
Abstract
Using density functional theory (B97-D/ECP2/PCM//RI-BP86/ECP1 level), we have studied the effects of ligand variation on OH− uptake by transition-metal carbonyls (Hieber base reaction), i.e., LnM(CO) + OH− → [LnM(CO2H)]−, M = Fe, Ru, Os, L = CO, PMe3, PF3, py, bipy, Cl, H. The viability of this step depends notably on the nature of the co-ligands, and a large span of driving forces is predicted, ranging from ΔG = −144 kJ/mol to +122 kJ/mol. Based on evaluation of atomic charges from natural population analysis, it is the ability of the co-ligands to delocalize the additional negative charge (through their π-acidity) that is the key factor affecting the driving force for OH− uptake. Implications for the design of new catalysts for water gas shift reaction are discussed.

ᅟ
Similar content being viewed by others

Introduction
Hydrogen is the cleanest fuel and a cost-effective energy carrier of the future [1,2,3,4,5,6], which produces three times more energy per unit mass than fossil fuels [7]. Industrially, hydrogen is generated from fossil fuels releasing higher amounts of greenhouse gas, CO2 [8, 9]. A sustainable supply of hydrogen from renewable resources, such as biomass, is highly desirable [4, 10]. Development of homogeneous transition metal catalysts for complete decomposition of polyhydroxy biomass constituents, i.e., carbohydrates, into H2 and CO2, could revolutionize H2 production from renewable resources. Methanol is the simplest model for such carbohydrates, but complete dehydrogenation of this compound according to
would be of considerable interest in its own right. This decomposition of methanol could follow a sequence of three reactions, (i) dehydrogenation, (ii) decarbonylation, and (iii) water–gas shift reaction (WGSR, see Scheme 1), all of which are well known.

Putative reaction sequence for complete methanol dehydrogenation
Development of homogeneous transition metal catalysts for H2 production from methanol [11, 12] has attracted much attention in the past few decades. Morton and Cole-Hamilton developed a ruthenium catalyst [Ru(H)2(X2)(PPh3)3] (X = N, H) for partial dehydrogenation of alcohols (including methanol) with notable turnover frequencies [13]. Aldehydes and ketones were the main products [formaldehyde in case of methanol, Scheme 1, step (i)]. During the conversion of ethanol, significant amounts of methane and a carbonyl complex, [RuH2(CO)(PPh3)3], were produced through decarbonylation [Scheme 1, step (ii)]. However, since no CO2 was noticed, apparently these Ru complexes are not active as WGSR catalysts [14].
Recently, based on density functional theory (DFT) calculations, we studied the mechanisms for the dehydrogenation [15] and decarbonylation [16] of aliphatic alcohols catalyzed by the Morton and Cole-Hamilton system, [RuH2(H2)(PPh3)3] [Scheme 1, steps (i) and (ii)]. When exploring the feasibility of WGSR with these complexes computationally [17], we noticed that the first step of WGSR, the attack of water (in the form of OH−) on the CO ligand (Scheme 2) is highly endergonic.

General mechanism for transition-metal-catalyzed WGSR under basic conditions, where OH− is the nucleophile
Significant research has been undertaken to reveal the mechanism of WGSRs catalyzed by homogeneous transition metal complexes, particularly the metal carbonyls of Fe, Ru, and Os [18,19,20,21,22,23,24,25,26,27,28,29,30]. Recently, Guo et al. have studied the WGSR mechanism catalyzed by hexacarbonyl complexes of Mo and W [31]. In all of these reactions, which are conducted under basic conditions, OH− is the nucleophile and its uptake to form a transient metallacarboxylic acid is considered as the initial step (in the grey box in Scheme 2). Such attack of OH− on carbonyl ligands is well known as Hieber base reaction [32].
