
Abstract
Cholecystokinin (CCK) receptors are overexpressed in numerous human cancers, like medullary thyroid carcinomas, small cell lung cancers and stromal ovarian cancers. The specific receptor-binding property of the endogenous ligands for these receptors can be exploited by labeling peptides with a radionuclide and using these as carriers to guide the radioactivity to the tissues that express the receptors. In this way, tumors can be visualized using positron emission tomography and single photon emission computed tomography imaging. A variety of radiolabeled CCK/gastrin-related peptides has been synthesized and characterized for imaging. All peptides have the C-terminal CCK receptor-binding tetrapeptide sequence Trp-Met-Asp-Phe-NH2 in common or derivatives thereof. This review focuses on the development and application of radiolabeled CCK/gastrin peptides for radionuclide imaging and radionuclide therapy of tumors expressing CCK receptors. We discuss both preclinical studies as well as clinical studies with CCK and gastrin peptides.
Similar content being viewed by others


Introduction on peptide receptor radionuclide imaging and therapy
Regulatory peptide receptors, such as somatostatin receptors, cholecystokinin (CCK) receptors, glucagon-like peptide-1 (GLP-1) receptors and gastrin-releasing peptide (GRP) receptors, are overexpressed in numerous human cancers. Endogenous ligands for the regulatory receptors are potent low molecular weight peptides that are mainly synthesized in the central nervous system and the gastrointestinal tract (Béhé and Behr 2002; Behr and Béhé 2002). Because of their low molecular weight, the regulatory peptides can rapidly penetrate tissues, except for the brain, because the peptides are too hydrophilic to pass through the blood–brain barrier. Therefore, the central nervous system and the gastrointestinal tract can use the same messenger molecules without mutual interference. The specific receptor-binding property of peptides can be exploited by labeling the peptides with a radionuclide and using these as carriers to guide the radioactivity to the tissues that express their specific receptors.
Imaging modalities that are used in nuclear medicine are positron emission tomography (PET) and single photon emission computed tomography (SPECT). PET involves the use of short-lived β+-emitting radionuclides, like 18F and 68Ga. Positrons annihilate with electrons, causing two γ photons of 511 keV to be emitted in opposite directions. A PET scanner detects these emissions coincidentally and provides high-resolution images. The SPECT imaging technique detects γ-photons directly from the radiotracer. SPECT imaging is less expensive than PET since different γ-emitting radionuclides can be used, which are mostly longer-lived and more easy to obtain than PET tracers.
One of the requirements of a diagnostic radiopharmaceutical is the ability to yield a high tumor-to-background ratio to obtain a high contrast image. A therapeutic radiopharmaceutical also requires high uptake in the tumor for prolonged time.
Various radionuclides are available for either imaging or therapeutic applications of peptides (Table 1). 99mTc is often used in SPECT because of its ideal physical properties (t 1/2 = 6 h, E γ = 140 keV) and its good, wide availability. The t 1/2 is long enough to synthesize the 99mTc-labeled radiopharmaceutical and perform imaging, and at the same time short enough to minimize the radiation dose to the patient (Fichna and Janecka 2003). In addition, 111In is often used. Due to its half-life of 2.8 days, 111In can be used to perform scans at later time-points post-injection (p.i.). 123I is used as a tracer in SPECT for scanning of thyroid.
18F is an important isotope in PET imaging. The radionuclide is used in 2-fluoro-2-deoxy-d-glucose (FDG), the most widely used PET tracer. A cyclotron is needed for the production of 18F and the half-life of the isotope (110 min) makes rapid and automated chemistry necessary. In recent years, the use of 68Ga for PET imaging has been evaluated in several studies (Maecke et al. 2005). 68Ga is eluted from an in-house 68Ge/68Ga generator and can be coupled to a chelator-conjugated biomolecule. Because of the long half-life of the parent radionuclide 68Ge (t1/2 = 271 days), 68Ga-based radiopharmaceuticals may become a very cost-effective alternative for cyclotron-based tracers.
Peptide receptor-targeted radiotherapy (PRRT) requires radionuclides with a high cytotoxic potential. Good cytotoxic agents are β-particle emitters, with an intermediate half-life. β-particles with a high energy transfer, such as 90Y, are considered to be appropriate in the treatment of larger tumors, whereas radionuclides with a lower energy, such as 177Lu or 131I, are more suitable for treating tumors with a relatively smaller diameter (O’Donoghue et al. 1995).
To allow radiolabeling with a radiometal, a chelator conjugated to the peptide is required. Several cyclic and acyclic bifunctional chelators have been developed, of which N α-diethylenetriaminopentaacetic acid (DTPA) and 1,4,7,10-tetraazacyclododecane-N,N′,N″,N′′′-tetraacetic acid (DOTA) and their derivatives are most widely used. DTPA is useful for 111In labeling, but it is less suitable for therapeutic purposes, as the 90Y- or 177Lu-DTPA complex is not stable in vivo over longer time (Fichna and Janecka 2003). DOTA conjugates are especially suitable for radionuclide therapy, as they can be radiolabeled with 90Y, 177Lu and 111In to yield a stable radiometal–chelator complex. However, the labeling procedure is more difficult, requiring a heating step that may damage the peptide.
This review will focus on the development and application of radiolabeled CCK/gastrin peptides for imaging and therapy of tumors expressing CCK receptors. We will discuss preclinical studies as well as clinical (imaging and therapy) studies with CCK and gastrin peptides.
