
Abstract
A capacitive sensor based on S-{12-[1-(pyridin-4-ylmethyl)-1H-1,2,3-triazol-4-yl]dodecyl} ethanethioate (FT), a compound with a functional group exhibiting selective affinity towards Cr(VI) ions, was developed. FT was mixed with shorter-chain thiol-decanethiol (DT), to obtain an Au electrode surface well covered by a thiol monolayer. The composition and high quality of self-assembled monolayers (SAMs) were crucial factors influencing the performance of the capacitive sensor. In this work, SAMs formed from FT and DT mixtures with different compositions were studied. For physicochemical characterization of SAMs X-ray photoelectron spectroscopy (XPS), contact angle measurements as well as atomic force microscopy (AFM) were used. Cyclic voltammetry was employed to estimate an electrode surface coverage. Based on the obtained results, the composition of thiol layer providing the best parameters for capacitive sensing of chromium(VI) was chosen. Moreover, the analytical performance of sensor was verified.
Similar content being viewed by others

Introduction
The ability of sensitive detection is of crucial importance in many areas of human life. Sensors of new generation seem to be ideal analytical tools for that purpose not only because of their highly sensitivity, but also due to their simplicity and minimal requirements for a sample pretreatment. Ultrasensitive sensors designated for medical diagnosis, food analysis, environmental analysis, and monitoring of air quality have been reported. In the trend of ultrasensitive detection, electrochemical methods are often used. The indicator-free DNA hybridization sensor based on graphene nanocomposite as the enhanced sensing platform for detection breakpoint cluster region gene and the cellular abl (BCR/ABL) [1] and non-enzymatic glassy carbon sensor modified with gold nanoparticles (GNPs) onto conductive polyaniline (PANI) for detection of glucose [2] can be given as examples. Electrochemical methods can be also adopted for the quantitative detection of medicament-diclofenac with the use of aptasensor based on bimetallic AgPt nanoparticle–decorated carbon nanotubes [3] or to sensitive impedimetric determination of melamine with molecularly imprinted polymer-based electrode [4].
Among electrochemical sensors, capacitive sensors can be distinguished. They are commonly known in two configurations: parallel-plate or interdigitated electrode (IDE) with various sensing layers [5]. They can be used in different areas of analysis. Zeinali et al. [5] and Homayoonia et al. [6] reported capacitive sensors designated for gas analysis. In both cases MOF was used as a component of capacitive sensor and volatile organic compounds (VOCs) were the analytes. A miniaturized sensor based on an interdigital capacitor (as the transducer) and a dual-imprinted titania-polyaniline composite film (as the receptor) for instant monitoring of ethanol in gasohol fuel blends were proposed by Irshad et al. [7]. For detection of 4-nitrophenol, manganese dioxide/reduced graphene oxide nanocomposite paper was fabricated for supercapacitor preparation [8]. Using the interdigitated electrode capacitors, glucose, based on solvatochromic-dye-containing sensing membrane [9], and foodborne pathogenic Escherichia coli [10] were detected. A supercapacitor-based biosensor for low lipoprotein detection was also proposed [11]. For this purpose, graphene oxide was functionalized with hyperbranched polyethylenimine and incorporated into chitosan to obtain a nanocomposite film on a Pt disk electrode.
Capacitive sensors are a different group of electrochemical sensors whose operation is based on the electrostriction phenomenon. The electrostriction, described in the literature in 1960s [12], consists in a change of dimensions of materials, influenced by electrical field, and is specific for dielectrics. This phenomenon is especially noticeable in nanomembranes, e.g., self-assembled monolayers (SAM) [13]. Applying to these membrane potential lower than 1 V causes an appearance of strong electrical field of 105–106 V cm−1 in intensity. Electrostatic forces change the dimensions of the dielectric. In this case, the membrane’s compression, manifested by a decrease in its thickness, occurs. As a result, an increase in the membrane capacitance is observed.
The electrostriction phenomenon is often found in layers formed by long-chain organic compounds organized on a surface of solid electrode being in contact with an electrolyte solution [13, 14]. A gold electrode covered with a self-assembled monolayer of long-chain thiols can be considered as a capacitor whose covers are the electrode surface from one side and the electrolyte (analyte) solution from the other side (Fig. 1). Functional groups of thiols exposed to the solution participate in different interactions with an analyte based on adsorption, association, formation of hydrogen or coordination bonds, redox reactions, or antigen-antibody interactions. The presence of analyte initiates phenomena on the membrane/solution phase boundary influencing the electrostriction of the double layer and affects the capacitance-potential characteristics. The measurement of capacitance changes can be used to determine different types of analytes.

