
Abstract
The reactivity of the 5-arylidenerhodanine and 5-arylideneisorhodanine derivatives in reactions with dimethyl maleate was computationally studied at the DFT(M06-2X)/6–311+G(d,p) theory level. Eight stereoisomers of the reaction products were considered. The effect of the solvent was taken into account by means of the continuous and discrete models for acetic acid (PCM and three CH3COOH molecules). Aromatic stabilization of the transition states was documented by the values of HOMA, NICS(0), and NICS(1) indices. The higher reactivity of the isorhodanine derivative was associated with a relatively low activation energy, ∆Ea (15.2–22.3 kcal mol–1), which is needed to cross the TS. For the rhodanine derivative, higher values of ∆Ea (34.1–36.1 kcal mol–1) were obtained. The reactivity was also studied from the perspective of the frontier molecular orbitals, the energy gaps between the HOMO and LUMO, the flux of electron density, the Fukui functions, f+(r), f−(r), and f0(r), and the global indexes defined in the conceptual DFT, i.e., the electronic chemical potential, chemical hardness, global electrophilicity, and empirical nucleophilicity index.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many researchers involved in the synthesis of organic compounds are focused on heterocyclic systems. These systems are studied because of their potential applications in pharmaceutical chemistry [1]. In the case of heterocyclic compounds, one of the synthesis methods is based on the Diels-Alder (DA) cycloaddition reaction. Formally, the official date of the discovery of the DA reaction is considered to be the year 1928, when a paper entitled “Synthesen in der hydroaromatischen Reihe” was published [2]. Since then, cycloaddition reactions have been widely used in organic synthesis [3]. They are used to obtain, inter alia, heterocyclic compounds with potential biological activity. One group of such compounds, exhibiting antitumor, anti-inflammatory, and antibacterial activity, are derivatives containing the pyrano[2,3-d]thiazole core [4].
Obtaining new compounds with therapeutic activity is a very important challenge for modern medical chemistry owing to the growing threat to public health. Important challenges include pathogenic microorganisms that acquire resistance to currently available drugs and pervasive neoplastic diseases. One of the reagents in the Diels-Alder reaction is the corresponding diene system. Very often, the 5-arylidene derivatives of 2-sulfanylidene-1,3-thiazolidine-4-one, commonly named rhodanine, act in this type of reaction [5]. Rhodanine, and its isomer, 4-sulfanylidene-1,3-thiazolidine-2-one, colloquially known as isorhodanine, have been studied for more than a century because of their interesting biological properties. The 5-arylidene derivatives of rhodanine and isorhodanine are shown in Scheme 1.

Diene moiety in the arylidene derivatives of rhodamine (Rd) and isorhodanine (IsRd)
It is intriguing that the diene system present in 5-arylidenerhodanine is rarely used for the synthesis of new heterocyclic compounds. Reactions occur only with very active dienophiles and their course depends on the nature and location of the groups present in the arylidene substituent. The electron-withdrawing group in the arylidene moiety facilitates the reaction [6]. On the other hand, the diene system present in 5-arylideneisorhodanine has significantly higher reactivity and is used much more frequently to carry out the DA-type cyclization reaction [7].
Our parallel experimental research indicates that the DA cyclization of 5-arylidenerhodanine derivatives with dienophiles, such as acrolein, maleic anhydride, dimethyl maleate, or acetylene dicarboxylic acid dimethyl ester, did not occur at all or occurred at very low yields. In contrast, cyclization reactions of 5-arylideneisorhodanine derivatives with the same dienophiles occurred with good or very good yields [8].
We decided to explain the difference in reactivity of both diene systems using computational methods. This study focused on the reaction of 5-arylidenerhodanine (rhodanine, Rd) and 5-arylideneisorhodanine (isorhodanine, IsRd) derivatives with dimethyl maleate (DMm), which were studied in both the gas phase and in solution.