CCK and CCK receptors
Cholecystokinin (CCK) is a peptide hormone, originally discovered in the gastrointestinal tract by Ivy and Oldberg (1928). In 1975, Vanderhaeghen et al. described CCK as a gastrin-like immunoreactive peptide and indicated that it is one of the most widespread neuropeptides in the central nervous system. CCK exerts a variety of physiological actions in the gastrointestinal tract and in the central nervous system. CCK was initially characterized as a 33-amino acid sequence, but the peptide showed to be present in a variety of biologically active molecular forms such as CCK39, CCK33, CCK8 and CCK4, all derived from a 115-amino acid precursor molecule. The most abundant peptide in the brain is CCK8 (Asp-Tyr-Met-Gly-Trp-Met-Asp-Phe-NH2) (Noble and Roques 1999; Noble et al. 1999).
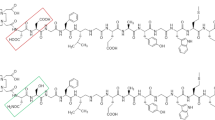
Receptors for CCK have been pharmacologically classified based on their affinity for the endogenous peptide CCK and gastrin. These two ligands share the same amidated C-terminal pentapeptide sequence but differ in sulfation of the tyrosine residue at position 6 (gastrin) or 7 (CCK) (Fig. 1) (Wank 1998; Behr et al. 1999).

Molecular structure of a CCK8 and b minigastrin: n = 0, MG11; n = 5, MG0
Three types of CCK receptors have been identified so far. The CCK1 (formerly known as CCK-A) receptor was first characterized in pancreatic acinar cells (Sankaran et al. 1980), and is mainly located in the periphery, but also in some regions of the brain. In contrast to the high expression of this receptor in various organs in the gastrointestinal tract in rodents, its expression in humans is limited. The CCK2 (formerly known as CCK-B) receptor was discovered in the brain (Innis and Snyder 1980) and is located in the brain and in the stomach, pancreas and gall bladder. In the gastrointestinal tract, activation of this receptor by gastrin stimulates gastric acid secretion (Noble and Roques 1999). The CCK1 and CCK2 receptors have been shown to differ by their relative affinity for gastrin binding, their differential distribution and their molecular structure. The CCK1 receptor binds sulfated CCK with a 500- to 1,000-fold higher affinity than non-sulfated CCK. The CCK2 receptor binds gastrin and CCK with almost the same affinity and does not discriminate between the sulfated and non-sulfated CCK analogs (Noble and Roques 1999). Gastrin receptors in the stomach and CCK2 receptors in the brain were initially thought to be distinct CCK receptors, based on their different relative affinities for CCK and gastrin-like peptides. However, subsequent cloning of both receptors revealed their identical molecular identity (Kopin et al. 1992; Wank 1995). The CCK2 receptor therefore is also referred to as gastrin receptor.
The third type of CCK receptors is the CCK2i4sv receptor, a splice variant of the CCK2 receptor, first isolated and characterized by Hellmich et al. (Hellmich et al. 2000). The CCK2i4sv receptor was discovered in human colorectal cancer cells and stimulates cell growth through a gastrin-independent mechanism. This receptor is generated by intron 4 retention during RNA processing, resulting in a 69-amino acid insert in the third intracellular loop domain of the receptor (Hellmich et al. 2000).
All CCK receptors belong to the superfamily of G-protein coupled receptors (GPCRs), which means that they consist of seven transmembrane domains and activate a second messenger pathway after internalization upon binding of a ligand (Béhé and Behr 2002). Several binding sites for ligand binding to CCK2 receptor have been identified in mutagenesis studies (Fig. 2) (Foucaud et al. 2008). Both the C-terminal phenylalanine of the CCK8 peptide and the tryptophan are located in a hydrophobic/aromatic pocket with residues from different transmembrane helices. The carbonyl group of the amide accepts a hydrogen bond from the hydroxyl oxygen of Tyr189 and the nitrogen amide donates a hydrogen bridge to Asn353. The sulfate of CCK8 binds to the receptor by two hydrogen bonds with Arg57 and Tyr61 in helix 1.

Tumor expression of CCK receptors
CCK1 and CCK2/gastrin receptors have been identified in several normal tissues and in various tumors. The group of Reubi identified an unexpected high incidence (>90%) of CCK2 receptors in medullary thyroid carcinomas (MTC), whereas differentiated thyroid cancers do not express CCK2 receptors (Reubi and Waser 1996). Medullary thyroid carcinomas comprise 3–12% of all thyroid cancers. The 10-year survival of MTC patients is approximately 30%, clearly worse than the survival rates of patients with other forms of differentiated thyroid cancers. One of the major reasons for this is probably that MTCs do not accumulate radioiodine, making radioiodine therapy useless in MTC patients (Béhé and Behr 2002).
CCK2 receptors are also frequently found in astrocytomas (65%) and stromal ovarian cancers (100%). Several other tumor types, such as meningiomas, endometrial and ovarian adenocarcinomas, breast carcinomas and gastroenteropancreatic tumors occasionally express CCK2 receptors. CCK1 receptors are rarely expressed in human tumors (Reubi et al. 1997).
Although earlier studies reported CCK2 receptors in colon cancers and gastric cancers (Upp et al. 1989), further research failed to find high-affinity CCK2 receptors in most of these tumors (Imdahl et al. 1995). Small cell lung cancers often express CCK2 receptors (incidence 89%), whereas non-small lung cancers do not express these receptors (incidence <6%) (Matsumori et al. 1995; Körner et al. 2009).
