containing at least one pathogen (84% averaged sensitivity) (Tables 2 and 3). Seven ETA samples, from patients 10 to 14, harbored two of the 6 targeted pathogens (6× H. influenzae/S. pneumoniae and 1× E. coli/S. pneumoniae) with CFU/mL ranging from 104 to 106. Both species were detected by SRM in two ETA samples (samples 11_1 and 12_2), one microorganism out two was detected in four other cases (samples 10_2, 11_2, 12_1, and 13_1) while no microorganism was identified in one case (sample 10_1) despite CFU/mL ≥ 105 for both. Finally, three samples (3_2, 8_1, 9_1) were declared negative following the VAP-6 proteomics assay while they were detected positive at ≥ 105 CFU/mL with the cultivation-dependent approach. Three of them grew only one species (S. aureus) and one was polymicrobial with H. influenzae and S. pneumoniae.
The overall sensitivity of SRM-based species identification was 19/25 (76%) for ETA samples (Table 3), 6/6 (100%), 4/6 (66.7%), 9/12 (75%), and 4/6 (66.7%) for E. coli, H. influenzae, S. aureus, and S. pneumoniae, respectively. The sensitivity was higher at the patient level (84.6%) and species were correctly identified in 1/1 (100%), 4/5 (80%), 8/8 (100%), and 4/5 (80%) patients, respectively.
Among the 11 samples showing a bacterial load around 104 CFU/mL, only 3 (27.3%) lead to species identification by the VAP-6 assay. This latter observation challenges the SRM use in clinics for early VAP diagnosis when the bacterial load is low. Further refinement in the sample preparation step may improve the LOD as well as the diagnosis robustness, particularly during the liquefaction step and the separation of bacteria from human cells. Indeed, we noticed that the false-negative identification for some ETA samples correlated with their difficulty to be processed. Our observation of variability in the physico-chemical properties of ETA samples may explain the lack of identification in sample 10_1 and in monoinfected 3_2, 8_1, and 9_1 samples, despite a CFU/mL value estimated from pure cultures well beyond the SRM LOD. The variability of the physico-chemical properties of ETA matrix between patients can impact the sample preparation, the trypsin digestion yield, and consequently the success of identification. The second cause of missed identification at a bacterial load of around 105 CFU/mL may be attributed to the greatest resistance of Gram-positive bacteria towards trypsin digestion since five out of the seven missed identifications were ETA harboring either S. aureus or S. pneumoniae. As stated above, this observation is an incentive for further improvement of sample preparation parameters.
Another and even more obvious option for improving the LOD is simply moving to a more recent mass spectrometer generation and to microflow format [24]. Interestingly, many mass spectrometer manufacturers already propose In Vitro Diagnostics-labeled MS platforms based on liquid chromatography coupled to a triple quadrupole instrument, which underline their interest for clinical diagnosis and suggest continued progress both in terms of instrument and software. Interestingly, quantitation using SRM and cultivation-dependent analysis were in agreement during patient monitoring (Fig. 4).

Patient monitoring by SRM and quantitative culture analysis. ETA samples from one patient, infected by E. coli, were obtained from day 1 to day 8. The semi-quantitative cultures are referred on the vertical left axis and represented by vertical bars. The integration area of a SRM chromatographic peak from an E. coli peptide is referred on the vertical right axis and represented by a black line
In conclusion, SRM-based assay could be an effective and non-invasive way to regularly monitor VAP by identifying mixed pathogens on a reduced 4 h time scale from ETA samples. Depending on the species, the LODs achieved with the assay in ETA samples range from 104 CFU/mL to 106 CFU/mL, with 76% of sensitivity for a clinical threshold value of 105 CFU/mL. These results provide an incentive to expand the pathogen panel and to move on a more sensitive chromatographic format for next performing extensive clinical evaluation of VAP diagnosis based on targeted MS with regard with the standard of care.
Data availability
The data conservation and the experimental protocol were conducted according to national regulations and approved by national ethical committee CCTIRS (French Consultative Committee of Information Treatment concerning health Research) (File no 12.001) and the CNIL (National commission of Informatics and freedom) (file no EGY/VCS/AR125816).