Computational details
All calculations were carried out using the Gaussian 16 (G16) programme [9]. The exchange-correlation functional from the Minnesota group, M06-2X [10] and standard Pople basis set, 6–311+G(d,p) [11, 12] were used as encoded in G16. The calculations were performed for 0 K. Two different environments were considered, i.e., the gas phase and using acetic acid as a solvent. The PCM model (ε = 6.2528) was used for the solvent simulation [13, 14]. Optimized structures were examined by IR-frequency calculation, yielding no imaginary frequencies for the reagents and products, and one imaginary frequency for all structures of the transition states (TS).
The activation energy (∆Ea) was calculated as the difference between the total energies (Etot) of the structures of the TS and the optimized structures of the reagents. The values of Etot were corrected using the zero-point vibrational energy. The energy of the reaction (∆Er) was calculated as the difference of Etot with the zero-point vibrational energy correction between the products and reagents.
The values of the transferred charge, Δq, for TS were calculated with the natural bond orbital (NBO) method 3.1 [15] implemented in G16 as the difference between the natural charge populations of the atoms for the diene and the dienophile.
The condensed Fukui functions [16] were calculated with Bader’s interpretation of atoms in molecules [17] using the TopMod09 package [18]. The wavefunction files for the topological analysis of the electron density field were obtained by single-point calculations carried out for the optimized structures. The electron density was evaluated with a cubical grid with a step size of 0.05 Bohr.
The aromaticity of the rhodanine and isorhodanine rings was tested using the harmonic oscillator model of aromaticity (HOMA) index [19, 20]. This popular index, using a simple formula based on the length of bonds in the ring under examination, provides an estimate aromaticity in a simple and reliable way [21]. For a fully aromatic ring, the HOMA index value is 1.00, for a nonaromatic ring it is about 0.00, and negative values are characteristic for anti-aromatic systems. Aromaticity is considered as a multidimensional phenomenon [22]. Owing to the expected aromaticity of rhodanines and isorhodanines, the alternative method based on magnetic properties of molecule has been investigated. The nucleus independent chemical shift, NICS, is defined as a negative value of the chemical shift calculated in characteristic points of a molecule [23]. Two NICS sub-indices, NICS(0) — a chemical shift calculated in the center of the ring [23], and NICS(1) — a chemical shifts calculated one angstrom above the center of the ring [24], were used.
Graphical representations of the highest occupied molecular orbitals (HOMO) were prepared using the Chimera programme [25].
Results and discussion
The formal mechanism (see Scheme 2) assumes that DA product is formed during cycloaddition between a diene and a conjugated alkene (the dienophile). The three π-bonds break and form two single σ-bonds and one π-bond, regardless of substituents on diene + dienophile. The DA reaction is classified as a [4 + 2] cycloaddition due to orbital symmetry considerations. This is allowed by the interaction of the 4π electron system (the diene structure) with the 2π electron system (the dienophile structure).

The Lewis formula representing the chemical bonds in the substrates, TS, and the product of the reaction of 5-arylideneisorhodanine and 5-arylidenerhodanine derivatives with dimethyl maleate. The stereocenters are denoted by stars
Both investigated reactions are characterized by a single transition structure (TS) and begin with the formation of the pre-reaction complex. The comparison of the relative energies for both reactions is presented in Fig. 1. The product of the reaction has three asymmetric carbon atoms yielding eight stereoisomers. The most stable stereoisomers, denoted as 8 for IsRd and 5 for Rd, are shown in Fig. 2a and b. The relative energies (ΔEtot) of all stereoisomers are collected in Table 1, while the values of Etot for the substrates and products are presented in the supplementary materials (Table S1). The formation of each stereoisomer is related to the cis or trans form of the DMm. In the case of discussed stereoisomers 8 (IsRd) and 5 (Rd), the DMm reacts in the cis and trans forms, respectively. If not specifically marked only the most stable stereoisomers, 8 for IsRd and 5 for Rd, are used in the calculations and discussed in the text.