The splice variant of the CCK2 receptor, the CCK2i4sv receptor, is expressed in human colorectal cancers and pancreatic cancers, but not in normal colorectal mucosa (Hellmich et al. 2000; Smith et al. 2002). However, expression levels of the CCK2i4sv receptor may be too low to allow sufficient targeting with radiolabeled peptides, as a recent study showed rare CCK2i4sv mRNA expression in pancreatic, gastric and colorectal carcinomas (Körner et al. 2009). This study showed high incidence of both the CCK2 and the CCK2i4sv receptor in insulinomas (both 100%), gastrointestinal stromal tumors (both 100%) and SCLC (resp. 89 and 67%).
CCK and gastrin peptides
A lot of research has been done on developing suitable radioligands for targeting the CCK2 receptor in vivo. A variety of radiolabeled CCK/gastrin-related peptides has been synthesized and characterized by different research groups (Table 2). All peptides have the C-terminal CCK receptor-binding tetrapeptide sequence Trp-Met-Asp-Phe-NH2 in common or derivatives thereof. The presence of an intact C-terminal sequence showed to be crucial for receptor binding, although the methionine may be replaced by leucine or norleucine (Behr et al. 1998, 1999). CCK receptor-targeting peptides are discussed in the following paragraphs. The peptides can be categorized based on the sequence of their parent peptide (gastrin or CCK) and on their form, i.e., linear, cyclic, multimers. (Pre)clinical studies with CCK and gastrin receptor-binding peptides are reviewed below.
Preclinical studies with linear CCK2R binding ligands
One of the endogenous ligands for the CCK2 receptor is gastrin, a peptide that has been an important lead compound in the search for new ligands of the receptor. In the late 1990s, Behr et al. (1998) showed promising results for the diagnostic and therapeutic application of 131I-radioiodinated human gastrin-I. This synthetic heptadecapeptide has an affinity for CCK2 receptors in the low nanomolar range, whereas its affinity for CCK1 receptors was at least four orders of magnitude lower. Gastrin-I has a pyroglutamate moiety at its N-terminus, which is known to protect peptides from rapid enzymatic degradation. Behr et al. were able to demonstrate the feasibility of 131I-labeled gastrin-I to target CCK2 receptor-expressing tumors in a patient, as well as CCK receptor expression in normal tissues. Furthermore, Behr and his group investigated a series of 18 radioiodinated gastrin and CCK derivatives for targeting CCK receptors in vivo (Behr et al. 1999). Sulfated CCK analogs and some non-sulfated gastrin analogs had the highest affinities (IC50 values in the nanomolar range), whereas desulfation or the complete removal of the N-terminally located tyrosine of the peptide led to a loss of affinity. Replacement of the pentaglutamate sequence by similarly polar but uncharged pentaglutamine caused a reduction in affinity and a decreased uptake in CCK2R expressing tissues (Behr et al. 1999).
Behr and Béhé investigated two DTPA-conjugated peptides in this study as well. The first amino acid of minigastrin (MG), leucine, was exchanged for d-Glu. Both peptides (minigastrin and d-Glu1-minigastrin) were derivatized with the benzylisothiocyanate derivative of DTPA for labeling with 111In and 90Y. DTPA-d-Glu1-minigastrin showed an improved radiochemical stability over the corresponding DTPA-Leu-minigastrin (t 1/2 is 2.5 times higher) and was therefore used in the subsequent studies (Béhé and Behr 2002; Béhé et al. 2003). The advantage of conjugation of DTPA via an isothiocyanate is that the resulting peptide conjugate has five carboxyl functions available for coordination, whereas DTPA-conjugated peptides normally have four. The additional carboxyl moiety may provide better stability for chelating radiometals such as 90Y, which is known to form rather unstable complexes with conventional DTPA-coupled peptides (Fichna and Janecka 2003; Aloj et al. 2004a).
At the same time that Béhé and Behr published their results on gastrin analogs, Reubi et al. (1998) developed a series of non-sulfated CCK8 analogs. The peptides were N-terminally coupled to DTPA or DOTA to allow radiometal binding. A high specificity towards CCK2 receptors was reported, which was determined by the presence of non-sulfated tyrosine. Wank (1995) had already demonstrated that a sulfated tyrosine in position 27 of CCK-33 is essential for CCK and its derivatives to display affinity for either CCK1 or CCK2 receptors. A CCK8 analog with a non-sulfated tyrosine results in selective binding to the CCK2 receptor. Analogs in which methionine in position 3 and 6 was replaced by norleucine—to prevent oxidation causing a loss of affinity—had similar binding properties (IC50 1.5 nM) as native CCK8 (IC50 2.3 nM) and showed increased plasma stability. These analogs showed promising results in healthy rats: rapid clearance by renal excretion, increased plasma stability and low uptake and retention in the main peripheral soft tissues. Based on this study, Reubi et al. concluded that non-sulfated CCK analogs are highly promising for CCK2 receptor scintigraphy.