In all studies of metal carbonyl catalyzed WGSR, this OH− uptake appeared to be highly exothermic and essentially barrierless. In contrast, in our study of WGSR in the Morton and Cole-Hamilton system [17], this step is predicted to be highly endergonic. The driving force for formation of the metallacarboxylic acid depends notably on the co-ligands that are present, in particular on the number of CO ligands. While large negative enthalpies and free energies are computed for the OH− uptake of Ru(CO)5 [28, 29] ΔG = 127.7 kJ/mol and 81.6 kJ/mol are predicted for [RuH2(CO)(PPh3)3] and [RuH2(CO)2(PPh3)2], respectively. It therefore appears that suitable ligand design, by varying the steric or electronic properties of the ligands, could make the process of OH− uptake feasible. In this work, we now report DFT-computed driving forces for OH− uptake in a number of metal-carbonyl complexes of Ru, Fe, and Os. Along with CO, we have made the choice of trimethylphosphine (comparable to triphenylphosphine), trifluorophosphine (a strong π-acceptor ligand, comparable to CO) [33], pyridine, and bipyridine ligands. For a perfect catalytic system, the OH− entry into the cycle should be facile and should not produce a very low-lying intermediate on the reaction profile that would eventually deactivate the catalytic system. This work can lead to the rational design of better catalysts for WGSR and, eventually, towards the complete decomposition of alcohols by dehydrogenation, decarbonylation, and the finally WGSR, which could facilitate entry into a hydrogen-based economy.
Results and discussion
Ligand effects on the initial uptake of OH− to form the metallacarboxylic acid
In metal carbonyls, OH− uptake is usually considered a fast process, and the resulting metallacarboxylic acid is usually too reactive to be isolated and decarboxylates under CO2 evolution to form a hydride (Scheme 2). Protonation of this hydride intermediate takes the system uphill on the free energy reaction profile and the H2 evolution tends to be associated with the highest energy transition state. Our work focuses on the ligand effects on the initial uptake of OH− to metal carbonyls (Eq. 2, corresponding to the first step in the grey box in Scheme 2), taking M(CO)5 pentacarbonyls as prototypical representatives (M = Fe, Ru, Os). Step by step, we replaced each of the CO in the metal pentacarbonyl system with selected ligands, namely trimethylphosphine (PMe3), trifluorophosphine (PF3), pyridine (py), and bipyridine (bipy). We compared the free energies of the reactants (metal carbonyls) and the products (metallacarboxylic acids) to see how such a ligand change affects the driving force for the OH- uptake to the system. We have included the results for Ru carbonyls and metallacarboxylic acids in the main paper, whereas the results for the Fe and Os analogs are included within the supporting information (SI).
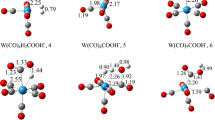
Unlike CO, PMe3 is a weak π-acceptor ligand and experiences weak backbonding with the metal center. At our chosen level of theory, B97-D/ECP2//RI-BP86/ECP1, Ru(CO)5 has a driving force of ΔG = −93.5 kJ/mol for the initial OH− uptake. This driving force decreases (i.e., ΔG increases) as we increase the number of PMe3 ligands that replace CO. On substituting one CO with one PMe3 ligand at the axial position, the free energy increases to −38.9 kJ/mol, which further increases to 49.2 kJ/mol on replacing the second CO on the axial position with another PMe3 ligand [34]. The OH− uptake to Ru(CO)2(PMe3)3 is unfavorable by a free energy of 87.8 kJ/mol, that of Ru(CO)(PMe3)4 by 122.2 kJ/mol (Fig. 1). Since the subsequent steps on the WGSR reaction profile (Scheme 2) add additional barriers, the latter two complexes are expected to be only weakly active or unproductive as WGSR catalysts.