Scheme of membrane-covered electrode in contact with an electrolyte solution
The crucial factors influencing the behavior of capacitive sensor are composition and quality of thiol SAM. To achieve the best analytical parameters of developed sensors, the self-assembled monolayer of thiols should expose the biggest number of functional groups to the analyte solution simultaneously providing the favorable wetting properties. Furthermore, the well-ordered thiol film enables the repeatable occurrence of electrostriction phenomenon beneficially influencing the sensor functioning.
Self-organizing layers have found applications in many areas of nanotechnology, molecular electronics, and (bio)sensors because of the possibility of changing the surface. Recently, modification of electrodes by self-assembled monolayers has become very attractive due to simplicity and quickness of this approach [15]. Well-defined self-assembled monolayer of thiols is formed as a result of strong chemisorption between metal surface (usually gold) and sulfur atoms of thiols [16]. SAMs of alkanethiols on the gold surface, Au(111), are the most commonly studied because of their well-packed structure, stability, and especially flexibility in design of the chain length [17]. For this reason, electrochemical and surface studies of different SAMs [18], blocking behavior [19], potential dependence of the kinetics of thiol self-assembling on gold [20] or impact of 1-decanethiol formation and removal cycles on the surface properties of polycrystalline Au and SAM quality [21] were reported. Moreover, these structures have found many applications in electrochemistry as sensors [22, 23] and biosensor platforms [24], in catalysis [25] and surface science [26]. The main advantage of using SAM is the possibility of fabricating and modifying the functional, active groups of thiols according to their purposes.
The present work deals with a capacitive sensor for Cr(VI) determination [27]. Chromium is believed to be one of the most common pollutants at hazardous waste sites. Chromium exists in the environment in the form of trivalent Cr(III) and hexavalent Cr(VI). The first one is considered to be essential for the maintenance of glucose, protein, and lipid human metabolism. The second form, Cr(VI), has an undesirable effect on the lungs, liver, and kidneys. The examples of electrochemical chromium(VI) determination include the use of a glassy carbon electrode modified with silicomolybdate doped polypyrrole film [28], the preparation of hybrid and electroactive thin films containing microcrystalline cellulose (MC), or MC modified with a phosphate group (PC) in conjunction with polyaniline (PANI) and poly(vinyl sulfonic acid) (PVS) by the layer-by-layer technique [29] or determination in estuarine waters by catalytic cathodic stripping voltammetry using a vibrating silver amalgam microwire electrode [30]. The determination of Cr(VI) employing the screen-printed carbon electrode (SPCE) modified with the use of organic compound (PANI) and graphene quantum dot [31] or inorganic nanoparticles like AuNPs [32] have been reported.
The presented capacitive sensor was formed by S-{12-[1-(pyridin-4-ylmethyl)-1H-1,2,3-triazol-4-yl]dodecyl} ethanethiol, with the terminal groups exhibiting the affinity towards CrO42− and Cr2O72− anions, assembled on a gold electrode surface [33]. Because of steric hindrance, attributed to the relatively large functional groups, to obtain the well-ordered thiol monolayer, which in turn minimizes non-specific adsorption of analyte, shorter-chain thiol-decanethiol (DT) was used as an additional component of thiolating solution [34, 35]. The main goal of the study was the characterization of thiol layers of various compositions supported on the gold surface. X-ray photoelectron spectroscopy (XPS) and contact angle measurements as well as atomic force microscopy (AFM) techniques were employed. An electrode surface coverage with the layer of thiols was calculated using cyclic voltammetric measurements. The research undertaken also dealt with optimization of measuring procedure and its influence on analytical performance of the sensor.
Experimental
Chemicals
All chemicals, unless mentioned otherwise, were analytical-grade reagents. Potassium dichromate(VI), K2Cr2O7 (> 99.5%), ethanol (EtOH), nitric acid, potassium chloride, sodium nitrate, and potassium hexacyanoferrate(III) were purchased from Avantor (Poland). 1-Decanethiol from Alfa Aesar (USA), sulfuric acid (95–97%) from Merck (Germany), acetone from Chempur (Poland), potassium hydroxide from Centro-chem (Poland), and 0.3 μm alumina powder, used for polishing working electrodes surface from Buehler Micropolish (USA), were supplied. Water solutions were prepared using ultrapure water (conductivity < 0.5 μS cm−1) from an HLP 5 system (Hydrolab, Poland), and EtOH/H2O mixture (1:1, v/v) was used for preparation of 0.2 mmol L−1 both decanethiol (DT) and functionalized thiol (FT) solutions.
1-Decanethiol 99% (DT) was purchased from Aldrich (USA), while S-{12-[1-(pyridin-4-ylmethyl)-1H-1,2,3-triazol-4-yl]dodecyl} ethane-thiol (FT) was synthesized according to the procedure described in Supporting information. The structure of FT was confirmed by NMR and ATR-IR spectroscopy as well as elemental analysis.
