
Abstract
Insulin-like 3 (INSL3) is a novel circulating peptide hormone that is produced by testicular Leydig cells and ovarian thecal and luteal cells. In males, INSL3 is responsible for testicular descent during foetal life and suppresses germ cell apoptosis in adult males, whereas in females, it causes oocyte maturation. Antagonists of INSL3 thus have significant potential clinical application as contraceptives in both males and females. Previous work has shown that the INSL3 receptor binding region is largely confined to the B-chain central α-helix of the hormone and a conformationally constrained analogue of this has modest receptor binding and INSL3 antagonist activity. In the present study, we have employed and evaluated several approaches for increasing the α-helicity of this peptide in order to better present the key receptor binding residues and increase its affinity for the receptor. Analogues of INSL3 with higher α-helicity generally had higher receptor binding affinity although other structural considerations limit their effectiveness.
Similar content being viewed by others
Introduction
Insulin-like peptide 3 (INSL3) was discovered in the early 1990s (Adham et al. 1993) and shown to belong to the insulin–relaxin superfamily of polypeptide hormones. It was originally named Leydig cell insulin-like peptide (Ley-IL) because it was found in the Leydig cells of the testis (Burkhardt et al. 1994) and has also been referred to as RLF (relaxin-like factor) due to its relaxin-like activity in a mouse interpubic ligament bioassay (Büllesbach and Schwabe 1995). In the male, INSL3 acts as a marker for fully differentiated adult-type Leydig cells (Ivell and Einspanier 2002) and is also expressed by ovarian follicles and in the corpus luteum in the female but at lower levels compared to the male (Roche et al. 1996; Tashima et al. 1995).
INSL3 is a circulating hormone which has important reproductive and non-reproductive roles. During foetal life it is principally involved in mediation of the transabdominal phase of testicular decent as INSL3 or its receptor, RXFP2, knockout male mice have been shown to have a similar phenotype in which both are cryptorchid, i.e. they retain their testes in the abdominal cavity, which leads to impaired spermatogenesis and infertility (Bachelot et al. 2000; Bogatcheva et al. 2003; Feng et al. 2004; Foresta and Ferlin 2004; Nef and Parada 1999; Spiess et al. 1999; Zimmermann et al. 1999). In adults, the INSL3 and RXFP2 system acts as a paracrine factor in mediating gonadotropin actions (Kawamura et al. 2004). Luteinizing hormone (LH), which is released by the anterior pituitary gland, stimulates INSL3 transcripts in ovarian theca and testicular Leydig cells. INSL3 successively binds RXFP2 expressed in germ cells to activate the inhibitory G protein, thus leading to decreases in cAMP production. This, in turn, leads to the initiation of meiotic progression of arrested oocytes in preovulatory follicles in vitro and in vivo and suppresses male germ cell apoptosis in vivo (Kawamura et al. 2004).
A recent study has shown that in males the INSL3/RXFP2 signalling system is also involved in bone metabolism as RXFP2 −/− knockout mice showed a considerable reduction in their bone mass, mineralizing surface and bone formation compared to wild type mice (Ferlin et al. 2008). This study also showed that 64% of young men with RXFP2 mutations had significant reduction in bone mass density, a sign of osteoporosis (Ferlin et al. 2008). INSL3 may also play a role in the pathobiology of some forms of human cancers, such as thyroid carcinoma, as its expression is upregulated in hyperplastic and neoplastic human thyrocytes (Klonisch et al. 2005).
INSL3 is expressed as a preprohormone with an N-terminal signal peptide for secretion, a B-chain, a C-peptide, and a C-terminal A-chain. The preprohormone is subsequently processed into a mature peptide through cleavage of the signal peptide and formation of two inter-chain and an intra-A-chain disulfide bond followed by proteolytic removal of the C-peptide (Adham et al. 1993; Hsu 2003). Mature human INSL3 consists of an A- and B-chain of 26 and 31 amino acids, respectively, and its tertiary structure has recently been solved using solution NMR spectroscopy (Rosengren et al. 2006) (Fig. 1a). INSL3 adopts a core structure similar to that found in insulin and relaxin, especially in the region confined by the disulfide bonds.