Subsequently, de Jong et al. (1999) investigated the potential of 111In-DOTA-CCK8[Nle3,6] for peptide receptor radionuclide therapy. This study concerned internalization, biodistribution and tumor targeting and demonstrated a receptor-specific and time- and temperature-dependent internalization of 111In-DOTA-CCK8 in AR42J cells (rat pancreatic tumor). Evaluation in a syngeneic rat tumor model showed good targeting of CA20948 tumors (rat pancreatic tumor). There was a specific uptake of 111In-DOTA-CCK8[Nle3,6] in CCK2 receptor-expressing tissues and a low uptake in receptor-negative organs. Also, much higher tumor-to-blood ratios were found for the 111In-labeled peptide when compared to a 131I-labeled peptide, showing that residualizing radionuclides such as 111In have advantages over 131I. After internalization and subsequent metabolism of the radiolabeled ligand in the lysozomes, 111In is retained in the cell, resulting in a prolonged residence time in the tumor cells. In contrast, 131I is released from the cell after internalization.
We studied two 99mTc-labeled CCK8 analogs for scintigraphic imaging of CCK receptors, non-sulfated CCK8 (nsCCK8) and sulfated CCK8 (sCCK8) (Laverman et al. 2004). We demonstrated that uptake of the sulfated analog, 99mTc-HYNIC-sCCK8, in both CCK1 and CCK2 receptor-expressing tumors in mice was approximately 15-fold higher than that of the non-sulfated analog. More recently, we showed that 111In-labeled sCCK8 and minigastrin also has affinity for the splice variant of the CCK2 receptor, the CCK2i4sv receptor (Laverman et al. 2007). Tumor uptake of 111In-labeled sCCK8 in CCK2i4sv receptor-positive tumors was similar to the uptake in CCK2 receptor-expressing tumors.
In 2004, Aloj et al. published results obtained with 111In-DTPA-Glu-Gly-CCK8, using DTPA as a chelator (Aloj et al. 2004a, b). They used the same chelator that Behr and Béhé used in their earlier studies with minigastrin (Béhé et al. 2003). A glycine residue was introduced as a spacer between the chelator and the peptide. The peptide conjugates showed good affinity for the CCK2 receptor. In line with the results of Béhé, chelation of 111In was found to be more stable (6% transchelation rate at 24 h in fetal bovine serum), resulting in low background accumulation and fast blood clearance. Tumor uptake of 111In-DTPAGlu-G-CCK8 in A431-CCK2R xenografts was 4% ID/g at 30 min p.i. Renal retention was 5.5% ID/g (Aloj et al. 2004a).
Three 99mTc-labeled minigastrin analogs were investigated by Nock et al. (2005) for targeting the CCK2/gastrin receptor. They derivatized minigastrin with an open-chain tetraamine either directly (Demogastrin 1) or via different spacers (Demogastrin 2 and 3), to achieve stable labeling with 99mTc. After injection in mice, tumor-to-nontarget ratios were especially favorable for 99mTc-Demogastrin 2 (with a glycine between the chelator and the peptide). The high kidney uptake could be reduced by coinjection of poly-Glu-containing peptides. Similar observations were reported by Béhé et al. (2005) after intraperitoneal injection of (Glu)x peptides in rats. Scintigraphy in one MTC patient revealed all known lesions at 90 min after injection, providing high quality images after 4 h.
Mather et al. (2007) aimed to identify a radioligand that combined the relatively high tumor uptake of peptides belonging to the gastrin family with the low renal uptake seen with CCK derivatives. They prepared a library of peptide-DOTA and peptide-DTPA conjugates based on the C-terminal structure of minigastrin. Removal of the pentaglutamate sequence present in minigastrin resulted in a strong reduction in kidney uptake from 60 to 3% ID/g, but also reduced tumor uptake by a factor of 3. Replacement of the pentaglutamate sequence in minigastrin with a hexahistidine tag resulted in a similar reduction in kidney and tumor uptake. They found that a dihistidine analog (DOTA-His-His-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) showed the best results in terms of tumor-to-kidney ratios, with a Ki value of 3.9 nM. Radiolabeling of these analogs, however, needed heating up to 100°C (instead of 70°C), resulting in a high level of oxidation of the methionine residues. The most effective, clinically acceptable antioxidant to overcome this problem was monothioglycerol (MTG). Both the in vitro receptor affinity and the in vivo tumor uptake were reduced by substitution of the methionine residue with nonoxidizable norleucine. These data are in contrast with that as reported by Reubi et al. in their study on CCK8 analogs (Reubi et al. 1998) and with what we found (Roosenburg et al. 2009). In the search for stabilized sCCK8 analogs we synthesized peptides in which the methionine residues were replaced by either norleucine or homopropargylglycine to prevent oxidation. Furthermore, the sulfated tyrosine was replaced by a stable synthetic isostere, phenylalanine sulfonate. In vitro studies showed that the peptides were resistant to oxidation, whereas the affinity was retained in the low nanomolar range. Biodistribution studies in AR42J-tumor-bearing mice showed a tumor uptake of 111In-DOTA-sCCK8[Phe2(p-CH2SO3H), Nle3,6] comparable to 111In-DOTA-sCCK8. Imaging was performed with 111In-DOTA-sCCK8[Phe2(p-CH2SO3H), HPG3,6] (Fig. 3). These compounds are currently being further investigated for its potential in PRRT.

Representative animal SPECT/CT image showing biodistribution of 111In-DOTA-sCCK8[Phe2(p-CH2SO3H), HPG3,6] at 1 h after administration. Radiotracer uptake is clearly visible in the CCK2R-transfected A431 tumor (left shoulder, arrow) and kidneys, whereas no uptake is noted in the mock-transfected A431 tumor (right flank, arrow head)
In mice, Good et al. (2008) reported a decreased circulatory half-life and lower tumor uptake of 111In-DOTA-MG11 (d-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) in comparison with 111In-DTPA-MG0 (d-Glu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2). Although tumor-to-kidneys ratios were higher for 111In-DOTA-MG11, the absolute tumor uptake in AR42J tumors was lower. Béhé et al. (2005) showed that the high renal uptake of 111In-DTPA-MG0 in mice could be significantly reduced by coinjection of polyglutamic acids, whereas tumor uptake was not impaired. Therefore, the diagnostic efficacy of this compound may be better than that of 111In-DOTA-MG11.
