
Abstract
Factor VIII, a human blood plasma protein, plays an important role during the intrinsic pathway of blood coagulation cascade after its activation by thrombin. The activated form of FVIII acts as cofactor to the serine protease Factor IXa, in the conversion of the zymogen Factor X to the active enzyme Factor Xa. The Ser558–Gln565 region of the A2 subunit of Factor VIII has been shown to be crucial for FVIIIa–FIXa interaction. Based on this, a series of linear peptides, analogs of the 558–565 loop of the A2 subunit of the heavy chain of Factor VIII were synthesized using the acid labile 2-chlorotrityl chloride resin and biologically evaluated in vitro by measuring the chronic delay of activated partial thromboplastin time and the inhibition of Factor VIII activity, as potential anticoagulants.
Similar content being viewed by others


Introduction
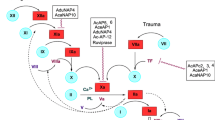
Cardiovascular diseases (CVDs) are the most common cause of morbidity and mortality worldwide (Mahan and Fanikos 2011). Hemostasis, from Greeks meaning blood-stop, is the essential human mechanism to prevent blood loss in case of blood vessel injury, whereas the pathological clot formation, that is thrombosis, is the mechanism when hemostasis is excessively activated in the absence of bleeding. In these abnormal conditions, clot formation is prevented using anticoagulant drugs. Current anticoagulant therapies (Vitamin K antagonists, heparins, direct inhibitors of Factor Xa, etc.) have their limitations, so identification of new targets for blocking thrombosis without increasing the risk of severe and fatal bleeding is the major goal in anticoagulation therapy. These include inhibitors of the factors VIIa, Xa, IXa and VIIIa (Minors 2007; Lin et al. 2006; Howard et al. 2007; Franchini and Mannucci 2011).
Clotting is the result of a combined cascade of two biochemical pathways: “intrinsic pathway”, so called because all components are present in blood, and “extrinsic pathway”, in which tissue factor (TF) is required in addition to circulating components. Although during the previous decades the intrinsic pathway was postulated to play a less significant role; recently, its important participation has been shown in sustaining thrombus development in vivo (Gailani and Renne 2007; Woodruff et al. 2010). FVIIIa forms a heterotrimer complex with FIXa, Ca2+ (which come from activated platelets) and negatively charged phospholipids of the cells membrane. This complex, called tenase, converts Factor X to the activated form FXa, another protease that binds to cofactor Va to generate a complex known as prothrombinase. The latter complex converts prothrombin to thrombin which acts on fibrinogen to generate fibrin monomer, which is polymerized rapidly to form fibrin clot.
Factor VIII is composed of a large number of amino acid residues arranged within six domains known as A1–A2–B–A3–C1–C2 (Vehar et al. 1984). The FVIII protein is secreted as a heterodimer including a heavy chain (A1–A2–B) and a light chain (A3–C1–C2). Both chains are highly associated with a lipoprotein receptor related protein (LRP). FVIII that circulates in blood as a complex multimeric protein with von Willebrand factor is synthesized by endothelial cells (Fay 2004). This protein, vWF, is present in plasma and has been released in the subendothelial tissue of human saphenous and umbilical veins (Terraube et al. 2010). Its main role is the engagement in other proteins, particularly in factor VIII and consequently it is important in the adherence of platelets at the points of wounded vessels (Terraube et al. 2010). Factor VIII is actually inactive as a cofactor in blood coagulation, but is converted into its active cofactor form by proteolytic cleavage (Fang et al. 2007). The A2 subunit includes an epitope, residues 558–565, which interacts with FIXa, residues 330–338, and it is likely the major catalytic interaction between the FVIIIa and FIXa (Fay et al. 1994; O’Brien et al. 1995; Bajaj et al. 2001; Shen et al. 2008; Ngo et al. 2008).