a Solution NMR structure of native human INSL3 showing the important receptor binding residues (H12, R16, V19, R20 and W27). b Analogue 30 (Table 1) in which a truncated INSL3 A-chain (from residue CysA15 to CysA24) is linked via a disulfide bond to the truncated B-chain
To determine the residues involved in receptor binding, recent structure–activity studies by our group using single Ala substitution have shown that substituting ArgB16 and ValB19 significantly reduced receptor binding affinity (Rosengren et al. 2006). On the other hand, multi-Ala substitution showed that HisB12 and ArgB20 have a strong synergistic effect with ArgB16, suggesting that HisB12 and ArgB20 may play a role in the initial step of receptor recognition, involving electrostatic interactions between basic residues of the peptide and acidic residues on the receptor (Rosengren et al. 2006). In addition to these residues, TrpB27 toward the C-terminus of the B-chain has also been shown to be crucial for binding of INSL3 as the mutation or deletion of TrpB27 leads to loss of receptor binding affinity (Büllesbach and Schwabe 1999; Rosengren et al. 2006). These B-chain residues collectively form a receptor binding motif (HB12, RB16, V19, R20 and W27). A-chain N-terminal truncation studies of INSL3 have shown that truncation of the INSL3 peptide to CysA10 results in a peptide with high receptor binding affinity but which is devoid of signalling activity, i.e. an antagonist (Büllesbach and Schwabe 2005; Hossain et al. 2008).
Despite knowledge of the region of the peptide that is involved in receptor signalling, there is no clear understanding of the mechanism of receptor activation. A recent study has shown that the mechanism of receptor activation by INSL3 is independent of the amino acid side chains and is a function of certain peptide bonds at the N-terminus of the A-chain (Büllesbach and Schwabe 2007). These authors proposed the backbone amide bond around ArgA8 and TyrA9 to be crucial for receptor activation, as the replacement of these residues with alanine does not affect signalling whereas their deletion or replacement with d-Pro has no impact on receptor binding but severely retards receptor activation (Büllesbach and Schwabe 2007). In contrast, a more recent study on a relaxin-2, which also binds to INSL3 receptor (RXFP2), has shown that there are other residues in the A-chain which are involved in receptor activation. These authors have shown that KA17 is an important residue for receptor activation as its mutation to alanine enhances RXFP2-activation activity of relaxin-2 as a result of inducing active conformational transformation. On the other hand, the replacement of this residue with a polar or negatively charged residue reduces the receptor activation activity of relaxin-2 (Park et al. 2008).
INSL3, due to its role in germ cell maturation in adults, has enormous potential as a clinical agent in the area of fertility management; in particular, antagonists of this peptide may have significant clinical promise for use as both a male and female contraceptive. As discussed above, INSL3 has been shown to bind to its receptor using the residues primarily located on the α-helical region of the B-chain. In an attempt to develop mimetics of INSL3 B-chain with high receptor binding affinity and antagonistic activity, our group recently designed and synthesized shortened analogues of the INSL3 B-chain that had antagonistic activity in vitro (Del Borgo et al. 2006; Shabanpoor et al. 2007). In vivo administration of one of these antagonists into the testes of rats resulted in a substantial decrease in testis weight probably due to the inhibition of germ cell survival (Del Borgo et al. 2006). However, these peptides have receptor binding affinities within the micromolar range compared to the nanomolar affinity of the native INSL3. This is due, in part, to the lack of INSL3-like native α-helical structure in these peptides, which is thought to be important for the presentation of binding residues in the correct orientation to the binding pocket of the receptor. Therefore, the aim of this study is to systematically examine known methods, including introduction of disulfide and lactam constraints or α-helix-inducing residues and N-caps, to induce additional α-helicity in the B-chain mimetics of INSL3 and to evaluate their effectiveness as INSL3 antagonists.
Materials and methods
9-Fluroenylmethoxycarbonyl (Fmoc) protected l-α-amino acids, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), N,N-dimethylformamide (DMF), piperidine and trifluoroacetic acid (TFA) were obtained from Auspep (West Melbourne, Australia). Fmoc-Aib-OH, Fmoc-Asp(O-2-PhiPr)-OH, Fmoc-Dab(Mtt)-OH, Fmoc-Glu(O-2-PhiPr)-OH, Fmoc-Lys(Mtt)-OH and PyBOP were obtained from Novabiochem (Melbourne, Australia). Fmoc-PAL-PEG-PS and Fmoc-L-Ala-PEG-PS resins with substitution of 0.20 mmol/g were purchased from Applied Biosystems (Melbourne, Australia). Methanol, diethylether, dichloromethane (Merck, Melbourne, Australia); 3,6-dioxa-1,8-octanedithiol (DODT), triisopropylsilane (TIPS), diisopropylethylamine (DIPEA), 1,2,4,5-benzene-tetracarboxylic dianhydride (Sigma-Aldrich, Sydney, Australia); 2,2′-dipyridyl disulfide (DPDS), (Fluka-Switzerland); acetonitrile and NH4HCO3, (NH4)2CO3 (BDH Laboratory Supplies, Poole, UK); and trifluoromethanesulfonic acid (TFMSA) (MP Biomedicals, Sydney, Australia). Dulbecco’s modified Eagles’ medium (DMEM), RPMI 1640 medium, 2 mM l-glutamine, foetal calf serum and penicillin/streptomycin were all obtained from Trace Biosciences (Sydney, Australia). All other reagents were obtained from Sigma-Aldrich (Sydney, Australia).