Preclinical studies with cyclic peptides
De Luca et al. (2006) designed a library of 14 cyclic CCK8 analogs, based on the structural details of the complex between CCK8 and the CCK2 receptor fragment. They identified structural requirements for preferential binding to the CCK2 receptor. The results of the binding assays with this series of 14 peptides confirmed that the tetrapeptide Trp30-Met31-Asp32-Phe33 is significant to binding, as was already stated earlier by Béhé and Behr (Behr et al. 1998, 1999). De Luca et al. found that, in fact, the most active molecules maintain the turn region centered around Trp30-Met31 and that the Phe33 side chain must be conserved. Nevertheless, the IC50 values of all the tested cyclic analogs were much higher (1,000-fold or more) than that of CCK8 itself.
Linear gastrin analogs exist under various folded conformations in solution. As cyclization showed to improve the in vivo characteristics of other peptides such as RGD analogs (Dijkgraaf et al. 2006; Haubner 2006), Von Guggenberg et al. (2009) designed and evaluated two cyclic minigastrin analogs (cyclo-MG), based on MG11. They synthesized two MG analogs containing unnatural amino acids in the peptide chain and a cyclic constraint that was introduced through an internal amide bond (Table 2). In position 1, d-Glu was incorporated in the peptide through the γ carboxylic group. In position 9, Gly was replaced by d-Lys. These modifications allowed cyclization by lactamization between the side-chain amino group of d-Lys9 and the α-carboxylic group of d-Glu1. To avoid oxidation during the radiolabeling process, Met was replaced by Nle in position 11. The peptide was derivatized with HYNIC on the N-terminus to allow radiolabeling with 99mTc. In vitro studies showed that receptor binding was impaired by cyclization, possibly related to the reduced flexibility of the peptide backbone. However, internalization in AR42J cells of both cyclic MG analogs was similar to that of linear HYNIC-MG11. Tumor uptake of >3% ID/g at 1 h p.i. was observed in nude mice with s.c. AR42J tumors for both analogs, whereas the linear MG1 (99mTc-HYNIC-γ-d-Glu-Ala-Tyr-d-Lys-Trp-Met-Asp-Phe-NH2) analog showed a very low tumor uptake of <0.3% ID/g. The overall biodistribution of 99mTc-HYNIC-cyclo-MG1 was similar to that of 99mTc-HYNIC-MG11. It was claimed that cyclization is important to maximize tumor uptake of this peptide, but the overall pharmacokinetic profile was not improved. Stabilization of the peptide requires further optimization to obtain a radioligand suitable for diagnostic and/or therapeutic applications. Stabilization could possibly also be achieved by dimerization of MG11, which was shown to be effective in increasing tumor uptake while maintaining low kidney retention (Sosabowski et al. 2008).
Clinical studies with CCK and gastrin analogs
Several studies in MTC patients have been performed with both gastrin-like (Behr et al. 1999; Béhé and Behr 2002; Nock et al. 2005; Gotthardt et al. 2006; Fröberg et al. 2009) and CCK-like (Kwekkeboom et al. 2000) peptides in the past few years. A direct comparison of the results of these studies is difficult, as different protocols were used.
The evaluation of the CCK8 analog 111In-DTPA-CCK8[Nle3,6] in patients was reported by Kwekkeboom et al. (2000). The results showed high background activity levels in the scintigraphic images, relatively low uptake in the strongly CCK receptor-positive stomach and a rapid degradation of 111In-DTPA-CCK8[Nle3,6] in serum. Although confirmed MTC lesions could be visualized in two patients, small MTC lesions could not be detected.
Behr et al. (1999) reported a pilot clinical study with 111In-DTPA-MG0 in four MTC patients. They found CCK2 receptor-targeting in physiologically CCK2 receptor-expressing tissues (e.g. the stomach) as well as in metastatic MTC lesions. In 2002, Behr and Béhé published a clinical study with 111In-DTPA-MG0 for both imaging and therapy in 75 and 8 MTC patients, respectively (Béhé and Behr 2002). In the imaging study, 185–259 MBq of 111In-DTPA-MG0 was injected and whole body scans were performed at several time-points after injection. They found that normal organ uptake was restricted to the stomach, caused by high physiological CCK2 receptor expression, and to the kidneys, due to tubular reabsorption. In patients known to have the disease, all of the tumors that were found with conventional imaging modalities were already visualized at 1 h p.i., although optimal scans were obtained at 24 h p.i. More importantly, in 29 out of 32 MTC patients with occult disease, at least one lesion was visualized. Uptake in the liver and spleen was very low.