Sensor preparation
Prior to formation of thiol monolayers, a gold electrode was cleaned by mechanical polishing with alumina slurry followed by cyclic voltammetry in 0.5 mol L−1 KOH and 0.5 mol L−1 H2SO4 solutions, in potential between of − 0.2 to 1.1 V and − 0.3 to 1.5 V vs. Ag/AgCl reference electrode, respectively. The measurements were carried out until repeatable cyclic voltammograms were obtained. Solution of K3[Fe(CN)6] (5 mmol L−1 with 20 mmol L−1 NaNO3) was used for verifying cleanliness of an electrode surface. A pair of well-defined redox peaks correlated to Fe(III) + e− ↔ Fe(II) reaction manifested a desired result of cleaning. The efficiency of the processes was evaluated based on peak potential separation, ΔEp [36]. The smaller was the ΔEp value and the closer to theoretical one (for one-electron process 0.058 V), the cleaner was the gold electrode surface. In our experiments, the value of ΔEp in the range of 0.07–0.08 V was accepted.
Self-assembled monolayers of thiols were spontaneously formed on a gold electrode surface by immersing the electrode in a thiol solution at 30 °C for 30 min, and subsequently at ambient temperature (ca. 20 °C) for 90 min. Then, the electrode was rinsed with a large amount of EtOH/H2O (1:1, v/v) solution. The prepared sensors were stored in the EtOH/H2O (1:1, v/v) solution and carefully rinsed with the EtOH/H2O (1:1, v/v) solution before capacitive measurements. Because of steric hindrance, attributed to relatively large functional groups, to obtain a high structural quality of the thiol membrane, FT was mixed with shorter-chain DT. In the thiolation process, the mixtures obtained by mixing of 0.2 mmol L−1 solutions of both thiols at various FT/(FT + DT) volume ratios (0.00, 0.05, 0.15, 0.25, 0.33, 0.50, 0.67, and 1.00) were examined.
Surface characterization
For surface investigation, gold layers with thickness of ca. 150 nm were evaporated onto crystal silicon wafers (ITME, Warsaw) consolidated with a 5-nm chromium adhesion layer. Evaporations were carried out in an oxygen-free glovebox (MBRAUN, Germany). SAMs of thiols were formed by immersing the freshly prepared wafers in the thiol mixture of different composition at ambient temperature for 24 h. Before measurements, the wafers were brought out from the thiol solution and carefully rinsed with EtOH/H2O (1:1, v/v). Before the AFM experiments, they were additionally dried using nitrogen.
Contact angle measurements were carried out at room temperature using a Standard Contact Angle Goniometer ramé-Hart Model 2000 (ramé-hart instrument co., USA). They were performed to study hydrophobicity of SAMs supported onto an evaporated gold layer. One microliter of deionized water was put on a gold surface covered by SAMs and a needle tip was in contact with a drop. For each SAM, the measurements were repeated five times.
X-ray photoelectron spectroscopy measurements were performed with a Prevac system equipped with a hemispherical analyzer VG SCIENTA R3000 (Scienta Omicron, Sweden). The spectra were taken using a monochromatized aluminum source AlKα (Eb = 1486.6 eV). The base pressure in the analytical chamber was about 5 × 10−9 mbar. The binding energy scale was calibrated using the Au 4f7/2 line of a cleaned gold sample at 84.0 eV. The surface composition was analyzed taking into account the areas and binding energies of Au 4f, C 1s, N 1s, S 2p, and O 1s. The spectra were fitted using the Casa XPS software.
Atomic force microscopy images were obtained with a Dimension Icon+ atomic force microscope (Bruker, Germany) working in air in PeakForce Tapping mode. Silicon nitride cantilevers (Bruker, Germany) with nominal normal spring constant of 0.58 N m−1 were used for all the measurements.
Electrochemical measurements
All electrochemical experiments were carried out at room temperature in a three-electrode arrangement consisting of a working gold electrode covered by SAM, a platinum counter electrode, and a Ag/AgCl (3 mol L−1) reference electrode. The gold electrodes were made from a 1.00-mm-diameter rod of pure gold (99.99%, Mint of Poland) placed into a Teflon holder. Both, measurement KSP-2 module, composed of a galvanostat, capacitance meter CMTR-243 and A/C-C/A converters, and the software used to control measurements, acquisition, and analysis of the capacitance-potential characteristics, were designed and produced by KSP Elektronika Laboratoryjna (Poland).
Capacitance measurements were performed using the capacitance-to-frequency conversion method [13, 37, 38]. In this approach, the investigated capacitor, an electrode covered with SAM of functionalized thiols, being in contact with analyte solution, is periodically charged and discharged by a current in a defined range of potential, ΔU, and the current is constant in its absolute value, but with alternating sign. The voltage change over the time is triangular as is presented in Fig. 2.

