
Abstract
Charcot-Marie-Tooth disease (CMT) refers to a group of clinically and genetically heterogeneous inherited neuropathies. Ganglioside-induced differentiation-associated protein 1 GDAP1-related CMT has been reported in an autosomal dominant or recessive form in patients presenting either axonal or demyelinating neuropathy. We report two Sri Lankan sisters born to consanguineous parents and presenting with a severe axonal sensorimotor neuropathy. The early onset of the disease, the distal and proximal weakness and atrophy leading to major disability, along with areflexia, and, most notably, vocal cord and diaphragm paralysis were highly evocative of a GDAP1-related CMT. However, sequencing of the coding regions of the gene was normal. Whole-exome sequencing (WES) was performed and revealed that the largest region of homozygosity was around GDAP1 with several variants, mostly in non-coding regions. In view of the high clinical suspicion of GDAP1 gene involvement, we examined the variants in this gene and this, along with functional studies, allowed us to identify an alternative splicing site revealing a cryptic in-frame stop codon in intron 4 responsible for a severe loss of wild-type GDAP1. This work is the first to describe a deleterious mutation in GDAP1 gene outside of coding sequences or intronic junctions and emphasizes the importance of interpreting molecular analysis, and in particular WES results, in light of the clinical and electrophysiological phenotype.




Similar content being viewed by others
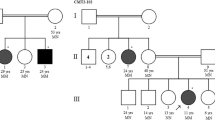
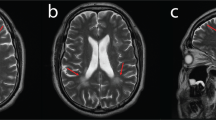
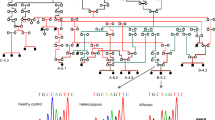
Abbreviations
- CMT :
-
Charcot-Marie-Tooth disease
- AR-CMT :
-
autosomal recessive CMT
- GDAP1 :
-
ganglioside-induced differentiation-associated protein 1
- WES :
-
whole-exome sequencing
References
Baets J, De JP, Timmerman V (2014) Recent advances in Charcot–Marie–Tooth disease. Curr Opin Neurol 27:532–540
Stojkovic T (2016) Hereditary neuropathies: an update. Rev Neurol (Paris) [online serial]. Elsevier Masson SAS; Epub 2016:4–7. Accessed at:. https://doi.org/10.1016/j.neurol.2016.06.007
Banchs I, Casasnovas C, Albert A et al (2009) Diagnosis of Charcot-Marie-Tooth disease. J Biomed Biotechnol 2009:985415
De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N (2002) Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 70(3):726–736. https://doi.org/10.1086/339274
Leal A, Huehne K, Bauer F, Sticht H, Berger P, Suter U, Morera B, Del Valle G, Lupski JR, Ekici A, Pasutto F, Endele S, Barrantes R, Berghoff C, Berghoff M, Neundörfer B, Heuss D, Dorn T, Young P, Santolin L, Uhlmann T, Meisterernst M, Sereda MW, Stassart RM, Meyer zu Horste G, Nave KA, Reis A, Rautenstrauss B (2009) Identification of the variant Ala335Val of MED25 as responsible for CMT2B2: molecular data; functional studies of the SH3 recognition motif and correlation between wild-type MED25 and PMP22 RNA levels in CMT1A animal models. Neurogenetics 10(4):275–287. https://doi.org/10.1007/s10048-009-0183-3
Higuchi Y, Hashiguchi A, Yuan J, Yoshimura A, Mitsui J, Ishiura H, Tanaka M, Ishihara S, Tanabe H, Nozuma S, Okamoto Y, Matsuura E, Ohkubo R, Inamizu S, Shiraishi W, Yamasaki R, Ohyagi Y, Kira J, Oya Y, Yabe H, Nishikawa N, Tobisawa S, Matsuda N, Masuda M, Kugimoto C, Fukushima K, Yano S, Yoshimura J, Doi K, Nakagawa M, Morishita S, Tsuji S, Takashima H (2016) Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol 79(4):659–672. https://doi.org/10.1002/ana.24612
Tazir M, Bellatache M, Nouioua S, Vallat J (2013) Autosomal recessive Charcot-Marie-Tooth disease: from genes to phenotypes. J Peripher Nerv Syst 129:113–129
Hoyle JC, Isfort MC, Roggenbuck J, Arnold WD (2015) The genetics of Charcot-Marie-Tooth disease: current trends and future implications for diagnosis and management. Appl Clin Genet 19:235–243
Claramunt R, Pedrola L, Sevilla T, López de Munain A, Berciano J, Cuesta A, Sánchez-Navarro B, Millán JM, Saifi GM, Lupski JR, Vílchez JJ, Espinós C, Palau F (2005) Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J Med Genet 42(4):358–365. https://doi.org/10.1136/jmg.2004.022178
Baxter RV, Ben OK, Rochelle JM et al (2002) Ganglioside-induced differentiation-associated protein 1 is mutant in Charcot-Marie-Tooth type 4A/8q21. Nat Genet 30(1):21–22. https://doi.org/10.1038/ng796
Chung KW, Hyun YS, Lee HJ, Jung HK, Koo H, Yoo JH, Kim SB, Park CI, Kim HN, Choi BO (2011) Two recessive intermediate Charcot-Marie-Tooth patients with GDAP1 mutations. J Peripher Nerv Syst 16(2):143–146. https://doi.org/10.1111/j.1529-8027.2011.00329.x