Computed free energies (B97-D level, kJ/mol) for the OH− uptake of the carbonyl reactant (note that this reactant is different for each product). The number of PMe3 ligands increases from left to right (see Fig. S1 in the ESI for a plot showing three-dimensional representations of the complexes)
Based on the results obtained for the PMe3 ligand exchange with CO, one can assume that the driving force for the OH− uptake should be affected by replacing CO ligands with ligands of slightly greater π-acceptor strength, e.g., PF3. A slight increase of the driving force for OH− uptake is observed on replacing one CO at the axial position with a PF3 ligand, from −93.5 kJ/mol in Ru(CO)5 (Fig. 1) to −101.8 kJ/mol for Ru(CO)4(PF3) (Fig. 2). On exchanging both axial CO ligands with PF3, the free energy decreases by 31.7 kJ/mol for the OH− uptake as compared to the free energy of the OH− uptake in Ru(CO)5. One would expect that exchanging three CO ligands, two at the axial and one at the equatorial position, should further favor OH− uptake, but this is not the case. After the OH− uptake by Ru(CO)2(PF3)3, the metalla-acid, [Ru(CO)(COOH)(PF3)3]−, is obtained with a free energy of −112.8 kJ/mol, which is higher by 12.5 kJ/mol than the free energy for the OH− uptake in Ru(CO)2(PF3)3, possibly because of the trans influence of CO. When employing another, higher-lying isomer of Ru(CO)2(PF3)3, where one of the CO ligands is positioned trans to PF3, the resulting metalla-acid is obtained at a ΔG of −121.4 kJ/mol. This value is lower by 8.6 kJ/mol compared to the ΔG for the most stable isomer included in Fig. 2. There is thus a noticeable trans influence on the driving force under scrutiny, although it does not seem to override other electronic effects.

Relative free energy (kJ/mol) for the OH− uptake with that of the respective carbonyl reactant set to 0.0 kJ/mol in each case. The number of PF3 ligands increases from left to right (see Fig. S2 in the ESI for a plot showing three-dimensional representations of the complexes)
Ru(CO)(PF3)4 follows the expected trend in terms of free energy for the OH− uptake and has the largest predicted affinity for OH− of all complexes studied here (−143.7 kJ/mol, Fig. 2). It should be noted that such a large driving force for OH− uptake does not necessarily make this complex a good target for a WGSR catalyst, because a correspondingly higher energy needs to be invested to close the cycle and re-form the initial catalyst.
A variety of [Ru]-CO2H complexes are known, some of which have been structurally characterized, notably with bidentate aromatic N-donor ligands [35, 36]. We therefore included a couple of model complexes with aromatic N-donor ligands, namely pyridine (py) and bipy. Both are coordinated through the lone pair of an electronegative N atom providing inductive donation, with the aromatic backbone allowing for significant π-backbonding interaction. As the inductive donation from the nitrogen lone pair is counteracted by the backbonding into the aromatic system, on exchanging one CO at the axial position in Ru(CO)5 with a pyridine (py), the OH− uptake becomes unfavorable as compared to that in parent Ru(CO)5, but not by that much as in case of PMe3 ligand. On replacing two CO with a bipy bidentate ligand, the OH− uptake becomes further unfavorable as compared to that in parent Ru(CO)5, but remains favorable by 63.4 kJ/mol as compared to the OH− uptake in the Ru(CO)3(PMe3)2 system (Fig. 3).

Relative free energy (kJ/mol) for the OH− uptake on replacing one CO ligand with py (left) and two CO ligands with bipy (right) (see Fig. S3 in the ESI for a plot showing three-dimensional representations of the complexes)
Finally, in addition to the pentacoordinate Ru(0) species, we considered the OH− uptake in a few selected octahedral Ru(II) complexes including Ru(CO)(H)2(PMe3)3, Ru(CO)(H)2(PF3)3, and [Ru(CO)3Cl3]− (Fig. 4). The driving force for OH− uptake in Ru(CO)(H)2(PMe3)3, endergonic by 82.7 kJ/mol, is comparable to that in the Morton and Cole-Hamilton system, Ru(CO)(H)2(PPh3)3, where it is endergonic by 127 kJ/mol at essentially the same level (at a higher temperature, however, 150 °C) [17, 37]. On replacing PMe3 ligands with PF3 ligands, the product is obtained at a free energy of −61.2 kJ/mol, obviously because of strong π-backbonding interaction.