Cyclic changes of the capacitor voltage during measurements
While the capacitor is charged from the voltage U+ to U−, the voltage changing rate, V, is influenced by the current, i, and the capacitance, C, according to the equation:
Owing to the fact, that the current value is the same in both directions of voltage change, charging of the capacitor from the voltage U+ to U− lasts the same as in opposite direction, from U− to U+ and can be expressed by the equation:
Since the total period of the resulting signal, T, is given as T = T+ + T−, it is equal to
As a consequence, the capacitance can be described as follows
The method employed for measurement enabled recording of capacitive characteristics in the form of capacitance-potential curves. The measurements were carried out in the potential range from − 200 to 800 mV with scan rate of 50 mV s−1 and potential step of 2 mV. Every day, before capacitance experiments, the measurement device was calibrated using a 10.00 nF capacitor. For each sensor, the capacitance measurements in solution of supporting electrolyte (0.1 mol L−1 NaNO3) were performed. After addition of a proper volume of the sample solution (or analyte standard solution), the mixture was stirred for 1 min using a magnetic stirrer MS 11HS (Wigo, Poland) and then measuring cycles were done registering capacitance-potential curves.
Results and Discussion
Electrochemical characterization of SAMs
Assuming that redox reactions can take place only on parts of electrode surface uncovered by thiols and that diffusion to bare surface spots is planar, characterization of SAMs can be done using fractional coverage of the monolayer on a gold surface, ΘiCV estimated from cyclic voltammetry measurements [39]:
where \( {i}_{\mathrm{p}}^{\mathrm{bare}} \) is a peak current recorded on the bare gold electrode in a K3[Fe(CN)6] solution, and \( {i}_{\mathrm{p}}^{\mathrm{SAM}} \) is a respective peak current recorded on the electrode covered by thiol at the same measuring conditions. Based on the CV measurements, carried out using the gold electrodes covered with thiol monolayers of varying composition, the ΘiCV values were calculated (see Table 1). It can be found that the best electrode surface coverage was achieved at the FT/(FT + DT) ratio of 0.67 (FT:DT ratio 2:1, v/v).
Physicochemical characterization of SAMs
X-ray photoelectron spectroscopy was used to follow the composition of the thiol monolayer formed on the Au electrode. Apart from the Au 4f photoemission observed as a doublet at 84.0 eV (Au 4f7/2) and 87.7 eV (Au 4f5/2) with spin-orbit splitting of 3.7 eV, the peaks related to C 1s, N 1s, S 2p, and O 1s are found in the recorded survey XPS spectra. The presence of oxygen impurities can be attributed to partially oxidized electrode surface and other contaminants (such as unremoved solvents) existing in the thiol layer. The remaining elements correspond mainly to thiols (FT and DT) introduced onto the Au surface. The determined amounts of the components of SAMs are listed in Table 1. Obviously, the packing density of the thiol layer on the Au electrode is very similar regardless to the composition of the thiolating mixture, and the S/Au atomic ratio of 0.05–0.06 was determined for all the studied membranes.
The S 2p spectra have a doublet structure due to the presence of the S 2p3/2 and S 2p1/2 peaks. Their positions at 162.0 ± 0.1 eV and 163.3 ± 0.2 eV suggest that sulfur exists exclusively in the thiol groups chemically bonded to the Au surface, and no significant amounts of free or oxidized thiols are identified [40]. Furthermore, the N and S contents were used to calculate the composition of the thiol layers formed at the various thiolating mixtures. In Fig. 3, % at. of FT in SAMs determined as
is shown. As can be found, at lower FT/(FT + DT) ratios in the thiolating mixture, FT is predominantly attached to the metal electrode. An increase in the FT content during the thiolating process results in obtaining the monolayer characterized by the chemical compositions very close to the intended ones.

Composition of thiol monolayer determined by XPS
The wetting behavior of the Au surface after modification with the thiol monolayer was studied by contact angle measurements. Examples of images captured for the thiol mixtures of different composition are shown in Fig. 4. The determined values of static contact angle (Table 1) clearly decrease with raising the FT content due to its higher hydrophilicity compared to DT. It should be kept in mind that both thiols differ in the length of hydrocarbon chain and the layer is formed mainly by the functional groups of FT. It is not therefore surprising that this surface becomes hydrophilic even at the low contents of FT in the formed membrane.