References
Bekaert M, Timsit J-F, Vansteelandt S, Depuydt P, Vésin A, Garrouste-Orgeas M et al (2011) Attributable mortality of ventilator-associated pneumonia. Am J Respir Crit Care Med 184:1133–1139. https://doi.org/10.1164/rccm.201105-0867OC
American Thoracic Society, Infectious Diseases Society of America (2005) Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 171:388–416. https://doi.org/10.1164/rccm.200405-644ST
Davies J, Davies D (2010) Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev MMBR 74:417–433. https://doi.org/10.1128/MMBR.00016-10
Heyland DK, Cook DJ, Marshall J, Heule M, Guslits B, Lang J et al (1999) The clinical utility of invasive diagnostic techniques in the setting of ventilator-associated pneumonia. Canadian Critical Care Trials Group. Chest 115:1076–1084
Khilnani GC, Arafath TKL, Hadda V, Kapil A, Sood S, Sharma SK (2011) Comparison of bronchoscopic and non-bronchoscopic techniques for diagnosis of ventilator associated pneumonia. Indian J Crit Care Med Peer-Rev Off Publ Indian Soc Crit Care Med 15:16–23. https://doi.org/10.4103/0972-5229.78218
Papazian L, Thomas P, Garbe L, Guignon I, Thirion X, Charrel J et al (1995) Bronchoscopic or blind sampling techniques for the diagnosis of ventilator-associated pneumonia. Am J Respir Crit Care Med 152:1982–1991. https://doi.org/10.1164/ajrccm.152.6.8520766
Torres A, Fàbregas N, Ewig S, de la Bellacasa JP, Bauer TT, Ramirez J (2000) Sampling methods for ventilator-associated pneumonia: validation using different histologic and microbiological references. Crit Care Med 28:2799–2804
Luna CM, Sarquis S, Niederman MS, Sosa FA, Otaola M, Bailleau N et al (2013) Is a strategy based on routine endotracheal cultures the best way to prescribe antibiotics in ventilator-associated pneumonia? Chest 144:63–71. https://doi.org/10.1378/chest.12-1477
Chastre J, Fagon J-Y (2002) Ventilator-associated pneumonia. Am J Respir Crit Care Med 165:867–903. https://doi.org/10.1164/ajrccm.165.7.2105078
Mahé P, Arsac M, Chatellier S, Monnin V, Perrot N, Mailler S et al (2014) Automatic identification of mixed bacterial species fingerprints in a MALDI-TOF mass-spectrum. Bioinform Oxf Engl 30:1280–1286. https://doi.org/10.1093/bioinformatics/btu022
Saleh S, Staes A, Deborggraeve S, Gevaert K (2019) Targeted proteomics for studying pathogenic bacteria. Proteomics 19:e1800435. https://doi.org/10.1002/pmic.201800435
Karlsson R, Thorsell A, Gomila M, Salvà-Serra F, Jakobsson HE, Gonzales-Siles L et al (2020) Discovery of species-unique peptide biomarkers of bacterial pathogens by tandem mass spectrometry-based proteotyping. Mol Cell Proteomics 19(3):518–528. https://doi.org/10.1074/mcp.RA119.001667
Charretier Y, Köhler T, Cecchini T, Bardet C, Cherkaoui A, Llanes C et al (2015) Label-free SRM-based relative quantification of antibiotic resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Front Microbiol 6:81. https://doi.org/10.3389/fmicb.2015.00081
Cecchini T, Yoon E-J, Charretier Y, Bardet C, Beaulieu C, Lacoux X et al (2017) Deciphering multifactorial resistance phenotypes in Acinetobacter baumannii by genomics and targeted label-free proteomics Mol Cell Proteomics mcp.RA117.000107. https://doi.org/10.1074/mcp.RA117.000107
Strich JR, Wang H, Cissé OH, Youn J-H, Drake SK, Chen Y et al (2019) Identification of the OXA-48 carbapenemase family by use of tryptic peptides and liquid chromatography-tandem mass spectrometry. J Clin Microbiol 57. https://doi.org/10.1128/JCM.01240-18
Wang H, Chen Y, Strich JR, Drake SK, Youn J-H, Rosenberg AZ et al (2019) Rapid detection of colistin resistance protein MCR-1 by LC-MS/MS. Clin Proteomics 16:8. https://doi.org/10.1186/s12014-019-9228-2
Wang H, Strich JR, Drake SK, Chen Y, Youn J-H, Rosenberg AZ et al (2019) Rapid identification of New Delhi Metallo-β-lactamase (NDM) using tryptic peptides and LC-MS/MS. Antimicrob Agents Chemother 63. https://doi.org/10.1128/AAC.00461-19