The comparison of the relative energies for the pre-reaction complex, the structure of the transition state, TS, and the product of the reaction of 5-arylidenerhodanine (Rd) and 5-arylideneisorhodanine (IsRd) derivatives with the dimethyl maleate (DMm). ∆Ea — the activation energy from the side of the substrates, ∆Er — the reaction energy. The value of ∆Ea has been calculated in relation to the energy of the pre-reaction complex. Each stereoisomer of the product, associated TS and the pre-reactive complex are denoted with the same color

The reactions start with the formation of the pre-reaction complexes IsRd...DMm and Rd...DMm, which are stabilized by noncovalent interactions. The values of the interaction energy (Eint), corrected by the difference of the zero-point vibrational energies (ΔZPVE) and the basis set superposition error (BSSE) removed using the counterpoise correction (EintCP) [26], are presented in Table 2. Depending on the considered stereoisomer, the values of EintCP + ΔZPVE ranged between −7.86 and −11.71 kcal mol–1 for the IsRd...DMm complexes and between −8.48 and −11.42 kcal mol–1 for the Rd...DMm complexes.
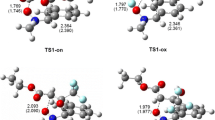
The most important aspect for understanding the reactivity of 5-arylidenerhodanine and 5-arylideneisorhodanine is the properties of the transition structures for the reacting molecules. The lengths of the C1–C3, S–C2 and C1–C3, O–C2 bonds for the optimized structures of TS for IsRd (Fig. 3a) and Rd (Fig. 3b) are shown in Table 3. The S–C2 and O–C2 bond length in the gas phase study of IsRd and Rd varied from 2.267–2.451 Å and 1.780–2.088 Å, respectively, i.e., in the typical range of sulfur–carbon and oxygen–carbon interactions [27]. In the presence of the solvent, the bond lengths were similar to those obtained in the gas phase, i.e., 2.284–2.448 Å and 1.822–2.065 Å, respectively.

a) The optimized structure of the transition state for the reaction of 5-arylideneisorhodanine (IsRd) with DMm, b) 5-arylidenerhodanine (Rd) derivative with dimethyl maleate (DMm). The ν1 value corresponds to the imaginary frequency for selected transition state
The values of the activation energy, ∆Ea, and the reaction energy, ∆Er, for both reactions are presented in Table 4. The value of ∆Ea was computed with respect to the Etot of the respective pre-reaction complex. In the case of the reaction of IsRd with DMm (gas phase), the value of ∆Ea ranged from 15.19 to 22.25 kcal mol–1, depending on the considered stereoisomer. The activation energy from the side of the product was significantly higher than that of ∆Ea; therefore, the product of the reaction was stabilized and the probability of the reaction occurring in both directions was small. The reaction energy, ∆Er, was negative for all stereoisomers and ranged from −9.78 to −27.40 kcal mol–1. To conclude this part, the cycloaddition of IsRd with DMm is an exothermic process in the gas phase and the formation of the product is favored.
In the presence of the solvent, the value of ∆Ea decreased to 15.20–20.40 kcal mol–1. The value of the reaction energy ranged between −10.60 and −27.82 kcal mol–1, and the values were more negative than those computed in the gas phase. Thus, the interaction with the solvent increased the probability of the reaction occurring and decreased the probability of the reverse process.
The analysis of the energetics of the reaction of Rd with DMm (gas phase) shows that the activation energy from the side of the reagents were similar to the values from the side of the products, i.e., 34.08–36.12 kcal mol–1. Thus, the probability of product formation and its decomposition into the reagents was comparable, and the reaction may be reversible. Furthermore, the values of ∆Ea were clearly larger than those obtained for the reaction of IsRd, so significantly more energy was needed to cross the TS. The values of ∆Er were in the range of −8.34 to 8.95 kcal mol–1 and for three stereoisomers, they were positive (see Table 4). Thus, formation of the product was less preferred than in the reaction of IsRd, for which the values of ∆Er were negative for all stereoisomers.