Synthetic peptides or peptidomimetics which show that block the interaction–formation of the FVIIIa–FIXa complex could be used as leading compounds for novel anticoagulant drugs. Here, we present the synthesis and biological evaluation of linear peptides, analogs of the sequence 558–565 of the A2 subunit, aiming at the inhibition of FVIIIa–FIXa interaction, and studying their anticoagulant activity in vitro.
Reagents and methods
The peptides were prepared by solid-phase peptide synthesis techniques. The 9-fluorenylmethoxycarbonyl (Fmoc) protected amino acids and coupling reagents (HOBt, DIC, PyBOP, etc.) as well as 2-chlorotrityl chloride resin (CLTR-Cl) and MBHA Rink amide linker, used as solid support, were purchased from CBL-Patras (Patras, Greece). All the solvents and reagents used for solid-phase synthesis were of analytical grade and were used without further purification. Dichloromethane (DCM) was distilled over CaH2 before usage. Thin layer chromatography (TLC) was carried out on precoated silica gel F-254 plates (Merck, Darmstadt, Germany). HPLC-grade solvents were also purchased from Merck (Darmstadt, Germany) whereas ESI–MS spectra were recorded on a Waters Micromass ZQ 4000 mass detector (positive mode), controlled by MassLynx 4.1 software.
Synthesis
Anchoring on the resin
The examined peptides were synthesized using CLTR-Cl or MBHA Rink amide linker resins by Fmoc solid-phase peptide synthesis protocol. The esterification of the first Fmoc-amino acid onto the CLTR-Cl was carried out as first described by Barlos et al. (1991). CLTR-Cl (1 g) was inflated in DCM (10 mL) for 10 min. Fmoc-amino acid (1 mmol) was then added and the mixture was stirred gently in the presence of DIPEA (2.5 mmol) for 45 min at RT. The remaining active sites of the resin were capped using a mixture of MeOH/DIPEA/DCM (10:5:85) (3 × 5 mL × 10 min) at RT. The Fmoc-amino acid-resin was filtered and washed successively with DCM (3 × 5 mL), DMF (3 × 5 mL), 2-propanol (3 × 5 mL) and n-hexane (1 × 5 mL) and dried over P2O5 under vacuum. Subsequently, the Fmoc group was removed by treatment with a solution of 20 % piperidine in DMF for 30 min.
Coupling procedure
For the coupling reactions the method of carbodiimides was applied. In particular, Fmoc-amino acid (3 equiv of the resin substitution) and HOBt (4.5 equiv of the resin substitution) were dissolved in DMF (1–3 mL) and DIC (3.3 equiv of the resin substitution) was added. The progress and the completeness of each coupling was verified by the Kaiser’s test and TLC, using MeCN/H2O (5:1), CHCl3/MeOH/AcOH (85:10:5) or Tol/MeOH/AcOH (7:1.5:1.5) as development solvents. (The TLC procedure is as follows: a few beads of resin were placed in a very small flask and 2–5 drops of TFA cocktail were added. After a short mixing, the mixture was left at room temperature for 15–20 min. TLC was then applied and the plate was visualized under UV lamp at 254 nm followed by ninhydrin spraying. In case of uncompleted reaction a magenta spot was near the start line of TLC.) The Fmoc group deprotection was performed as described above. All coupling and deprotection steps have been repeated until the expected sequence to be complete.
Cleavage from the resin
The synthesized protected peptide was cleaved from the resin in two steps: firstly, the peptide–resin ester was cleaved with a mixture of DCM/TFE (70:30) for 1 h at RT. The resin was filtered off and the solvent was removed on a rotary evaporator. The obtained crude protected peptide was washed with DCM and precipitated by adding cold and dried diethyl ether as a white solid. In a second step, the side chain deprotection of the linear protected peptide was achieved using a mixture of TFA/DCM/TES (90:5:5) and stirred gently for 3 h at RT. The solvent was evaporated under vacuum; the crude free peptide was precipitated with cold dried diethyl ether and collected by filtration (Fig. 1).