Cornaglia G, Courcol R, Herrmann J-L, Kahlmeter G (2012) European manual of clinical microbiology. European Society for Clinical Microbiology and Infections Diseases
Zelenin S, Hansson J, Ardabili S, Ramachandraiah H, Brismar H, Russom A (2015) Microfluidic-based isolation of bacteria from whole blood for sepsis diagnostics. Biotechnol Lett 37:825–830. https://doi.org/10.1007/s10529-014-1734-8
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Rasko DA, Rosovitz MJ, Myers GSA, Mongodin EF, Fricke WF, Gajer P et al (2008) The Pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 190:6881–6893. https://doi.org/10.1128/JB.00619-08
Lan R, Reeves PR (2002) Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect Inst Pasteur 4:1125–1132
Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB et al (2016) Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 Clinical Practice Guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis 63:e61–e111. https://doi.org/10.1093/cid/ciw353
Bache N, Geyer PE, Bekker-Jensen DB, Hoerning O, Falkenby L, Treit PV et al (2018) A novel LC system embeds analytes in pre-formed gradients for rapid, ultra-robust proteomics. Mol Cell Proteomics MCP 17:2284–2296. https://doi.org/10.1074/mcp.TIR118.000853
Acknowledgments
Authors gratefully acknowledge the VALIBI study group for their contribution to the VALIBI clinical study. The members of this group were Limoges: Nicolas Pichon, Philippe Vignon, Roselyne Droual, Cecile Duchiron, Julie Vignaud, Delphine Chainier; Brive: Mathieu Mattei, Andre Sommabere; Tours: Emmanuelle Mercier, Cecile Le Brun; Angouleme: Arnaud Desachy, Caroline Garandeau; bioMerieux: Marc Rodrigue, Morgane Lacroix, Sandrine Prudent, Marie-Astrid Jestin, Javier Yugueros-Marcos. We thank Isabelle Herafa from Inserm CIC 1435 for managing operations during the whole VALIBI clinical study.
Funding
C.B. was funded by the Association Nationale de la Recherche et de la Technologie (ANRT), grant No 2011/0456. This project was part of the Advanced Diagnostics for New Therapeutic approaches, a program dedicated to personalized medicine, coordinated by Mérieux Alliance and supported by the French public agency, OSEO.
Author information
Authors and Affiliations
Consortia
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Chloé Bardet, Olivier Barraud, Marc Clavel, Tanguy Fortin, Jean-Philippe Charrier, Bruno François, Jérôme Lemoine, and Marie-Cécile Ploy. The first draft of the manuscript was written by Chloé Bardet, Jérôme Lemoine, and Marie-Cécile Ploy and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
BF, OB, and MCP institutions have received funding from bioMérieux to run the VALIBI study. C.B., T.F., J.P.C., M.R., and J.Y.M. are, or were, employed by bioMérieux. C.B., T.F., and J.P.C. have filed patents assigned to bioMérieux.
Ethics approval
The data conservation and the experimental protocol were conducted according to national regulations and approved by national ethical committee CCTIRS (French Consultative Committee of Information Treatment concerning health Research) (File no 12.001) and the CNIL (National commission of Informatics and freedom) (file no EGY/VCS/AR125816).
Consent to participate
Informed consent for inclusion in the study was obtained from an authority member of the family. A non-opposition certificate was transmitted to the patient.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jerome Lemoine and Marie-Cécile Ploy have participated equally to the work.
Olivier Barraud, Marc Clavel, and Tanguy Fortin have participated equally to the work
Supplementary information
ESM 1
(DOCX 127 kb)
Rights and permissions
About this article

Cite this article
Bardet, C., Barraud, O., Clavel, M. et al. Early and specific targeted mass spectrometry-based identification of bacteria in endotracheal aspirates of patients suspected with ventilator-associated pneumonia. Eur J Clin Microbiol Infect Dis 40, 1291–1301 (2021). https://doi.org/10.1007/s10096-020-04132-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-020-04132-y