The application of the continuous model (PCM) of the solvent for the reaction of Rd with DMm resulted in a decrease in ∆Ea to 32.44–34.89 kcal mol–1 and an increase of the activation energy from the side of the products. Thus, the solvent exerted a stabilizing effect on product formation. However, a comparison of the energy activation barriers from both side shows that the values were rather similar with a maximal difference of 5.71 kcal mol–1. Thus, in solution, the reaction of Rd with DMm may also proceed in both directions. Unlike the results obtained for the gas phase, all values of ∆Er calculated using the solvent model were negative and ranged from −1.14 to −10.37 kcal mol–1. The interaction with the solvent increased the probability of product formation.
In order to further investigate the role of solvent molecules on the energetics of both reactions, three CH3COOH molecules were gradually added to the interacting reagents in the discrete model of the solvent (microsolvation). The calculations were carried out at the DFT(M06-2X)/6–311+G(d,p) computational level, with the PCM solvent model (acetic acid). The values of the ∆Ea and ∆Er are shown in Table 5. The optimized structures for the TS and the products of IsRd and Rd reacting with DMm in the presence of three CH3COOH molecules are shown in Fig. 4.

The optimized structures of the transition state and the product for the reaction of 5-arylideneisorhodanine (IsRd) and 5-arylidenerhodanine (Rd) derivatives with dimethyl maleate (DMm) with three molecules of the acetic acid. The ν1 value corresponds to the imaginary frequency for transition state
The presence of one, two, and three molecules of CH3COOH led to a decrease in the activation energy ∆Ea for IsRd by 2.68, 2.80, and 2.87 kcal mol–1, respectively, while for Rd, the value of ∆Ea increased by 2.48, 1.90, and 2.57 kcal mol–1, respectively. The reaction energy, ∆Er, for IsRd seemed to stabilize with an increase with one and a decrease with two and three acetic acid molecules, by 4.33, 0.24, and 0.06 kcal mol–1, respectively. However, different results were obtained for Rd. The reaction energy increased by 0.49 kcal mol–1 with one CH3COOH molecule. Two acetic acid molecules decreased the ∆Er by 0.27 kcal mol–1 and three CH3COOH molecules increased ∆Er, by 1.69 kcal mol–1. The interaction with acetic acid molecules favored the DA reaction of isorhodanine and made the reaction with the rhodanine less likely to occur.
The possible origin of these differences in the reactivity of Rd and IsRd in the DA reaction may be explained on the basis of the analysis of the frontier molecular orbitals [28]. The 3D plots of the HOMO orbitals for Rd and IsRd are shown in Fig. 5. For the interaction of IsRd with DMm, the most effective interaction between the orbitals occurred for the 3px atomic orbital at the S atom and the 2px atomic orbital at the C2 atom. The respective values of the coefficients are similar: −0.217 and −0.377. The effective overlapping of the orbitals explains the relative ease of the formation of the S–C bond. However, for the reaction of Rd with DMm, the interaction of similar atomic orbitals, i.e., the 2px atomic orbital at the O atom and the 2px atomic orbital at the C2 atom, is hindered. The respective values of the coefficients differ by approximately one order of magnitude, i.e., –0.094 and −0.549. Because of the weak overlapping of the atomic orbitals of the HOMO of the diene with the LUMO of the dienophile, the formation of the O–C2 bond is less effective and it cannot be formed easily.

a The 3D plot of the HOMO for IsRd, b the 3D plot of the HOMO for Rd. The isosurfaces plotted for 0.05 au
The difference between the reactivity of IsRd and Rd may also be correlated with the values of the energy gaps between the HOMO (diene) and LUMO (dienophile) calculated for the optimized geometries. The energies of the MOs are schematically compared for IsRd and DMm in Fig. 6a and for Rd and DMm in Fig. 6b. The difference between the energies of the LUMOdienophile and HOMOdiene (6.793 eV and 6.157 eV) is smaller than the energy gap between the LUMOdiene and HOMOdienophile (7.441 eV and 7.895 eV). Thus, the results indicate the normal electronic demand (NED) character of both reactions. Furthermore, Rd and IsRd can be considered as the nucleophile and DMm as the electrophile.

