
Abstract
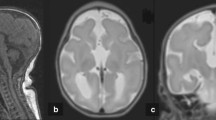
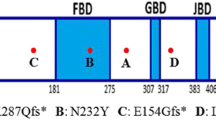
The Forkhead box G1 (FOXG1) is a transcription factor that is critical for forebrain development, where it promotes progenitor proliferation and suppresses premature neurogenesis. Recently, the FOXG1 gene was implicated in the molecular aetiology of the congenital variant of Rett syndrome. So far, 15 FOXG1 molecular alterations, including only eight point mutations, have been reported. We screened the FOXG1 gene in a cohort of 206 MECP2 and CDKL5 mutation negative patients (136 females and 70 males) with severe encephalopathy and microcephaly. The screening was negative in all males, but two de novo mutations (c.1248C>G, p.Y416X and c.460_461dupG, p.E154GfsX300) were identified in two unrelated girls. Both patients showed neurological symptoms from the neonatal period with poor reactivity, hypotonia, and severe microcephaly. During the first year of life, both patients had feeding difficulties and made slow developmental progress. At 5 years old, the girls were significantly neurologically impaired with gross hypotonia, no language, convergent strabismus, and no voluntary hand use. Moreover, they presented a combination of jerky movements, hand-mouthing, and hand-washing stereotypies. Hence, FOXG1 mutation patients demonstrate severe encephalopathy compatible with the congenital variant, as well as additional features such as absent eye contact, inconsolable crying during the perinatal period, and delayed myelination with thin to hypoplastic corpus callosum. Although the overall frequency of mutations in FOXG1 in females with severe mental retardation and microcephaly appears to be low (1.5%), our findings suggest the requirement to investigate both point mutations and gene dosage in the FOXG1 gene in patients with severe encephalopathy with microcephaly and some Rett-like features.



Similar content being viewed by others

References
Shoichet SA, Kunde SA, Viertel P et al (2005) Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet 117:536–544
Bisgaard AM, Kirchhoff M, Tumer Z et al (2006) Additional chromosomal abnormalities in patients with a previously detected abnormal karyotype, mental retardation, and dysmorphic features. Am J Med Genet 140:2180–2187
Papa FT, Mencarelli MA, Caselli R et al (2008) A 3 Mb deletion in 14q12 causes severe mental retardation, mild facial dysmorphisms and Rett-like features. Am J Med Genet 146A:1994–1998
Mencarelli MA, Kleefstra T, Katzaki E et al (2009) 14q12 microdeletion syndrome and congenital variant of Rett syndrome. Eur J Med Genet 52:148–152
Ariani F, Hayek G, Rondinella D et al (2008) FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 83:89–93
Huppke P, Laccone F, Kramer N et al (2000) Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Hum Mol Genet 9:1369–1375
Monros E, Armstrong J, Aibar E et al (2001) Rett syndrome in Spain: mutation analysis and clinical correlations. Brain Dev 23(Suppl 1):S251–S253
Smeets E, Schollen E, Moog U et al (2003) Rett syndrome in adolescent and adult females: clinical and molecular genetic findings. Am J Med Genet 122A:227–233
Jacob FD, Ramaswamy V, Andersen J, Bolduc FV (2009) Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet (in press)
Mencarelli M, Spanhol-Rosseto A, Artuso R et al (2009) Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet (in press)
Philippe C, Amsallem D, Francannet C et al (2009) Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. J Med Genet (in press)
Temudo T, Oliveira P, Santos M et al (2007) Stereotypies in Rett syndrome: analysis of 83 patients with and without detected MECP2 mutations. Neurology 68:1183–1187
Bahi-Buisson N, Kaminska A, Boddaert N et al (2008) The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia 49:1027–1037
Bahi-Buisson N, Nectoux J, Rosas-Vargas H et al (2008) Key clinical features to identify girls with CDKL5 mutations. Brain 131:2647–2661
Hagberg B, Skjeldal OH (1994) Rett variants: a suggested model for inclusion criteria. Pediatr Neurol 11:5–11
Hagberg B (1995) Clinical delineation of Rett syndrome variants. Neuropediatrics 26:62
Acknowledgements
We thank the patients, clinicians, and families who helped the study. This work was supported by the Institut National de la Santé et de la Recherche Médicale (ANR e-RARE EURORett).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Video electroencephalogram showing abnormal dyskinetic and non-epileptic movement disorder. (WMV 1857 kb)
Rights and permissions
About this article
Cite this article
Bahi-Buisson, N., Nectoux, J., Girard, B. et al. Revisiting the phenotype associated with FOXG1 mutations: two novel cases of congenital Rett variant. Neurogenetics 11, 241–249 (2010). https://doi.org/10.1007/s10048-009-0220-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-009-0220-2