Synthetic procedure of linear analogs of the Sequence Ser558–Gln565
For the peptides attached onto the MBHA Rink amide resin the coupling and deprotection steps were performed as mentioned above. After the completeness of the desired sequence, the cleavage of the peptide from the resin as well as the deprotection of the side protected groups were performed in one step using a mixture of TFA/DCM/TES (90:5:5) by stirring gently for 3 h at RT. The solvent was evaporated under vacuum and the crude free peptide was precipitated with cold and dried diethyl ether and collected by filtration.
Purification and characterization of the crude peptides
The crude intermediates as well as the final compounds were purified on a semi-preparative RP-HPLC (Marathon IV pumps combined with a Fasma 500 UV detector, Rigas Labs, Greece) using a RP-HPLC Nucleosil C-18 column (250 mm × 10 mm, 7 μm) by gradient elution of 5–85 % solvent B (solvent A: 0.1 % ΤFA in H2O; solvent B: 0.1 % ΤFA in MeCN) over 70 min at a flow rate of 2.5 mL/min at 214 and 254 nm. All analytical chromatograms and mass spectra (ESI–MS) were recorded on a Waters Alliance 2695LC HPLC system with a Waters 2966 Photodiode Array detector coupled to a Waters Micromass ZQ mass spectrometer using a RP-Nucleosil C-18 column (250 × 4.6 mm, 5 μm) at 214 and 254 nm. Separation was achieved by gradient elution under the following conditions: S1, 0–30 % solvent B over 30 min at a flow rate of 1 mL/min; S2, 0–40 % solvent B over 30 min at a flow rate of 1 mL/min (solvent A: 0.1 % TFA in water; solvent B: 0.1 % TFA in MeCN).
Biological: anticoagulant assays
Measurements were performed on an automatic analyzer ACL Elite Pro whereas hemostasis reagents were purchased from the Instrumentation Laboratory, UK and recombinant FVIII (rFVIII) was purchased from Baxter AG, USA. The assays were performed in triplicate and the results were expressed as the average of measurements for each sample. Blood from healthy individuals was collected into plastic tubes containing 3.8 % trisodium citrate as anticoagulant (blood/trisodium citrate, 9:1). Citrated blood was immediately centrifuged at 13,000 rpm for 10 min at room temperature to obtain platelet poor plasma (PPP). Citrated samples can be run within 2 hours of sample collection.
Measurement of activated partial thromboplastin time (aPTT) assay
An aliquot of PPP (200 μL) was transferred into a vial and peptide solution (200 μL, 1 mg/mL in Owren–Koller buffer, pH 7.4) was added and the mixture was incubated at 37 °C for 30 min. The control solution was comprised of PPP (200 μL) and buffer (200 μL) and was incubated under the same conditions. After that, the incubated peptide solution (10 μL) was transferred to the instrument cuvette as well as cephalin–kaolin solution (10 μL, 20 mg/mL kaolin and a 1:10 dilution of bovine brain cephalin in 154 mmol/L NaCl), acting as aPTT contact activator (Brunnee et al. 1993). The cuvettes were transferred to the measuring position and CaCl2 (10 μL, 25 mM) was added to prolong further reaction. Time required for clot formation was then reported and the divergent time was extracted by the instrument. Phospholipids as well as Ca2+ are essential for activation of FX and fibrin clot formation.
Prothrombin time (PT) assay tests the overall efficiency of the extrinsic pathway. PT reagent (tissue factor and phospholipids) and calcium ions (CaCl2) are added to pre-incubated plasma and clotting time is recorded directly.
Inhibition of FVIII activity assay
If Factor VIII is added to plasma containing a FVIII antagonist and the mixture is incubated, then the FVIII is progressively inactivated. This assay can be performed using human or porcine recombinant FVIII. The presence of the antagonist is associated with the reduced activity of FVIII, which was determined using aPTT assay with FVIII deficient platelet poor plasma (dPPP) instead of normal one.