The comparison of the energies of HOMO and LUMO (frontier molecular orbitals) for IsRd and DMm (a) and for Rd and DMm (b). The calculations carried out for isolated monomers. NED – the normal electron demand Diels–Alder reaction, IED – the inverse electron demand Diels–Alder reaction
The character of DA reactions is related to charge transfer (CT) between interacting molecules [29]. The values of the transferred charge, Δq, for the reactions of IsRd with DMm and Rd with DMm, calculated for TS for eight stereoisomers, are shown in Table 6. The comparison reveals qualitative differences between the properties of IsRd and Rd. In the case of isorhodanine, all values of Δq were positive and spanned a range between 0.226e (stereoisomer 8) and 0.348e (stereoisomer 2). The flux of electron density goes from the nucleophile, IsRd, to the electrophile, DMm, in accordance with the polar character of the DA reaction. On the other hand, for rhodanine, the negative values of Δq were computed in the range from −0.215e (stereoisomer 8) to −0.063e (stereoisomer 2), with the exception of stereoisomer 1 for which Δq was small and positive (0.013e). The negative sign of Δq indicates a possible inversion of the electron density flux, where the electron density goes from DMm to Rd. This result emphasizes the difference in the physical nature of IsRd and Rd in relation to DMm. It is also worth noting that the absolute values of the transferred charge, |Δq|, were smaller for the reaction with Rd than for IsRd. The polar character of the reaction of Rd with DMm was less than that of IsRd with DMm.
The DA reactions were also analyzed using the global indexes defined in the conceptual DFT. Recent studies, carried out for cycloaddition reactions with a polar character, have shown that these indexes are powerful tools to study both reactivity and regioselectivity [30,31,32].
In Table 7, the values of the electronic chemical potential μ, chemical hardness η, global electrophilicity ω, and empirical nucleophilicity index N [33, 34] for the Rd, IsRd, and DMm (cis, trans) molecules are presented. Since the electronic chemical potential of IsRd (μ = −0.183 a.u.) and Rd (μ = −0.179 a.u.) is higher than that of the cis-DMm (μ = −0.195 a.u.) and trans-DMm (μ = −0.211 a.u.), the net CT during a polar DA reaction occurs from the diene to the dienophile. It is worth noting that the obtained result suggests an opposite net CT between Rd and trans-DMm than that calculated with the NPA method. According to the interpretation of the ω and N indexes proposed by Domingo et al. [35, 36], IsRd can be considered as a moderate nucleophile and strong electrophile. DMm can be considered as a strong electrophile and a marginal nucleophile. However, it was found that the global electrophilicities of IsRd (2.267 eV) and Rd (2.096 eV) were higher than those of cis- and trans-DMm (1.602 eV and 1.967 eV, respectively). Therefore IsRd and Rd will act as electrophiles, while DMm acts as a nucleophile. From this point of view, the studied reaction may be classified as the inverse electronic demand (IED) type.
Another look at the reactivity of IsRd, Rd, and DMm was obtained from the analysis of the values of the Fukui function [37], f(r), for the atoms taking part in the formation of C1–C3, S–C2, and O–C2 bonds. The S, C3 atoms in IsRd, the O, C3 atoms in Rd, and the C1, C2 atoms in DMm (cis, trans) were considered. The Fukui function is widely used in the prediction of reactive sites in molecules [38,39,40]. The values of the f+(r), f−(r), and f0(r) functions, which can be interpreted as indicators of the preferred place of the nucleophilic, electrophilic, and radical attack in molecules, are presented in Table 8.
The S atom in IsRd exhibited similar values of the f+(r), f−(r), and f0(r) functions (0.22–0.34). Thus, isorhodanine can act as a nucleophile and an electrophile (stronger) in chemical reactions. In the case of the C3 atom, where the value of f−(r) was much smaller (0.07) than that calculated for the S atom (0.34), lower electrophilic character is observed than for the sulfur atom. Interesting results were obtained for the O atom in Rd. It has less nucleophilic and electrophilic character than the S atom due to very small values of f+(r) and f−(r), i.e., 0.11 and 0.01, respectively. Thus, the O atom in Rd seems to be a less preferred place for either nucleophilic or electrophilic attack than the S atom. This result supports the interpretation based on the values of ∆Ea, i.e., that the reaction of Rd with DMm needs much a higher activation energy than the reaction with IsRd.