Relative free energy (kJ/mol) for the OH− uptake with that of the respective octahedral carbonyl reactant set to 0.0 kJ/mol in each case (see Fig. S4 in the ESI for a plot showing three-dimensional representations of the complexes)
The results for the analogous Os complexes are very similar to those for the Ru species just discussed, with individual driving forces for OH− uptake within typically 10 kJ/mol of each other (ca. 20 kJ/mol for the bipy complex, compare Tables S1 and S3 in the SI). On going from Ru to Fe congeners, the changes in this driving force become somewhat more variable (up to ca. 30 kJ/mol, compare Tables S1 and S2 in the SI), but overall the same trends are obtained irrespective of the group 8 metal.
The metallacarboxylic acid arising from Hieber base reaction of [Ru(CO)3Cl3]− has been implicated as a key reactive intermediate in a complex variety of reactions [32]. Indeed, despite forming a dianion from two monoions, OH− uptake of [Ru(CO)3Cl3]− affording [Ru(CO)2(CO2H)Cl3]2− is highly exergonic, with a free energy of −119.0 kJ/mol. This large driving force is fully consistent with the fact that this complex is a reactive intermediate that can be formed through Hieber base reaction [32]. Experimentally, [Ru(CO)2(CO2H)Cl3]2− appears to lose a chloride ion consistent with our calculations as at our level, as this process is computed to be slightly exergonic, by −5.9 kJ/mol.
Natural population analysis
What is the origin of the huge variation in driving forces for OH− uptake in these complexes? Hypothesizing that a key factor should be delocalization of the additional negative charge brought into the complex, we used natural population analysis (NPA) [38] to evaluate the extent of charge transfer from OH− upon attack on the carbonyl ligand. To this end, we simply calculated the natural charge on the OH− fragments in the ruthenacarboxylic acid products, assessing how it changes from the value in free OH−, where it is −1. A substantial reduction from this absolute value is found in the complexes, indicating that most of the charge is actually delocalized into the complex, but there is still a notable variation of this charge, between −0.32 and −0.19 (see Table S4 in the SI).
The PMe3 ligand has σ-donating abilities, which pushes electron density to the metal center, which increases the amount of backbonding interaction between the filled metal d-orbital and the empty π*- orbital of the carbon atom of CO. The overall affect makes it difficult for the OH− fragment to delocalize electron density over the metal complex. As we replace more CO ligands with PMe3 ligands, the natural charge at the OH− fragment decreases. In [Ru(CO)2(CO2H)(PMe3)2]−, presence of a CO ligand at the axial position trans to the –CO2H− fragment increases its distance from the metal center, making it less available for OH− fragment to accommodate the charge density. Here the trans influence dominates the electronic nature of the ligands and a small discrepancy in the natural charges of the OH− fragment occurs when we move from [Ru(CO)2(CO2H)(PMe3)2]− to [Ru(CO)(CO2H)(PMe3)3]−, similar is the case with py and bipy ligands. For the PF3 ligands, on replacing each with the CO ligands, the natural charge at the OH− fragment gradually increases, as expected (Table S4).
A plot of the computed driving forces vs. OH− charges indeed reveals an overall trend towards more favorable OH− uptake with decreasing charge on this fragment (see Fig. 5; essentially the same correlation is obtained when enthalpies are used instead of free energies, see Fig. S5 in the SI). No strict relationship is apparent, and there are a few outliers, notably the bipy complex, but overall our results are consistent with the charge delocalization being a key factor for the driving force of this reaction step. For instance, the PF3 ligands, which are predicted to strongly promote Hieber base reaction (Fig. 2), are indicated to do so because they are very efficient in delocalizing the incoming negative charge (see red triangles in the lower right of Fig. 5). Arguably, the extent of this charge delocalization will depend on the balance between σ-donating and π-backdonating capabilities of the co-ligands, which should allow for a rational tailoring of complexes toward Hieber base reaction and, eventually, for designing new WGSR catalysts.