Cuesta A, Pedrola L, Sevilla T, García-Planells J, Chumillas MJ, Mayordomo F, LeGuern E, Marín I, Vílchez JJ, Palau F (2002) The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30(1):22–25. https://doi.org/10.1038/ng798
Stojkovic T, Latour P, Viet G, de Seze J, Hurtevent JF, Vandenberghe A, Vermersch P (2004) Vocal cord and diaphragm paralysis, as clinical features of a French family with autosomal recessive Charot-Marie-Tooth disease, associated with a new mutation in the GDAP1 gene. Neuromuscul Disord 14:261–264
Crehalet H, Latour P, Bonnet V, Attarian S, Labauge P, Bonello N, Bernard R, Millat G, Rousson R, Bozon D (2010) U1 snRNA mis-binding: a new cause of CMT1B. Neurogenetics 11(1):13–19. https://doi.org/10.1007/s10048-009-0199-8
Ben Othmane K, Hentati F, Lennon F et al (1993) Linkage of a locus (CMT4A) for autosomal recessive Charcot-Marie-Tooth disease to chromosome 8q. Hum Mol Genet 2:1625–1628
Nelis E, Erdem S, Van den Bergh PYK, Belpaire-Dethiou MC, Ceuterick C, van Gerwen V, Cuesta A, Pedrola L, Palau F, Gabreels-Festen AAWM, Verellen C, Tan E, Demirci M, van Broeckhoven C, de Jonghe P, Topaloglu H, Timmerman V (2002) Mutations in GDAP1. Autosomal recessive CMT with demyelination and axonopathy. Neurology 59(12):1865–1872. https://doi.org/10.1212/01.WNL.0000036272.36047.54
Senderek J, Bergmann C, Ramaekers VT et al (2003) Mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot-Marie-Tooth neuropathy. Brain 126(3):642–649. https://doi.org/10.1093/brain/awg068
Birouk N, Azzedine H, Dubourg O et al (2003) Phenotypical features of a Moroccan family with autosomal recessive Charcot-Marie-Tooth disease associated with the S194X mutation in the GDAP1 gene. Arch Neurol 60:598–604
Azzedine H, Ruberg M, Ente D, Gilardeau C, Périé S, Wechsler B, Brice A, LeGuern E, Dubourg O (2003) Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul Disord 13(4):341–346. https://doi.org/10.1016/S0960-8966(02)00281-X
Sivera R, Frasquet M, Lupo V, García-Sobrino T, Blanco-Arias P, Pardo J, Fernández-Torrón R et al (2017) Distribution and genotype-phenotype correlation of GDAP1 mutations in Spain. Sci Rep 7:66–77
Sivera R, Espinós C, Vílchez JJ, Mas F, Martínez-Rubio D, Chumillas MJ (2010) Phenotypical features of the p.R120W mutation in the GDAP1 gene causing autosomal dominant Charcot-Marie-Tooth disease. Peripher Nerv Syst 15(4):334–344. https://doi.org/10.1111/j.1529-8027.2010.00286.x
Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, Day J, Feely S, Finkel RS, Grider T, Kirk CA, Herrmann DN, Laurá M, Li J, Lloyd T, Sumner CJ, Muntoni F, Piscosquito G, Ramchandren S, Shy R, Siskind CE, Yum SW, Moroni I, Pagliano E, Zuchner S, Scherer SS, Shy ME, Inherited Neuropathies Consortium (2015) CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry 86(8):873–878. https://doi.org/10.1136/jnnp-2014-308826
Tomaselli PJ, Rossor AM, Horga A, Jaunmuktane Z, Carr A, Saveri P, Piscosquito G, Pareyson D, Laura M, Blake JC, Poh R, Polke J, Houlden H, Reilly MM (2017) Mutations in noncoding regions of GJB1 are a major cause of X-linked CMT. Neurology 88(15):1445–1453. https://doi.org/10.1212/WNL.0000000000003819
Funding
JP is funded by Association pour le Développement de la Neurogénétique (Hospices Civils de Lyon DRCI 69HCL15_0734).
Author information
Authors and Affiliations
Contributions
MM, TS, and PL were involved in the conception and design of the research project, analysis and interpretation of the data, and writing of the first draft of the manuscript. TS was involved in the acquisition of clinical data, and PL, JP, and GPL were involved in the acquisition of molecular data. All authors read and approved the final manuscript. RYC was involved in the acquisition and interpretation of whole-body MRI.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors declare that this work received all necessary ethical approvals and that all the patients involved consented to participate.
Consent for publication
Consent for publication was provided for this study.
Conflict of interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Masingue, M., Perrot, J., Carlier, RY. et al. WES homozygosity mapping in a recessive form of Charcot-Marie-Tooth neuropathy reveals intronic GDAP1 variant leading to a premature stop codon. Neurogenetics 19, 67–76 (2018). https://doi.org/10.1007/s10048-018-0539-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-018-0539-7