Finally, the issue of the aromaticity of the TS for the reactions of Rd and IsRd with DMm is discussed. A high degree of aromaticity for Diels–Adler reactions has been postulated and studied theoretically in the past [41,42,43]. However, aromaticity changes during DA reactions for complicated heterocyclic systems such as Rd and IsRd have not been examined yet. In order to gain insight into this problem, we calculated the HOMA index values for “pure” rhodanine and isorhodanine rings and their derivatives with exocyclic =CH2 and =CHPhe substituents (Rd and IsRd) for both reagents (diene and dienophile) with geometrical structures optimized for the pre-reaction complexes (Substrates, Sub) and finally for the TS of the DA reactions.
The calculated HOMA values are presented in Table 9 for both the gas phase and in the presence of the solvent (acetic acid). The aromaticity of “pure” rhodanine and isorhodanine rings was very poor, as indicated by the negative HOMA values. As mentioned previously, the higher the HOMA index, the stronger the predicted aromaticity, with a value of 1.00 defining a fully aromatic ring. Thus, rhodanine and isorhodanine with “pure” rings can be considered as slightly antiaromatic compounds. They both gain a very small amount of aromaticity when the exocyclic substituent with a double bond (=CH2) is attached to the ring. For such a derivative, the HOMA index has a positive value of about 0.18. The HOMA index increased again, by about 0.04 units, in the derivatives with a =CHPhe substituent instead of =CH2. Another slight increase in aromaticity was observed for the pre-reaction complexes where the dienophile and the diene interacted through noncovalent interactions (with a small exception for Sub1). Then, the reaction started and the corresponding TS was formed. For this step, a large increase in aromaticity was noted. Aromaticity increased to HOMA values of 0.60 (IsRd) and 0.75 (Rd). Thus, the aromaticity of the transition states was significantly higher than that of the simpler systems. On the other hand, the aromaticity of the TS for the DA reactions of Rd and IsRd derivatives was not perfect and was still far from a HOMA value of 1.00. However, it is still quite high for a not fully conjugated compound. It was clearly observed that the aromaticity of the studied heterocyclic ring was a bit higher in the acetic acid environment than in the gas phase. However, the influence of the solvent was small, typically about 0.05 HOMA units. All main aromatic features evaluated above from the HOMA data are followed by the results of NICS calculations. Thus, we gathered NICS data in Table S2 and stored it as supplementary data.
Aromaticity is an important factor for molecular energies. Thus, in conclusion, we can say that the aromatic stabilization of studied reaction transition states is important but is not a deciding factor. If so, reactions with Rd, where the TS usually showed slightly stronger aromatic stabilization, would have been easier to conduct. In addition, there was no correlation between the degree of TS aromatic stabilization and the ∆Ea values presented in Table 1.
Conclusions
A quantum chemical study, carried out for the DA reaction of 5-arylidenerhodanine and 5-arylideneisorhodanine with dimethyl maleate (in the gas phase and in solution), enabled us to explain experimentally observed differences in the reactivity of both molecules. The high reactivity of isorhodanine is associated with a relatively low activation energy, ∆Ea, that is needed to cross the TS, and a much larger value of the barrier, which separates the TS from the product. The lower reactivity of rhodanine in the DA reaction is due to a higher value of ∆Ea, i.e., 35.06 kcal mol–1 (gas phase) and 33.75 kcal mol–1 (acetic acid), with similar values of energy barriers from both side. The probability of the reversible process is much greater for the reaction with rhodanine.
The reaction energy for the reaction of isorhodanine with dimethyl maleate was clearly smaller than for the reaction of rhodanine with dimethyl maleate. Thus, the formation of the product in the latter case is thermodynamically less favored.