An aliquot of rFVIIIa (200 μL, diluted (1:1) in Owren–Koller buffer pH 7.4, 1 U/mL) was transferred into a vial containing peptide solution (200 μL, 1 mg/mL in Owren–Koller buffer, pH 7.4) and incubated at 37 °C for 30 min. Control 1 was comprised of rFVIIIa (400 μL, diluted (1:1) in Owren–Koller buffer pH 7.4, 1 U/mL) whereas Control 2 contained rFVIIIa (200 μL diluted (1:1) in Owren–Koller buffer pH 7.4, 1 U/mL) and buffer (200 μL). All the sample vials were incubated at 37 °C for 30 min. Then, the incubated peptide solution (10 μL) was transferred to the instrument cuvette as well as cephalin–kaolin solution (10 μL), acting as aPTT contact activator and dPPP (deficient PPP to FVIII, 10 μL). The cuvette was transferred to the measuring position and CaCl2 (25 mM, 10 μL) was added to prolong further reactions. Inhibition values were reported and extracted automatically by the instrument (Barrowcliffe et al. 2002).
Results and discussion
Anticoagulants inhibit thrombin and fibrin formation and are used widely for the treatment of venous thromboembolism. The main anticoagulants used by physicians over 60 years are heparin and warfarin. However, the limitations of these medicines have led the pharmaceutical industry to search for new targets and develop more efficient compounds without the side-effects of current anticoagulants (like bleeding complications, interactions with other drugs, dietary implications, etc.) (Gailani and Renne 2007). Current knowledge highlights the importance of FVIIIa–FIXa interaction in clot formation. Thus, the interaction sites represent excellent targets for the anticoagulant therapy (Lenting et al. 2010; Mahan and Fanikos 2011).
Our research effort is focused on the synthesis of peptides (Sarigiannis et al. 2006), analogs of the sequence Ser558–Gln565 of the A2 subunit of FVIIIa that interacts with FIXa, aiming to block the FVIIIa–FIXa complex formation, to suspend platelets adhesion and furthermore clot formation.
Ser558-Val559-Asp560-Gln561Arg562-Gly563-Asn564-Gln565
Here, we present a series of synthetic linear peptides incorporating Gln, Glu, Asp and their side chain benzyl and methyl esters instead of Asn564 as well as the Nα-acetyl peptidic analogs and their biological effect acting as anticoagulant compounds. They are based on point mutations within the 558–565 region resulting in severe or mild hemophilia A, such as Asp560–Ala, mild; Val559–Ala, mild; Ser558–Phe, mild; etc. (Fay and Jenkins 2005; Jenkins et al. 2002).
All the analogs shown in Table 1 were synthesized manually using as solid support the 2-chlorotrityl chloride resin or Rink amide MBHA resin and standard coupling procedures for Fmoc/But methodology. The nature of the resins used for peptide synthesis as well as the efficient protection of the side chain of Gln residues with Trt group yielded final products with high purity. Nevertheless, these crude products were purified by semi-preparative HPLC as reported above. The appropriate fractions containing the desired products were lyophilized to give a white fluffy solid. The final verification of the peptide sequence was achieved by ESI-MS (Micromass, UK). An example of analytical RP-HPLC chromatogram of purified peptide and ESI-MS spectrum is shown in Figs. 2 and 3, respectively for the analog 7.

RP-HPLC of the purified analog Ser-Val-Asp-Gln-Arg-Gly-Asn-Gln-NH2

ESI-MS of the purified analog Ser-Val-Asp-Gln-Arg-Gly-Asn-Gln-NH2
The synthesized analogs were investigated for their anticoagulant activity by measuring their activated partial thromboplastin time (aPTT) and prothrombin time (PT) assay, a sensitive screen method for the evaluation of the intrinsic, extrinsic and common coagulation pathways. PT and aPTT tests are the suggested protocols when a coagulation disorder is suspected, for monitoring anticoagulant therapy, as well as for a preoperative screening. Light transmission is used to determine PT and aPTT times. A change in the light transmittance due to clot formation is converted through a microcomputer as determined coagulation time.