Plot of driving forces for OH− uptake vs. natural charges of the OH− fragments in the products (B97-D level)
Conclusions
In summary, using an appropriate DFT level, we have computed the driving forces for formation of metallacarboxylic acids from group 8 carbonyl complexes through uptake of OH−. This reaction (Eq. 2), known as Hieber base reaction, is the first step of water–gas shift reaction (WGSR) that can be catalyzed by transition metal complexes under basic conditions. According to our findings, the driving force for this step is surprisingly sensitive to the nature of the co-ligands at the metal, and can range from ΔG = −144 kJ/mol to +122 kJ/mol [for R = F and Me, respectively, in Ru(CO)(PR3)4]. Far from being innocent spectator ligands, these co-ligands actively take part in OH− uptake through delocalization of the negative charge, as apparent in the computed atomic charges from natural population analysis. Fe and Ru pentacarbonyls are prototypical WGSR catalysts; it is remarkable how replacement of CO ligands with electron-rich phosphines (which are ubiquitous in modern transition metal chemistry) can impede the first step of this WGSR catalytic cycle. In that case, use of phosphines with electron-withdrawing substituents (where we have used PF3 as extreme example) or aromatic N-donor ligands can increase the driving force for Hieber base reaction. Compared to these ligand effects, the nature of the metal (Fe, Ru, or Os) or its oxidation state [e.g., Ru(0) vs. Ru(II)] seems to be of lesser importance for OH− uptake.
We are convinced that the tunability of the driving force for Hieber base reaction through appropriate choice of co-ligands can inform on the rational design of new WGSR catalysts. As this quantity, a simple reaction (free) energy, can be readily computed with modern DFT tools, large libraries of ligands can be screened computationally, opening up new avenues for applications of molecular modeling in homogeneous catalysis.
Computational methodology
In this work, we are mainly interested in calculating the driving force related to change in Gibbs free energy for the OH− uptake, which is considered as the initial step of the WGSR. Our calculations follow a computationally cost-effective protocol based on density functional theory (DFT) that had been validated [39] and fruitfully applied to mechanistic DFT studies of related Ru complexes [15, 16]. Geometry optimizations were carried out at a lower level, RI-BP86/ECP1, whereas the energies were refined at B97-D/ECP2 [40] level. All the metal complexes were fully optimized at the RI-BP86/ECP1 level, i.e., by the use of Becke [41] and Perdew [42] exchange and correlation functionals along with SDD [43] core potential and valence basis on the metal atoms, whereas all the other atoms were treated with the standard 6-31G(d,p) basis. The nature of all the minima was verified by frequency calculations within the harmonic approximation, which were further used to obtain the enthalpic and entropic corrections under standard conditions (1 atm and 298.15 K). Thermochemical correction terms δEG were obtained as:
where ΔERI-BP86/ECP1 is the reaction energy and ΔGRI-BP86/ECP1 is the corresponding Gibbs free energy (analogously for corrections to enthaly, δEH from ΔHRI-BP86/ECP1 - ΔERI-BP86/ECP1).
The energies of the optimized complexes were refined through single-point calculations at the B97-D/ECP2 level, i.e., using dispersion-corrected B97-D functional, which includes Grimme’s dispersion correction [40] along with 6-311+G(d,p) basis set for all the non-metal atoms (SDD on the metals). Solvent effects were included by a polarizable continuum model (PCM) [44, 45] using methanol as a model solvent with self-consistent reaction field (SCRF) method. The solvent energy correction, δEsolv, was performed as:
Difference between the reaction energy in the continuum is donated as ΔEPCM and ΔE is the difference between the reaction energy in gas phase at B97-D/ECP2 level. The final enthalpies and free energies, ΔH and ΔG, were obtained as sum of all energy correction terms:
where ΔE and δEsolv were calculated at the B97-D/ECP2 level, whereas δEH and δEG were obtained at RI-BP86/ECP1. Atomic charges from natural population analysis [38] were evaluated at the B97-D/ECP2/PCM level. All calculations were performed using the Gaussian 09 suite of programs [46].
In order to identify the most stable isomers and conformers of each reactant and product, an exhaustive screening of the possible stereoisomers was undertaken. Only the results for the most stable forms are reported. The conformation of the carboxylic acid group was uniformly taken as that where the hydrogen of the OH− fragment is pointing towards the metal center. We investigated the stability of these metalla-acids against those in which the hydrogen of the OH− fragments points away from the metal center, particularly in Fe complexes, the former complexes are appeared to be more stable (see Table S5).