Eight patients with advanced and rapidly progressing metastatic MTC were injected with high activity doses of 90Y-labeled DTPA-MG0. The study was designed starting at 1.11 GBq/m2 per injection with a maximum of four injections. Every fourth patient the dose was escalated by 1.11 GBq/m2. Three patients treated with the lowest activity dose did not show any signs of toxicity. All three patients treated at 1.48 GBq/m2 developed transient and usually mild (≤grade 3) hematologic toxicity. One of the two patients at 1.85 GBq/m2 developed transient grade 4 thrombocytopenia and grade 1 nephrotoxicity. Four patients experienced stabilization of their previously rapidly progressing disease, lasting for up to 36 months. However, two patients developed severe renal failure 1–2 years after radiopeptide therapy. In summary, the therapeutic effects of 90Y-DTPA-MG0 were encouraging, but renal toxicity in a few patients was rather severe. Therefore the authors aimed to continue the studies with Auger electron-emitting radionuclides such as 111In instead of β+-emitting 90Y, as these showed much less nephrotoxicity in preclinical settings.
Gotthardt et al. (2006) reported a tumor detection rate of 87% by means of scintigraphy using 111In-DTPA-MG0 in a group of 26 MTC patients. Most of the patients in that study, however, had known metastases and tumor lesions were found in only one patient in the group of patients with occult disease.
Recently, Fröberg et al. (2009) investigated and compared 99mTc-Demogastrin 2, evaluated earlier in vitro and in vivo by Nock et al. (2005), with 111In-DOTA-CCK8 and 111In-DOTA-MG11.An earlier study of this group (Breeman et al. 2008) involved optimizing the radiolabeling conditions and investigating the preclinical aspects of the compounds. 99mTc-Demogastrin 2 showed the best visualization, which may be due to better imaging properties of 99mTc compared to 111In. Figure 4 shows examples of the comparative images of the three radiopharmaceuticals studied. 99mTc-Demogastrin 2 visualized all known lesions in six MTC patients, whereas several known lesions were missed with the other two compounds. In addition, in four patients, new lesions in neck, brain, bone and liver were discovered with 99mTc-Demogastrin 2. Both 111In-DOTA-CCK8 and 111In-DOTA-MG11 showed to be less suitable for scintigraphy, as the sensitivity as well as the uptake in visible lesions was limited and appeared to be insufficient for radionuclide therapy in these tumors. Moreover, the stability of 111In-DOTA-MG11 was low. Although this peptide was reported to be stable in ex vivo human serum, an HPLC analysis of blood samples from patients in this study showed that only 10% of the original peptide was still intact 10 min after administration. 99mTc-Demogastrin 2 was more stable, illustrated by the fact that more than 60% of the radioligand was still intact at the same time point. Visual evaluation showed that renal uptake of 99mTc-Demogastrin 2 was in the same range as the uptake of 111In-DOTA-MG11 and 111In-DOTA-CCK8. Therefore, 99mTc-Demogastrin 2 appeared to be a promising diagnostic tool in patients with MTC.

Scintigrams of a patient (anterior and posterior views): first row 99mTc-Demogastrin 2 at 4 and 24 h, second row 111In-DOTA-MG11 at 4 and 24 h and thirrd row 111In-DOTA-CCK at 4 and 24 h. Tumor lesions (and stomach) are best visible on 99mTc-Demogastrin 2 scintigraphy; some lesions are marked with arrows [reprinted with kind permission of Springer Science + Business Media from Fröberg et al. (2009)]
Conclusion
We have discussed a range of gastrin and CCK analogs for targeting CCK2R positive tumors in this review. As is the case for all peptides used in peptide receptor radionuclide imaging and therapy, CCK/gastrin receptor-targeting peptides should meet some basic criteria, such as low nanomolar receptor affinity, rapid and efficient accumulation in the tumor, low uptake and rapid wash-out in/from normal tissues and good in vivo stability.
Of the peptides discussed in this paper, the minigastrin analog MG0 showed good tumor targeting, but this analog has the disadvantage of extremely high renal retention. The N-terminally truncated analog MG11 displayed a similar receptor affinity, with a much lower kidney accumulation. However, due to poor in vivo stability, the accretion of this peptide in the tumor is much lower. In an attempt to improve stability, a cyclic variant of this MG11 was synthesized. However, this did not improve the stability. In another approach, the dimerization of MG11 does lead to improved tumor-background ratios, while maintaining low kidney uptake. Further studies will be required to investigate the stability of this dimeric minigastrin analog. Finally, incorporation of synthetic amino acids may result in improved in vivo stability.
So far (unsulfated) CCK8 analogs combine good tumor targeting with low kidney retention, making these peptide suitable candidates for PRRT. Future studies are warranted to establish the optimal CCK2R targeting peptide.