Examples of contact angle images of DT monolayer (a), DT:FT monolayer at volume ratio 0.50 (b) and FT monolayer (c)
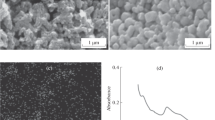
The coating of Au electrode with the thiol monolayer was also controlled by atomic force microscopy. In Fig. 5, an example of AFM image taken for the bare gold and gold covered with mixture of FT/(FT + DT) at molar ratio of 0.67 is shown. The obtained layer is continuous, remarkably smooth, and densely uniform (compared with bare Au surface), without any clear faults. For other compositions of thiol films, similar AFM pictures were collected.

AFM images of uncovered Au surface (a) and of thiol monolayer formed on Au electrode in solution containing FT/(FT + DT) molar ratio of 0.67 (b)
Verification of sensor functioning
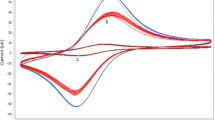
Based on the results of electrochemical and physicochemical studies performed, for further experiments, the mixture of FT/(FT + DT) at molar ratio of 0.67 was chosen for the sensor construction. For a series of prepared electrodes, similar in shape capacitance-potential curves usually with hysteresis were recorded, in both, supporting electrolyte and analyte solutions (see Fig. 6). The fluctuation of capacitive characteristic pattern and the level of measured capacitance were random, which could be clarified by unrepeatable assembling of thiols on the gold electrode surface being an effect of non-ideal smoothness of gold surface after the electrode preparing process.

Example of capacitance-potential curves recorded for developed sensors in supporting electrolyte (dotted line) and Cr(VI) solutions of increasing concentrations from 1 to 10 nmol·L−1, scan rate 20 mV·s−1
The important issue of proposed approach was the way of the analytical signal readout. From Fig. 6, it is seen that, with increasing concentration of Cr(VI), up to 500 nmol L−1, capacitance characteristics moved along the capacitance axis. It was noticed that in nearly all cases, the most significant signal changes were revealed at potentials of 0.70 V, accordingly that potential was selected for the analytical signal readout.
To improve the analytical parameters of proposed sensor, consideration about experiments performed in the solution of supporting electrolyte was taken. The most often employed procedure of electrochemical measurements includes recording signal in supporting electrolyte until its constant value is reached. During capacitive experiments it was observed that the biggest difference in capacitance-potential curves registered for supporting electrolyte occurred during the first three measuring cycles. Simultaneously, it was revealed that the sensor sensitivity decreased along with the increase in the number of measurements carried out in solution of supporting electrolyte It is likely that the thiol monolayer, after being exposed many times to variable electromagnetic field, was less flexible and, as results, electrostriction of SAMs was weaker. For that reason, it was decided that five measuring cycles in supporting electrolyte (0.1 mol L−1 NaNO3) for each sensor will be carried out, assuming that the further changes in the course of capacitance-potential curves could be neglected. After an introduction of each volume of standard Cr(VI) solution and mixture stirring, five consecutive capacitance-potential characteristics were recorded. Since, starting from the third measuring cycle, the curves overlapped; therefore, the fourth cycle was used for analytical purposes.
In order to verify if the modified procedure of capacitance measurements enables the determination of chromium(VI) at lower concentrations, the measurements were performed in three different Cr(VI) concentration ranges: up to 10, 50, and 500 nmol L−1. The following linear relationships of the capacity versus concentration (C–c) were obtained, as mean values for three sensors for each concentration range:
(standard deviations were calculated based on the law of propagation of uncertainty). Comparing the calibration relationships obtained for different concentration ranges, it was realized that the highest sensor sensitivity was received for the lowest analyte concentration, up to 10 nmol L−1, 119.4 nF cm−2 nmol−1 L1, whereas 13.0 and 7.0 nF·cm−2·nmol−1·L for the Cr(VI) concentrations up to 50 and 500 nmol L−1, respectively. In comparison to the results reported previously [27], it was noticed that reducing the number of measuring cycles recorded in supporting electrolyte allowed to increase the sensor sensitivity, its value corresponding to the highest studied Cr(VI) concentration range (up to 500 nmol L−1) increased by more than 50%, from 4.6 to 7.0 nF cm−2 nmol−1 L1 (0.036 and 0.0557 nF nmol−1 L1, respectively).
Due to fluctuations of the capacitive characteristics observed for various sensors, analytical signals for a sample and standard solutions should be measured utilizing the same sensor and in the same solution of a supporting electrolyte. Furthermore, measurements for growing analyte concentrations were necessary. Considering the above, the adequate calibration approach was applied—the modified standard addition method, called signal increment extrapolative calibration method [27, 41]. Calibration procedure was consisted into few steps. At the beginning, the one addition of the standard analyte was introduced into supporting electrolyte and the analytical signal was measured (R1) (see Fig. 7a). In the next step, the known volume of the sample solution with unknown concentration of the analyte was added and new analytical signal was recorded (R2). Afterwards, analytical signals (R3 ÷ R5) were measured for subsequent three standard additions of known volume and concentration. As a result, based on signals (R2 ÷ R5), the calibration graph could be created, and the concentration of the unknown sample could be calculated by extrapolation of the calibration plot to the signal of the first standard solution (R1).