References
Pena MA, Gomez JP, Fierro JLG (1996) New catalytic routes for syngas and hydrogen production. Appl Catal, A 144:7–57. https://doi.org/10.1016/0926-860X(96)00108-1
Armor JN (1999) The multiple roles for catalysis in the production of H2. Appl Catal, A 176:159–176. https://doi.org/10.1016/S0926-860X(98)00244-0
Trimm DL, Onsan ZI (2001) Onboard fuel conversion for hydrogen-fuel-cell-driven vehicles. Catal Rev - Sci Eng 43:31–84. https://doi.org/10.1081/CR-100104386
Zhang YHP (2011) Hydrogen production from carbohydrates: a mini-review. ACS Symp Ser 1067 (Sustainable Production of Fuels, Chemicals, and Fibers from Forest Biomass):203-216. https://doi.org/10.1021/bk-2011-1067.ch008
Ricci M, Bellaby P, Flynn R (2008) What do we know about public perceptions and acceptance of hydrogen? A critical review and new case study evidence. Int. J. Hydrog. Energy 33:5868–5880. https://doi.org/10.1016/j.ijhydene.2008.07.106
Momirlan M, Veziroglu T (1999) Recent directions of world hydrogen production. Renew. Sust. Energ. Rev. 3:219–231. https://doi.org/10.1016/S1364-0321(98)00017-3
Hemmes H, Driessen A, Griessen R (1986) Thermodynamic properties of hydrogen at pressures up to 1 mbar and temperatures between 100 and 1000 K. J. Phys. C Solid State Phys. 19:3571–3585. https://doi.org/10.1088/0022-3719/19/19/013
Navarro RM, Pena MA, Fierro JLG (2007) Hydrogen production reactions from carbon feedstocks: fossil fuels and biomass. Chem. Rev. 107:3952–3991. https://doi.org/10.1021/cr0501994
Veziroglu TN, Editor (1976) Conference Proceedings of the 1st World Hydrogen Energy conference, Vol. 1: March 1-3, 1976 Miami Beach, Fla. vol Copyright (C) 2018 American Chemical Society (ACS). All Rights Reserved. Univ. Miami Press,
Ishida Y, Kumabe K, Hata K, Tanifuji K, Hasegawa T, Kitagawa K, Isu N, Funahashi Y, Asai T (2009) Selective hydrogen generation from real biomass through hydrothermal reaction at relatively low temperatures. Biomass Bioenergy 33:8–13. https://doi.org/10.1016/j.biombioe.2008.04.004
Sordakis K, Tang C, Vogt LK, Junge H, Dyson PJ, Beller M, Laurenczy G (2018) Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem Rev (Washington, DC, US) 118:372–433. https://doi.org/10.1021/acs.chemrev.7b00182
Shen Y, Zhan Y, Li S, Ning F, Du Y, Huang Y, He T, Zhou X (2017) Hydrogen generation from methanol at near-room temperature. Chem. Sci. 8:7498–7504. https://doi.org/10.1039/C7SC01778B
Morton D, Cole-Hamilton DJ (1988) Molecular hydrogen complexes in catalysis: highly efficient hydrogen production from alcoholic substrates catalyzed by ruthenium complexes. J Chem Soc, Chem Commun:1154-1156. https://doi.org/10.1039/c39880001154
Morton D, Cole-Hamilton DJ, Utuk ID, Paneque-Sosa M, Lopez-Poveda M (1989) Hydrogen production from ethanol catalyzed by group 8 metal complexes. J Chem Soc, Dalton Trans, pp 489–495
Sieffert N, Bühl M (2010) Hydrogen generation from alcohols catalyzed by ruthenium-triphenylphosphine complexes: multiple reaction pathways. J. Am. Chem. Soc. 132:8056–8070. https://doi.org/10.1021/ja101044c
Sieffert N, Reocreux R, Lorusso P, Cole-Hamilton DJ, Bühl M (2014) On the importance of decarbonylation as a side-reaction in the ruthenium-catalysed dehydrogenation of alcohols: a combined experimental and density functional study. Chem. Eur. J. 20:4141–4155. https://doi.org/10.1002/chem.201303722