References
Aloj L, Caraco C, Panico M, Zannetti A, Del Vecchio S, Tesauro D, De Luca S, Arra C, Pedone C, Morelli G, Salvatore M (2004a) In vitro and in vivo evaluation of 111In-DTPAGlu-G-CCK8 for cholecystokinin-B receptor imaging. J Nucl Med 45:485–494
Aloj L, Panico M, Caraco C, Del Vecchio S, Arra C, Affuso A, Accardo A, Mansi R, Tesauro D, De Luca S, Pedone C, Visentin R, Mazzi U, Morelli G, Salvatore M (2004b) In vitro and in vivo characterization of indium-111 and technetium-99m labeled CCK-8 derivatives for CCK-B receptor imaging. Cancer Biother Radiopharm 19(1):93–98
Béhé MP, Behr TM (2002) Cholecystokinin-B (CCK-B)/gastrin receptor-targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers 66:399–418
Béhé MP, Becker W, Gotthardt M, Angerstein C, Behr TM (2003) Improved kinetic stability of DTPA-dGlu as compared with conventional monofunctional DTPA in chelating indium and yttrium: preclinical and initial clinical evaluation of radiometal labelled minigastrin derivatives. Eur J Nucl Med Mol Imaging 30:1140–1146
Béhé MP, Kluge G, Becker W, Gotthardt M, Behr TM (2005) Use of polyglutamic acids to reduce uptake of radiometal-labeled minigastrin in the kidneys. J Nucl Med 46:1012–1015
Behr TM, Béhé MP (2002) Cholecystokinin-B/gastrin receptor-targeting peptides for staging and therapy of medullary thyroid cancer and other cholecystokinin-B receptor-expressing malignancies. Semin Nucl Med 32(2):97–109
Behr TM, Jenner N, Radetzky S, Béhé MP, Gratz S, Yücekent S, Raue F, Becker W (1998) Targeting of cholecystokinin-B/gastrin receptors in vivo: preclinical and initial clinical evaluation of the diagnostic and therapeutic potential of radiolabelled gastrin. Eur J Nucl Med 25:424–430
Behr TM, Jenner N, Béhé MP, Angerstein C, Gratz S, Raue F, Becker W (1999) Radiolabeled peptides for targeting cholecystokinin-B/gastrin receptor-expressing tumors. J Nucl Med 40:1029–1044
Breeman WAP, Fröberg AC, De Blois E, van Gameren A, Melis M, De Jong M, Maina T, Nock BA, Erion JL, Maecke HR, Krenning EP (2008) Optimised labeling, preclinical and initial clinical aspects of CCK-2 receptor-targeting with 3 radiolabeled peptides. Nucl Med Biol 35:839–849
de Jong M, Bakker WH, Bernard BF, Valkema R, Kwekkeboom DJ, Reubi J-C, Srinivasan A, Schmidt M, Krenning EP (1999) Preclinical and initial clinical evaluation of 111In-labeled non-sulfated CCK8 analog: a peptide for CCK-B receptor-targeted scintigraphy and radionuclide therapy. J Nucl Med 40(12):2081–2087
De Luca S, Saviano M, Della Moglie R, Digilio G, Bracco C, Aloj L, Tarallo L, Pedone C, Morelli G (2006) Conformationally constrained CCK8 analogues obtained from a rationally designed peptide library as ligands for cholecystokinin type B receptor. Chem Med Chem 1:997–1006
Dijkgraaf I, Kruijtzer JA, Frielink C, Soede AC, Hilbers HW, Oyen WJG, Corstens FH, Liskamp RM, Boerman OC (2006) Synthesis and biological evaluation of potent alphavbeta3-integrin receptor antagonists. Nucl Med Biol 33:953–961
Fichna J, Janecka A (2003) Synthesis of target-specific radiolabeled peptides for diagnostic imaging. Bioconjug Chem 14:3–17
Foucaud M, Archer-Lahlou E, Marco E, Tikhonova IG, Maigret B, Escrieut C, Langer I, Fourmy D (2008) Insights into the binding and activation sites of the receptors for cholecystokinin and gastrin. Regul Pept 145:17–23
Fröberg AC, de Jong M, Nock BA, Breeman WAP, Erion JL, Maina T, Verdijsseldonck M, de Herder WW, van der Lugt A, Kooij PPM, Krenning EP (2009) Comparison of three radiolabelled peptide analogues for CCK-2 receptor scintigraphy in medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging 36:1265–1272
Good S, Walter MA, Waser B, Wang X, Müller-Brand J, Béhé MP, Reubi J-C, Maecke HR (2008) Macrocyclic chelator-coupled gastrin-based radiopharmaceuticals for targeting of gastrin receptor-expressing tumours. Eur J Nucl Med Mol Imaging 35:1868–1877
Gotthardt M, Béhé MP, Beuter D, Battmann A, Bauhofer A, Schurrat T, Schipper M, Pollum H, Oyen WJG, Behr TM (2006) Improved tumour detection by gastrin receptor scintigraphy in patients with metastasised medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging 33:1273–1279
Haubner R (2006) Aplhavbeta3-integrin imaging: a new approach to characterise angiogenesis? Eur J Nucl Med Mol Imaging 33:S54–S63
Hellmich MR, Rui X-L, Hellmich HL, Fleming RYD, Evers BM, Townsend CM Jr (2000) Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J Biol Chem 275:32122–32128
Imdahl A, Mantamadiotis T, Eggstein S, Farthmann EH, Baldwin GS (1995) Expression of gastrin, gastrin/CCK-B and gastrin/CCK-C receptors in human colorectal carcinomas. J Cancer Res Clin Oncol 121:661–666
Innis RB, Snyder SH (1980) Distinct cholecystokinin receptors in the brain and pancreas. Proc Natl Acad Sci USA 77:6917–6921
Ivy AC, Oldberg E (1928) A hormone reaction for gallbladder contraction and evacuation. Am J Physiol 86:599–613