Prolonged clotting time is observed in case of factor deficiencies, liver disease, vitamin K deficiency, heparin presence or other inhibitors, whereas a shortened aPTT is associated with the risk of venous thromboembolism. A long aPTT with normal PT indicates inhibition of coagulation process or bleeding disorders due to deficiencies of factors VIII, IX, XI and XII or the presence of a coagulation inhibitor. Abnormalities in the levels of factors VIII, IX, X, XI or XII have an impact on aPTT, which measures only the integrity of the intrinsic pathway. Short aPTT has been associated with a significantly increased risk of thrombotic events (CLSI 2008). A prolongation of aPTT over 4 s (aPTTsample−aPTTcontrol > 4 s) is remarkable.
The Fig. 4 summarizes the effect of the synthesized peptides on the aPTT assay. It is obvious that analogs 1, 2 and 7 are the most active. Analog 1 shows a prolonged aPTT of 5.8 s whereas analog 2, that incorporates Asp564 instead of Asn, displays similar activity (aPTT: 5.0 s). Furthermore, a quite analogous aPTT (5.1 s) is observed for analog 7. The results show that the native sequence incorporating either C-terminal carboxy or amide group retards clotting formation; more active is the C-terminal carboxy group by comparing analogs 1 and 7. On the opposite, analogs 3 and 4 result in a significant loss of activity in aPTT assay, likely due to the incorporation of aspartic acid β-methyl or β-benzyl ester, respectively. Especially, analog 3 accelerates aPTT more than all the other synthesized peptides, two to fourfold drastic loss of activity. Replacement of Asn564 with Gln (5) or Glu (6), e.g. elongation of the side chain of amino acid at the position 564, doesn’t have any significant result, thus Asn564 appears to be crucial for the prolongation of aPTT. In addition, all the Nα-acetyl analogs reduce aPTT, something rather not usually observed. Furthermore, all the tested peptides show normal PT as it was expected; in case of abnormal PT interaction of the peptide with the factors of extrinsic pathway have to be tested. This, associated with the above results strengthens further the evidence of the role of the peptides 1, 2 and 7 only in the intrinsic coagulation pathway to be evolved.

Divergence of activated partial thromboplastin time (Δt = aPTTsample − aPTTcontrol)
Another useful and more specific test for monitoring hyper-coagulable state is Factor VIII activity. We have examined the influence of these synthetic peptides on the interaction between FVIIIa and FIXa as a reduction of the FVIIIa activity using rFVIIIa in human plasma deficient to FVIII. In order to confirm the presence of the other coagulation factors at normal levels, PT assay applied first. rFVIII is added to deficient platelet poor plasma (dPPP) containing a synthetic peptide and the mixture is incubated. A reduced FVIIIa activity is observed when a synthetic peptide acts as inhibitor.
The Fig. 5 represents the % inhibition of the FVIIIa activity due to the synthesized peptides. Analogs 1, 6 and 12 cause the highest % inhibition of FVIIIa activity. However, it seems interesting that the replacement of Asn564 with Glu (6) enhances the inhibitory activity in comparison with the native peptide, whereas substitution with Asp in the same position (2) slightly reduces this activity. Substitution of Asn564 by Gln and acetylation at Nα-terminal site (12) yields an interesting result too. On the other hand, transformation of peptide 1 to the C-terminal carboxamide (7) decreases the inhibitory activity of the peptide. Nα-acetylation of the above peptide (14) results in a significant loss of the activity. Similar result is obtained by the Nα-acetylated analog of peptide 6, resultant the analog 13. Analogs 8 and 9, Nα-acetyl products of analogs 1 and 2, respectively, show satisfactory activity too. It is also clear that those modifications by introduction of Asp β-alkylated products at the position of Asn564 (3, 4, 10, and 11) show a rather insufficient activity.