Lorusso P, Ahmad S, Schmid K, Cole-Hamilton DJ, Sieffert N, Bühl M Manuscript in preparation
Laine RM, Rinker RG, Ford PC (1977) Homogeneous catalysis by ruthenium carbonyl in alkaline solution: the water gas shift reaction. J. Am. Chem. Soc. 99:252–253. https://doi.org/10.1021/ja00443a049
King RB, Frazier CC, Hanes RM, King Jr AD (1978) Active homogeneous catalysts for the water gas shift reaction derived from the simple mononuclear carbonyls of iron, chromium, molybdenum, and tungsten. J. Am. Chem. Soc. 100:2925–2927. https://doi.org/10.1021/ja00477a076
Ford PC (1981) The water gas shift reaction: homogeneous catalysis by ruthenium and other metal carbonyls. Acc. Chem. Res. 14:31–37. https://doi.org/10.1021/ar00062a001
Sunderlin LS, Squires RR (1993) Energetics and mechanism of the thermal decarboxylation of tetracarbonyl(carboxy)ferrate(1-) in the gas phase. J. Am. Chem. Soc. 115:337–343. https://doi.org/10.1021/ja00054a048
Torrent M, Sola M, Frenking G (1999) Theoretical study of gas-phase reactions of Fe(CO)5 with OH- and their relevance for the water gas shift reaction. Organometallics 18:2801–2812. https://doi.org/10.1021/om9810504
King RB (1999) Homogeneous transition metal catalysis: from the water gas shift reaction to nuclear waste vitrification. J. Organomet. Chem. 586:2–17. https://doi.org/10.1016/S0022-328X(99)00155-2
Barrows SE (2004) Theoretical study of the gas-phase Fe(CO)5 catalyzed water gas shift reaction: a new mechanism proposed. Inorg. Chem. 43:8236–8238. https://doi.org/10.1021/ic049159t
Rozanska X, Vuilleumier R (2008) Mechanisms of the water–gas-shift reaction by iron pentacarbonyl in the gas phase. Inorg. Chem. 47:8635–8640. https://doi.org/10.1021/ic8001866
Zhang F, Zhao L, Xu C, Chen Y (2010) Theoretical revisit of a Fe(CO)5-catalyzed water–gas shift reaction. Inorg. Chem. 49:3278–3281. https://doi.org/10.1021/ic902148y
Chen Y, Zhang F, Xu C, Gao J, Zhai D, Zhao Z (2012) Theoretical investigation of water gas shift reaction catalyzed by iron group carbonyl complexes M(CO)5 (M = Fe, Ru, Os). J. Phys. Chem. A 116:2529–2535. https://doi.org/10.1021/jp204776a
Schulz H, Goerling A, Hieringer W (2013) Mechanisms of the water–gas shift reaction catalyzed by ruthenium pentacarbonyl: a density functional theory study. Inorg. Chem. 52:4786–4794. https://doi.org/10.1021/ic301539q
Liu N, Guo L, Cao Z, Li W, Zheng X, Shi Y, Guo J, Xi Y (2016) Mechanisms of the water–gas shift reaction catalyzed by ruthenium carbonyl complexes. J. Phys. Chem. A 120:2408–2419. https://doi.org/10.1021/acs.jpca.6b00301
Schaper L-A, Herrmann WA, Kuehn FE Water-Gas Shift Reaction. In, 2018. Wiley-VCH Verlag GmbH & Co. KGaA, pp 1689-1698. https://doi.org/10.1002/9783527651733.ch38
Guo L, Cao Z, Liu N, An X, Li A, Li W, Zheng X (2016) Mechanisms of the water–gas shift reaction catalyzed by carbonyl complexes M(CO)6 (M = Mo, W). Int. J. Hydrog. Energy 41:2432–2446. https://doi.org/10.1016/j.ijhydene.2015.11.108
Hill AF (2000) "simple" carbonyls of ruthenium: new avenues from the Hieber base reaction. Angew. Chem. Int. Ed. 39:130–133. https://doi.org/10.1002/(SICI)1521-3773(20000103)39:1<130::AID-ANIE130>3.0.CO;2-6
Hill AF (2002) Organotransition metal chemistry. The Royal Society of Chemistry, Cambridge