The signal increment extrapolative calibration–general approach (a); exemplary calibration graphs received for three 2 nmol L−1 Cr(VI) synthetic solutions (b)
The developed methodology was verified in a synthetic solution containing different amounts of Cr(VI) within the lowest examined concentration range. The analyses of synthetic solutions were performed at four different concentrations. The results of determinations are presented in Table 2. Exemplary calibration graphs obtained for 2 nmol·L−1 chromium(VI) solutions according to the calibration method used are shown in Fig. 7b. The data in Table 2 reveal that relative errors are lower than 7%, even at very low chromium(VI) concentrations (1 and 2 nmol·L−1); therefore, the accuracy of the sensor can be regarded as satisfactory. The RSD values do not exceed 6%, indicating acceptable precision of the method.
Based on described-above calibration approach, the limit of detection was estimated. For this purpose, the formula LOD = tα, f ∙ s was employed, where tα, f was the critical Student’s t value read for the significant level α = 0.1 and f degrees of freedom for n measurements (f = n − 1), and s was the standard deviation of the obtained results [42]. The calculation performed based on ten analyses carried out for 1 nmol·L−1 Cr(VI) standard solutions gave the LOD value of 0.31 nmol·L−1. Reproducibility of sensor operation was estimated as 3.5% (RSD%) for the three independently prepared sensors employing for Cr(VI) determination in solution with concentration of 5 nmol·L−1. Repeatability obtained for three analyses carried out with the use of one sensor gave an RSD value of 9.1%.
Comparing the presented sensor to other electrochemical sensors designated for Cr(VI) determination (see Table 3), it can be noticed that in this case, linear response range was better than obtained for the sensors based on hybrid films of phosphate cellulose and electroactive polymer [29], polyaniline/graphene quantum dot–modified screen-printed carbon electrode [31] and screen-printed carbon electrode modified with gold nanoparticles [32]. The developed sensor exhibited lower limit of detection in comparison with all reported sensors except for vibrating silver amalgam microwire electrode demonstrating similar LOD values [30].
Conclusion
The electrochemical sensor of new generation for Cr(VI) detection, based on the electrostriction phenomenon of a thiol monolayer covalently bound on a gold electrode, was presented. To characterize the surface of thiol layers embedded on gold surface, electrochemical (CV) and physicochemical methods (XPS, AFM, and contact angle measurements) were employed. Based on the CV measurements, it was found that the best surface coverage of the gold electrode was obtained using the mixture of thiols at the FT/(FT + DT) ratio of 0.67. The contact angle measurements confirmed that hydrophilicity of the monolayer clearly increases due to the growth of FT content in comparison to DT. It can be noticed that even in the case of using two thiols with different lengths of hydrocarbon chain at different proportions (FT and DT), the external surface of SAMs is mainly formed by the terminal functional groups of FT. Therefore, the electrode surface becomes hydrophilic even at the low content of FT in the formed monolayer. According to the XPS measurements, the packing density of the thiol monolayer on the Au electrode is very similar regardless of the composition of the thiolating mixture, and the S/Au atomic ratio of 0.05–0.06 was determined for all the studied thiol films. An increase in the FT content during the thiolating process results in obtaining the membranes characterized by the chemical compositions very close to the intended ones.
Our results show that the modification of measuring procedure beneficially influenced the sensor performance. The experiments carried out demonstrated that reducing the number of measuring cycles recorded in supporting electrolyte allowed to increase the sensor sensitivity and made possible determination of Cr(VI) at lower concentration levels, in nanomolar range, in comparison with those reported formerly [27].