Kopin AS, Lee YM, McBride EW, Miller LJ, Lu M, Lin HY, Kolakowski LF, Beinborn M (1992) Expression, cloning and characterization of the canine parietal cell gastrin receptor. Proc Natl Acad Sci USA 89:3605–3609
Körner M, Waser B, Reubi JC and Miller LJ (2009) CCK2 receptor splice variant with intron 4 retention in human gastrointestinal and lung tumors. J Cell Mol Med. doi:10.1111/j.1582-4934.2009.00859.x
Kwekkeboom DJ, Bakker WH, Kooij PPM, Erion J, Srinivasan A, de Jong M, Reubi J-C, Krenning EP (2000) Cholecystokinin receptor imaging using an octapeptide DTPA-CCK analogue in patients with medullary thyroid carcinoma. Eur J Nucl Med 27:1312–1317
Laverman P, Béhé MP, Oyen WJG, Willems PHG, Corstens FHM, Behr TM, Boerman OC (2004) Two technetium-99m-labeled cholestokinin-8 (CCK8) peptides for scintigraphic imaging of CCK receptors. Bioconjug Chem 15:561–568
Laverman P, Roosenburg S, Gotthardt M, Park J, Oyen WJG, de Jong M, Hellmich MR, Rutjes FPJT, van Delft FL, Boerman OC (2007) Targeting of a CCK2 receptor splice variant with 111In-labelled cholecystokinin-8 (CCK8) and 111In-labelled minigastrin. Eur J Nucl Med Mol Imaging 35:386–392
Maecke HR, Hofmann M, Haberkorn U (2005) (68)Ga-labeled peptides in tumor imaging. J Nucl Med 46:172S–178S
Mather SJ, McKenzie AJ, Sosabowski JK, Morris TM, Ellison D, Watson SA (2007) Selection of radiolabeled gastrin analogs for peptide receptor-targeted radionuclide therapy. J Nucl Med 48:615–622
Matsumori Y, Katakami N, Ito M, Taniguchi T, Iwata N, Takaishi T, Chihara K, Matsui T (1995) Cholecystokinin-B/gastrin receptor: a novel molecular probe for human small cell lung cancer. Cancer Res 55:276–279
Noble F, Roques BP (1999) CCK-B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog Neurobiol 58:349–379
Noble F, Wank SA, Crawley JN, Bradwejn J, Seroogy KB, Hamon M, Roques BP (1999) Structure, distribution and functions of cholecystokinin receptors. Pharmacol Rev 51(4):745–781
Nock BA, Maina T, Béhé MP, Nikolopoulou A, Gotthardt M, Schmitt JS, Behr TM, Maecke HR (2005) CCK-2/gastrin receptor-targeted tumor imaging with 99mTc-labeled minigastrin analogs. J Nucl Med 46:1727–1736
O’Donoghue JA, Bardiès M, Wheldon TE (1995) Relationships between tumor size and curability for uniformly targeted therapy with beta-emitting radionuclides. J Nucl Med 36:1902–1909
Reubi JC, Waser B (1996) Unexpected high incidence of cholecystokinin B/gastrin receptors in human medullary thyroid carcinomas. Int J Cancer 67:644–647
Reubi J-C, Schaer J-C, Waser B (1997) Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res 57:1377–1386
Reubi J-C, Waser B, Schaer J-C, Laederach U, Erion J, Srinivasan A, Schmidt M, Bugaj JE (1998) Unsulfated DTPA- and DOTA-CCK analogs as specific high-affinity ligands for CCK-B receptor-expressing human and rat tissues in vitro and in vivo. Eur J Nucl Med 25:481–490
Roosenburg S, Laverman P, Joosten L, Eek A, Oyen WJG, de Jong M, Rutjes FPJT, van Delft FL, Boerman OC (2009) Stabilized 111In-labeled sCCK8 analogues for targeting CCK2-receptor positive tumors: synthesis and evaluation (submitted)
Sankaran H, Goldfine ID, Deveney CW (1980) Binding of cholecystokinin to high affinity receptors on isolated rat pancreatic acini. J Biol Chem 255:11849–11853
Smith JP, Verderame MF, McLaughlin P, Martenis M, Ballard E, Zagon IS (2002) Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int J Mol Med 10:689–694
Sosabowski J, Matzow T, Foster J and Mather S (2008) Targeting of CCK2 receptor expressing tumours using an 111In-labelled minigastrin dimer. Q J Nucl Med Mol Imaging 33 (S109)
Upp JR Jr, Singh P, Townsend CM Jr, Thompson JC (1989) Clinical significance of gastrin receptors in human colon cancers. Cancer Res 49:488–492
Vanderhaeghen JJ, Signeau JC, Gepts W (1975) New peptide in vertebrate CNS reacting with antigastrin antibodies. Nature (Lond) 257:604–605
von Guggenberg E, Salleger W, Helbok A, Ocak M, King R, Mather SJ, Decristoforo C (2009) Cyclic minigastrin analogues for gastrin receptor scintigraphy with technetium-99m: preclinical evaluation. J Med Chem 52:4786–4793
Wank SA (1995) Cholecystokinin receptors. Am J Physiol 269:G628–G646
Wank SA (1998) G Protein-coupled receptors in gastrointestinal physiology I. CCK receptors: an exemplary family. Am J Physiol 274:G607–G613
Acknowledgments
The authors thank Ciara Finucane, Julie Foster and Jane Sosabowski, Centre for Molecular Oncology and Imaging, Institute of Cancer, Barts and the London, Queen Mary’s School of Medicine and Dentistry, London, United Kingdom for performing animal SPECT/CT imaging (Fig. 3). This study was financially supported by Dutch Cancer Society grant KUN 2006-3575.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Roosenburg, S., Laverman, P., van Delft, F.L. et al. Radiolabeled CCK/gastrin peptides for imaging and therapy of CCK2 receptor-expressing tumors. Amino Acids 41, 1049–1058 (2011). https://doi.org/10.1007/s00726-010-0501-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0501-y