Ru(CO)3(PMe3)2,is a model for Ru(CO)3(PPh3)2,, also known as Roper’s catalyst: Cavit BE, Grundy KR, Roper WR (1972) Dicarbonyltris(triphenylphosphine)ruthenium and -osmium. Ethylene complex of ruthenium and a dioxygen complex of osmium. J Chem Soc, Chem Commun:60-61. https://doi.org/10.1039/c3972000060b
e.g. [Ru(phen)2(CO)(CO2H)]+ (phen = 1,10-phenanthroline): Gibson DH, Ding Y, Andino JG, Mashuta MS, Richardson JF (1998) Synthesis, Characterization, and Reactions of Ruthenium Phenanthroline Complexes Bearing C1 Ligands: Formyl, Metallocarboxylate, and CO2-Bridged Complexes. Organometallics 17:5178-5183. https://doi.org/10.1021/OM9806242
e.g. [Ru(bipy)2(CO)(CO2H)]+ (bipy = 2,2′=bipyridine): Toyohara K, Nagao H, Adachi T, Yoshida T, Tanaka K (1996) Crystal structure of [Ru(bpy)2(CO)(η1-C(O)OH)]+ (bpy = 2,2′-bipyridine) as a key intermediate in CO2/CO conversion. Chem Lett :27-28. https://doi.org/10.1246/cl.1996.27
In terms of reaction enthalpies (i.e. without the temperature-dependent entropy part) the driving forces are ΔH = 36.7 kJ/mol and 33.0 kJ/mol for PMe3 and PPh3, respectively. These values are thus rather similar for both phosphines, indicating little steric hindrance with the bulkier PPh3
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J. Chem. Phys. 83:735–746. https://doi.org/10.1063/1.449486
Sieffert N, Bühl M (2009) Noncovalent interactions in a transition-metal triphenylphosphine complex: a density functional case study. Inorg. Chem. 48:4622–4624. https://doi.org/10.1021/ic900347e
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27:1787–1799. https://doi.org/10.1002/jcc.20495
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A: Gen Phys 38:3098–3100. https://doi.org/10.1103/PhysRevA.38.3098
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 33:8822–8824. https://doi.org/10.1103/PhysRevB.33.8822
Andrae D, Haeussermann U, Dolg M, Stoll H, Preuss H (1990) Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 77:123–141. https://doi.org/10.1007/BF01114537
Cossi M, Barone V, Cammi R, Tomasi J (1996) Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 255:327–335. https://doi.org/10.1016/0009-2614(96)00349-1
Dong W, Yan M, Zhang M, Liu Z, Li Y (2005) A computational and experimental investigation of the interaction between the template molecule and the functional monomer used in the molecularly imprinted polymer. Anal. Chim. Acta 542:186–192. https://doi.org/10.1016/j.aca.2005.03.032
Frisch MJ et al (2009) Gaussian 09. Revision D.01 edn. Gaussian, Inc., Pittsburgh
Acknowledgements
We thank EaStCHEM and the School of Chemistry for support. Computations were carried out on a local Opteron PC cluster maintained by Dr. H. Früchtl.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Prof. Tim Clark on the occasion of his 70th birthday
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to the Topical Collection Tim Clark 70th Birthday Festschrift
Electronic supplementary material
ESM 1
(PDF 1045 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ahmad, S., Berry, E.A., Boyle, C.H. et al. Formation of metallacarboxylic acids through Hieber base reaction. A density functional theory study. J Mol Model 25, 45 (2019). https://doi.org/10.1007/s00894-018-3915-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-018-3915-1