References
Yang J, Zhang W (2014) Indicator-free impedimetric detection of BCR/ABL fusion gene based on ordered FePt nanoparticle-decorated electrochemically reduced graphene oxide. J Solid State Electrochem 18(10):2863–2868
Ahammad AJS, Mamun AA, Akter T, Mamun MA, Faraezi S, Monira FZ (2016) Enzyme-free impedimetric glucose sensor based on gold nanoparticles/polyaniline composite film. J Solid State Electrochem 20(7):1933–1939
Shiravand T, Azadbakht A (2017) Impedimetric biosensor based on bimetallic AgPt nanoparticle-decorated carbon nanotubes as highly conductive film surface. J Solid State Electrochem 21(6):1699–1711
Shamsipur M, Moradi N, Pashabadi A (2018) Coupled electrochemical-chemical procedure used in construction of molecularly imprinted polymer-based electrode: a highly sensitive impedimetric melamine sensor. J Solid State Electrochem 22(1):169–180
Zeinali S, Homayoonnia S, Homayoonnia G (2019) Comparative investigation of interdigitated and parallel-plate capacitive gas sensors based on Cu-BTC nanoparticles for selective detection of polar and apolar VOCs indoors. Sensor Actuat B-Chem 278:153–164
Homayoonia S, Zeinali S (2016) Design and fabrication of capacitor nanosensor based on MOF nanoparticles as sensing for VOCs detection. Sensor Actuat B-Chem 237:776–786
Irshad M, Mujahid A, Afzal A, Bajwa SZ, Hussain T, Zaman W, Latife U, Makshoof Athar M (2018) A miniaturized electronic sensor for instant monitoring of ethanol in gasohol fuel blends. RSC Adv 8(41):22952–22962
Haldorai Y, Giribabu K, Hwang SK, Kwak CH, Huh YS, Han YK (2016) Facile synthesis of α-MnO2 nanorod/graphene nanocomposite paper electrodes using a 3D precursor for supercapacitors and sensing platform to detect 4-nitrophenol. Electrochim Acta 222:717–727
Khan MR, Khalilian A, Kang SW (2016) Fast, highly-sensitive, and wide-dynamic-range interdigitated capacitor glucose biosensor using solvatochromic dye-containing sensing membrane. Sensor 16(2):265–278
Pandey A, Gurbuz Y, Ozguz V, Niazi JH, Qureshi A (2017) Graphene-interfaced electrical biosensor for label-free and sensitive detection of foodborne pathogenic E. coli O157:H7. Biosens Bioelectron 91:225–231
Rodriguez-Silva AA, Movil-Cabrera O, Oliveira dos Anjos CT, Staser JA (2016) Supercapacitor-Based biosensor for low density lipoprotein detection. J Electrochem Soc 163:256–263
Babakov AV, Ermishkin LN, Lieberman EA (1966) Influence of electric field on the capacity of phospholipid membranes. Nature 210(5039):953–955
Kalinowski S (2005) Electrochemical methods and their application. Adv Planar Lipid Bilayers Liposomes 2(C):1–47
Kilian J, Kalinowski S, Kochana J, Knihnicki P (2012) A new capacitive sensor based on electrostriction phenomenon. Application for determination of ionic surfactants. Procedia Eng 47:1338–1341
Schneider S, Partes C, Wiesner A, Terfort A (2018) Potential-induced phase transition of benzoxazole-2-thiol, naphthaleneoxazole-2-thiol and anthraceneoxazole-2-thiol monolayers on gold electrodes. Electrochim Acta 283:167–173
Shamsipur M, Kazemi SH, Mehdinia A, Mousavi MF, Sharghi H (2008) Electron transfer behavior through densely packed self-assembled monolayers of a novel heteroaromatic thiol derivative onto the gold surface. Electroanal 20(5):513–519
Mandler D, Kraus-Ophir S (2011) Self-assembled monolayers (SAMs) for electrochemical sensing. J Solid State Electrochem 15(7-8):1535–1558
Scharf J, Strehblow HH, Zeysing B, Terfort A (2001) Electrochemical and surface analytical studies of self-assembled monolayers of three aromatic thiols on old electrodes. J Solid State Electrochem 5(6):396–401
Shen H, Mark JE, Seliskar CJ, Mark HB Jr, Heineman WR (1997) Blocking behavior of self-assembled monolayers on gold electrodes. J Solid State Electrochem 1(2):148–154
Rohwerder M, de Weldige K, Stratmann M (1998) Potential dependence of the kinetics of thiol self-organization on Au(111). J Solid State Electrochem 2(2):88–93
Thal D, Kahlert H, Chinnaya J, Ahrens P, Hasse U (2018) Impact of gold-1-decanethiol-SAM formation and removal cycles on the surface properties of polycrystalline gold and SAM quality. J Solid State Electrochem 22(4):1149–1154
Dey RS, Gupta S, Paira R, Chen SM, Raj CR (2012) Flow injection amperometric sensing of uric acid and ascorbic acid using the self-assembly of heterocyclic thiol on Au electrode. J Solid State Electrochem 16(1):173–178
Kang J, Zhuo L, Lu X, Wang X (2005) Electrochemical behavior of dopamine at a quercetin-SAM-modified gold electrode and analytical application. J Solid State Electrochem 9(2):114–120