Inhibition (%) of the FVIIIa activity caused by the synthesized analogs. Inhibition (%) of the FVIIIa activity = [(100 % value FVIIIa activitycontrol − % value FVIIIa activitysample) / % value FVIIIa activitycontrol] × 100 %
Variations of the results between the two anticoagulant assays are rather expected as the aPTT assay is a general screening test, while monitoring of FVIII activity assay is a more specific test. These variations have to be investigated by examining the interaction of these peptides with other blood coagulation factors involved in the extrinsic pathway.
Considering all the above mentioned remarks, we can conclude that: (i) analogs of the sequence Ser558–Gln565 of the A2 subunit of FVIIIa that interacts with FIXa represent a promising epitope, acting as intrinsic anticoagulants targeting FVIII–FIX interaction; (ii) modifications at the position 564, according to the biological results, show that Asn564 is rather a crucial residue for its anticoagulant activity; (iii) both anticoagulant assays (general screening test as well as more specific assays, monitor of FVIII or other factors activity) are essential for studying the role of these peptides.
Abbreviations
- AcOH:
-
Acetic acid
- aPTT:
-
Activated partial thromboplastin time
- Boc:
-
Tert-butoxycarbonyl
- Bzl:
-
Benzyl group
- CLTR-Cl:
-
2-chlorotrityl chloride
- DCM:
-
Dichloromethane
- DIC:
-
N,N′-diisopropylcarbodiimide
- DIPEA:
-
Diisopropylethylamine
- DMF:
-
N,N′-dimethylformamide
- dPPP:
-
Deficient platelet poor plasma
- ESI-MS:
-
Electrospray ionization mass spectrometry
- Fmoc:
-
9-Fluorenylmethyloxycarbonyl group
- FVIII:
-
Factor VIII
- FIX:
-
Factor IX
- HOBt:
-
1-hydroxybenzotriazole
- i-PrOH:
-
2-propanol
- Me:
-
Methyl group
- MeCN:
-
Acetonitrile
- MeOH:
-
Methanol
- MBHA:
-
4-methylbenzhydrylamine
- PPP:
-
Platelet poor plasma
- PyBOP:
-
(Benzotriazol-1-yloxy)-tris(pyrrolidino)phosphonium hexafluorophosphate
- PT:
-
Prothrombin time
- rFVIII:
-
Recombinant Factor VIII
- tBu:
-
Tert-butyl group
- RP-HPLC:
-
Reversed phase high performance liquid chromatography
- TES:
-
Triethylsilane
- TFA:
-
Trifluoroacetic acid
- TFE:
-
2,2,2-trifluoroethanol
- TLC:
-
Thin layer chromatography
- Tol:
-
Toluene
- Trt:
-
Trityl group
References
Bajaj P, Schmidt A, Mathur A, Padmanabhan K, Zhong D, Mastrii M, Fay P (2001) Factor IXa:Factor VIIIa interaction. J Biol Chem 276:16302–16309
Barlos K, Chantzi O, Gatos D, Stavropoulos G (1991) 2-chlorotrityl chloride resin: studies on anchoring of Fmoc-amino acids and peptide cleavage. Int J Pept Protein Res 37:513–520
Barrowcliffe T, Raut S, Sands D, Hubbard A (2002) Coagulation and chromogenic assays of factor VIII activity: general aspects, standardization, and recommendations. Semin Thromb Hemostasis 28:247–255
Brunnee T, La Porta C, Reddigari S, Salerno V, Kaplan A, Silverberg M (1993) Activation of factor XI in plasma is dependent on factor XII. Blood 81:580–586
Clinical and Laboratory Standards Institute (CLSI) (2008) One stage prothrombin time (PT) test and activated partial thromboplastin time (aPTT) test: approved guide line, document H47-A2, 2nd edn
Fang H, Wang L, Wang H (2007) The protein structure and effect of factor VIII. Thromb Res 119:1–13
Fay P (2004) Activation of factor VIII and mechanisms of cofactor action. Blood Rev 18:1–15