The application of the PCM model of the solvent for the reaction of isorhodanine resulted in a decrease in ∆Ea and ∆Er, favoring the formation of the product. In the case of rhodanine, in the presence of the solvent, similar values of energy barriers were obtained.
Modeling microsolvation in the acetic acid phase with three CH3COOH molecules showed a decrease in the energy barrier ∆Ea for isorhodanine by 2.68, 2.80, and 2.87 kcal mol–1, respectively, while for rhodanine ∆Ea increased by 2.48, 1.90, and 2.57 kcal mol–1. Thus, the presence of acetic acid favors the DA reaction of isorhodanine and makes the reaction with the rhodanine less likely to occur.
The analysis of frontier molecular orbitals taking part in the formation of S–C2 and O–C2 bonds in the TS, showed effective overlapping of the atomic orbitals of the S and C2 atoms in isorhodanine and a small degree of overlapping between the atomic orbitals of the O and C2 atoms in rhodanine. This result corresponds to the smaller value of the energy barrier ∆Ea computed for the reaction of isorhodanine and the higher value of ∆Ea for rhodanine.
Aromatic stabilization of transition states has been documented. The rhodanine and isorhodanine rings in the TS of the studied DA reactions were aromatic. This type of stabilization clearly had an influence on the energy of the studied TS reactions. However, this aromatic stabilization is not responsible for determining which reaction is easier to perform with rhodanine or isorhodanine agents.
References
Al-Mulla A (2017) A review: biological importance of heterocyclic compounds. Der Pharma Chem 9:141–147
Diels O, Alder K (1928) Synthesen in der hydroaromatischen Reihe. Liebigs Annalen der Chemie 460:98–122
Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G (2002) The Diels-Alder reaction in total synthesis. Angew Chem Int Ed 41:1668–1698
Salem MA, Helalc MH, El-Gabyd MSA, Ammar YA, Gouda MA, Abbas SY (2018) Pyrano[2,3-d]thiazole: synthesis, reactions and biological applications. To Chem J 1:114–144
Yarovenko VN, Nikitina AS, Zayakin ES, Zavarzin IV, Krayushkin MM Kovalenko LV (2008) 2-thioxopyrano[2,3-d][1,3] thiazoles by Diels-Alder reaction of arylidenerhodanines under microwave irradiation. ARKIVOC 4:103–111
Rahman MA, Ghany HA, Ghattas ABAG (1989) Heterodiene synthesis I. reaction of 5-arylidenerhodanine derivatives with 1-morpholinocyclohexene enamine. Synth Commun 19:345–354
Atamanyuk D, Zimenkovsky B, Atamanyuk V, Nektegayev I, Lesyk R (2013) Synthesis and biological activity of new thiopyrano[2,3-d]thiazoles containing a naphtoquinone moiety. Sci Pharm 81:423–436
Lozynskyi A, Zimenkovsky B, Lesyk R (2014) Synthesis and anticancer activity of new thiopyrano[2,3-d]thiazoles based on cinnamic acid amides. Sci Pharm 82:723–733
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16, revision B.01. Gaussian, Inc., Wallingford
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor Chem Accounts 120:215–241
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72:650–654
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Barone V, Cossi M (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 102:1995–2001
Cossi M, Rega N, Scalmani G, Barone V (2003) Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comput Chem 24:669–681
Glendening ED, Reed AE, Carpenter JE, Weinhold F (1998) NBO Version 3.1. TCI, University of Wisconsin, Madison
Yang W, Mortier WJ (1986) The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J Am Chem Soc 108:5708–5711
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford University Press, Oxford
Noury S, Krokidis X, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23:597–604
Kruszewski J, Krygowski TM (1972) Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 13:3839–3842
Krygowski TM (1993) Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of π-electron systems. J Chem Inf Comp Sci 33:70–78