Foubert A, Beloglazova NV, Hedström M, Saeger SDE (2019) Antibody immobilization strategy for the development of a capacitive immunosensor detecting zearalenone. Talanta 191:202–208
Hao P, Pylypenko S, Schwartz DK, Medlin JW (2016) Application of thiolate self-assembled monolayers in selective alcohol oxidation for suppression of Pd catalyst deactivation. J Catal 344:722–728
Venäläinen A, Meinander K, Räisänen M, Tuboltsev V, Räisänen J (2018) Metallization of self-assembled organic monolayer surfaces by Pd nanocluster deposition. Surf Sci 677:68–77
Wieczorek M, Kochana J, Knihnicki P, Wapiennik K, Kościelniak P (2017) Novel electroanalytical method based on the electrostriction phenomenon and its application to determination of Cr(VI) by the flow injection technique. Talanta 166:383–390
Balamurugan A, Chen SM (2007) Silicomolybdate doped polypyrrole film modified glassy carbon electrode for electrocatalytic reduction of Cr(VI). J Solid State Electrochem 11(12):1679–1687
Teixeira PRS, do Nascimento Marreiro AS, Farias EAO, Dionisio NA, Filho ECS, Eiras C (2015) Layer-by-layer hybrid films of phosphate cellulose and electroactive polymer as chromium(VI) sensors. J Solid State Electrochem 19(7):2129–2139
Espada-Bellido E, Bi Z, van den Berg CMG (2013) Determination of chromium in estuarine waters by catalytic cathodic stripping voltammetry using a vibrating silver amalgam microwire electrode. Talanta 105:287–291
Punrat E, Maksuk C, Chuanuwatanakula S, Wonsawat W, Chailapakul O (2016) Polyaniline/graphene quantum dot-modified screen-printed carbon electrode for the rapid determination of Cr(VI) using stopped-flow analysis coupled with voltammetric technique. Talanta 150:198–205
Tu J, Gan Y, Liang T, Wan H, Wang P (2018) A miniaturized electrochemical system for high sensitive determination of chromium(VI) by screen-printed carbon electrode with gold nanoparticles modification. Sensors Actuators B Chem 272:582–588
Turyan I, Mandler D (1997) Selective determination of Cr(VI) by a self-assembled monolayer-based electrode. Anal Chem 69(5):894–897
Huang L, Reekmans G, Saerens D, Friedt JM, Frederix F, Francis L, Muyldermans S, Campitelli A, Van Hoof C (2005) Prostate-specific antigen immunosensing based on mixed self-assembled monolayers, camel antibodies and colloidal gold enhanced sandwich assays. Biosens Bioelectron 21(3):483–490
Jambrec D, Gebala M, Mantia FL, Schuhmann W (2015) Potential-assisted DNA immobilization as a prerequisite for fast and controlled formation of DNA monolayers. Angew Chem Int Ed 54(50):15064–15068
Fischer LM, Tenje M, Heiskanen AR, Masuda N, Castillo JL, Bentien A, Emneus J, Jakobsen MH, Boisen A (2009) Gold cleaning methods for electrochemical detection applications. Microelectron Eng 86(4-6):1282–1285
Kalinowski S, Three-electrode system for measuring the electrical capacitance of the membrane deposited on the electrode (2014), Polish Patent No. PL218407B1
Kalinowski S, Figaszewski Z (1995) A four-electrode system for measurement of bilayer liquid membrane capacitance. Meas Sci Technol 6:1043–1049
Campuzano S, Pedrero M, Montemayor C, Fatás E, Pingarrón JM (2006) Characterization of alkanethiol-self-assembled monolayers-modified gold electrodes by electrochemical impedance spectroscopy. J Electroanal Chem 586(1):112–121
Castner DG, Hinds K, Grainger DW (1996) X-ray photoelectron spectroscopy sulfur 2p study of organic thiol and disulfide binding interactions with gold surfaces. Langmuir 12(21):5083–5086
Kościelniak P, Wieczorek M (2016) Univariate analytical calibration methods and procedures. A review. Anal Chim Acta 944:14–28
Geiß S, Einax JW (2001) Comparison of detection limits in environmental analysis – is it possible? An approach on quality assurance in the lower working range by verification. Fresenius J Anal Chem 370(6):673–678
Miscoria SA, Jacq C, Maeder T, Martín Negri R (2014) Screen-printed electrodes for electroanalytical sensing, of chromium VI in strong acid media. Sensor Actuat B-Chem 195:94–302
Acknowledgements
Szczepan Zapotoczny and Sylwia Dąbrowska are kindly acknowledged for taking AFM pictures and helping with the experimental part work, respectively.
Funding
The study was supported by the National Science Centre, Poland, Project 2013/11/B/ST4/00864. The research was partially carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kochana, J., Starzec, K., Wieczorek, M. et al. Study on self-assembled monolayer of functionalized thiol on gold electrode forming capacitive sensor for chromium(VI) determination. J Solid State Electrochem 23, 1463–1472 (2019). https://doi.org/10.1007/s10008-019-04236-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-019-04236-2