Fay P, Beattie T, Huggins C, Regan L (1994) Factor VIIIa A2 subunit residues 558-565 represent a factor IXa interactive site. J Biol Chem 269:20522–20527
Fay P, Jenkins P (2005) Mutating Factor VIII: lessons from structure to function. Blood Rev 19:15–27
Franchini M, Mannucci P (2011) Inhibitors of propagation of coagulation (factors VIII, IX and XI): a review of current therapeutic practice. British J Clin Pharmacol 72:553–562
Gailani D, Renne T (2007) The intrinsic pathway of coagulation: a target for treating thromboembolic disease. J Thromb Haemostasis 5:1106–1112
Howard E, Becker K, Rusconi C, Becker R (2007) Factor IXa Inhibitors as Novel Anticoagulants. Arterioscler Thromb Vasc Biol 27:722–727
Jenkins PV, Freas J, Schmidt KM, Zhou Q, Fay PJ (2002) Mutations associated with hemophilia A in the 558–565 loop of the factor VIIIa A2 subunit alter the catalytic activity of the factor Xase complex. Blood 100:501–508
Lenting P, Christophe O, Gueguen P (2010) The disappearing act of factor VIII. Haemophilia 16:6–15
Lin J et al (2006) Design, synthesis, and biological evaluation of peptidomimetic inhibitors of factor XIa as novel anticoagulants. J Med Chem 49:7781–7791
Mahan C, Fanikos J (2011) New antithrombotics: the impact on global health care. Thromb Res 127:518–524
Minors D (2007) Haemostasis, blood platelets and coagulation. Anaesth Intensive Care Med 8:214–216
Ngo J, Huang M, Roth D, Furie B, Furie BR (2008) Crystal structure of human factor VIII: implications for the formation of the factor IXa–factor VIIIa complex. Structure 16:597–606
O’Brien L, Medved L, Fay P (1995) Localization of factor IXa and factor VIIIa interactive sites. J Biol Chem 270:27087–27092
Sarigiannis Y, Anastasopoulos C, Liakopoulou-Kyriakides M, Stavropoulos G (2006) Peptides 2006. In: Proceedings of the 29th EPS Gdansk, Poland 0344:694–695
Shen B, Spiegel P, Chang Ch, Huh J, Lee J, Kim J, Kim Y, Stoddard B (2008) The tertiary structure and domain organization of coagulation factor VIII. Blood 111:1240–1247
Terraube V, O’ Donnell J, Jenkins P (2010) Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia 16:3–13
Vehar G, Keyt B, Eaton D et al (1984) Structure of human factor VIII. Nature 312:337–342
Woodruff B, Sullenger B, Becker R (2010) Antithrombotic therapy in acute coronary syndrome: how far up the coagulation cascade will we go. Curr Cardiol Rep 12:315–320
Acknowledgments
Anticoagulant assays were performed under the supervision of Professor of Haematology, Dr Pantelis Makris, School of Medicine, Aristotle University of Thessaloniki, Greece. This Research Project is co-financed: 80 % by European Union—European Social Fund and 20 % by General Secretary of Research & Technology (PENED 03ED569). We also thank SANOFI S.A., Athens, Greece for the financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Abbreviations of common amino acids are in accordance with the recommendations of IUPAC-IUB Joint Commission on Biochemical Nomenclature: Arch Biochem Biophys 206 (1988) v–xxii, J Biol Chem 264 (1989) 668–673, J Peptide Sci 12 (2006) 1–8.
Rights and permissions
About this article
Cite this article
Anastasopoulos, C., Sarigiannis, Y. & Stavropoulos, G. A novel approach in potential anticoagulants from peptides epitope 558–565 of A2 subunit of factor VIII. Amino Acids 44, 1159–1165 (2013). https://doi.org/10.1007/s00726-012-1448-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-012-1448-y