Krygowski TM, Cyrański MK (2004) Two faces of the structural aspects of aromaticity. Phys Chem Chem Phys 6:249–255
Jug K, Köster AM (1991) Aromaticity as a multi-dimensional phenomenon. J Phys Org Chem 4:163–169
Schleyer PR, Maerker C, Dransfeld A, Jiao H, van Eikema Hommes NJR (1996) Nucleus-independent chemical shifts: a simple and efficient aromaticity probe. J Am Chem Soc 118:6317–6318
Schleyer PR, Manoharan M, Wang ZX, Kiran B, Jiao HJ, Puchta R, van Eikema Hommes NJR (2001) Dissected nucleus-independent chemical shift analysis of pi-aromaticity and antiaromaticity. Org Lett 3:2465–2468
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera-a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Bruno IJ, Cole JC, Edgington PR, Kessler M, Macrae C, McCabe P, Pearson J, Taylor R (2008) New software for searching the Cambridge structural database and visualizing crystal structures. Acta Crystallogr Sect B 58:389–397
Houk KN (1975) Frontier molecular orbital theory of cycloaddition reactions. Acc Chem Res 8:361–369
Domingo LR, Sáez JA (2009) Understanding the mechanism of polar Diels-Alder reactions. Org Biomol Chem 7:3576–3583
Domingo LR, Aurell J, Pérez P, Contreras R (2002) Origin of the synchronicity on the transition structures of polar Diels-Alder reactions. Are these reactions [4 + 2] processes? J Org Chem 68:3884–3890
Domingo LR, Arnó M, Contreras R, Pérez P (2002) Density functional theory study for the cycloaddition of 1,3-butadienes with dimethyl acetylenedicarboxylate. Polar stepwise vs. concerted mechanisms. J Phys Chem A 106:952–961
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the local electrophilicity of organic molecules. Understanding the regioselectivity on Diels-Alder reactions. J Phys Chem A 106:6871–6875
Parr RG (1999) Szentpály L v., Liu S. Electrophilicity index. J Am Chem Soc 121:1922–1924
Pérez P, Domingo LR, Duque-Noreña M, Chamorro E (2009) A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J Mol Struct THEOCHEM 895:86–91
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423
Jaramillo P, Domingo LR, Chamorro E, Pérez P (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct THEOCHEM 865:68–72
Parr RG, Yang W (1984) Density functional approach to the frontier-electron theory of chemical reactivity. J Am Chem Soc 106:4049–4050
Cerda-Monje A, Ormazábal-Toledo R, Cárdenas C, Fuentealba P, Contreras R (2014) Regional electrophilic and nucleophilic Fukui functions efficiently highlight the Lewis acidic/basic regions in ionic liquids. J Phys Chem B 118:3696–3701
Lee C, Yang W, Parr RG (1988) Local softness and chemical reactivity in the molecules CO, SCN− and H2CO. J Mol Struct THEOCHEM 163:305–313
Martinez C, Rivera JL, Herrera R, Rico JL, Flores N, Rutiaga JG, López P (2007) Evaluation of the chemical reactivity in lignin precursors using the Fukui function. J Mol Model 14:77
Manoharan M, Venuvanalingam P (1998) Gain or loss of aromaticity in Diels-Alder transition states and adducts: a theoretical investigation. J Phys Org Chem 11:133–140
Bachrach SM, White PB (2007) Toward assessing the aromaticity of the Diels-Alder transition state. J Mol Struct THEOCHEM 819:72–78
Corminboeuf C, Heine T, Weber J (2003) The change of aromaticity along a Diels-Alder reaction path. Org Lett 5:1127–1130
Acknowledgments
The authors are grateful to the Wroclaw Centre for Networking and Supercomputing for the generous allocation of computer time.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest with regard to the publication of this article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to the Topical Collection Zdzislaw Latajka 70th Birthday Festschrift
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tejchman, W., Michalski, M., Zborowski, K.K. et al. Why are rhodanines less efficient reagents in Diels–Alder reactions than isorhodanines? A quantum chemical study. J Mol Model 25, 190 (2019). https://doi.org/10.1007/s00894-019-4063-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4063-y