Molecular modelling
All molecular modelling was performed using SYBYL molecular modelling software (Tripos, version 7.0, St Louis, MO, USA) on a Silicon Graphics O2 workstation.
Design of disulfide constrained mimetics
All the single chain disulfide constrained mimetics were designed as described previously (Shabanpoor et al. 2007). Briefly, using the NMR structure of native human INSL3 as a template, the A-chain was deleted and a disulfide bond was inserted between β-carbon atoms of residues less than 10 Å apart on the strand and α-helical segments of the B-chain. The two chain disulfide constrained analogue 30 (Fig. 1b) was designed by truncating the B-chain strand from N-terminus up to LeuB9 and from the C-terminus until TrpB27, and the A-chain was truncated from N-terminus until CysA15 and from C-terminus until CysA24. In native INSL3, CysA24 forms a disulfide bond with CysB22 so, hence, there was no need for creation of a disulfide bond. CysB10, which pairs with CysA11 in an inter-chain disulfide bond, was mutated to serine (SerB10). On the other hand, CysA15 forms an intra-A-chain disulfide bond with CysA10 and in order to from the second disulfide bond, LeuB9, which points toward CysA15, was mutated to CysB9 and then a disulfide bond was created between this pair of cysteines. Finally, the two-chain disulfide constrained analogue was energy minimized in vacuo as described previously (Shabanpoor et al. 2007) using the Powell method with the Tripos force field, Gasteiger-Marsili charges and termination at a root mean square (RMS) gradient of less than 0.05 kcal/mol per Å.
Design of i to i + 4 lactam constrained mimetics
In designing lactam constrained mimetics, we first inspected the NMR structure of the INSL3 B-chain for an optimum place to introduce the lactam. PheB14 and LeuB18, spaced i and i + 4 on one face of the helix opposite to the side where the key receptor binding residues were located, was observed to be a suitable place to introduce a lactam constraint. We designed a series of lactam constrained analogues of INSL3 B-chain where we truncated the B-chain from the C-terminus until TrpB27 and to ProB1 at the N-terminus. Some of the lactam constrained analogues were truncated further from the N-terminus to GlyB11. Following this truncation, GlyB11 was mutated to AlaB11, then PheB14 was mutated to LysB14 or DabB14, and LeuB18 was mutated to GluB18or AspB18 (Fig. 3). Finally, an amide bond was created between the ε-amino group of the Lys or Dab side-chain and carbonyl group of either the Glu or Asp side-chain, and the resultant analogues were energy minimized as described earlier.
Incorporation of α-helix-inducing residues and N-caps
The INSL3 B-chain was truncated from the N-terminus up to GlyB11, which was then mutated to a more helix-favouring residue, AlaB11. AlaB17 was mutated to a more helix-inducing residue, α-aminoisobutyric acid (Aib). Valine residues along B-chain helix were mutated to either Ala or Aib. The N-cap, 2,4,5 benzenecarboxylate, which is known to stabilize helices by acting as a surrogate H-bond acceptor (Mimna et al. 2007), was coupled to the N-terminus of the INSL3 B-chain helix.
Solid-phase peptide synthesis
In order to increase the enzymatic stability of the analogues, all linear precursor peptides were synthesized as C-terminal amides (Werle and Bernkop-Schnürch 2006) on PAL-PEG-PS resin with 0.19–0.22 mmol/g loadings using Fmoc chemistry. The side chain protected amino acids used were: Arg(Pbf), Asp(OPip), Cys(Trt), Cys(Acm), Cys(tBu), Glu(OPip), Glu(OtBu), His(Trt), Lys(Boc), Lys(Mtt) and Trp(Boc). Peptides were synthesized on either a Pioneer peptide synthesizer (PerSeptive Biosystems, MA, USA) using continuous flow methodology or a microwave peptide synthesizer (CEM, Liberty, Matthews, USA). In continuous flow syntheses, the coupling of Fmoc protected l-α-amino acids was accomplished using HBTU (0.3 mmol) and DIPEA in DMF (5 ml) for 30 min and Fmoc protecting groups were removed by treating the resin-attached peptide with piperidine (20% v/v) in DMF for 20 min. For microwave-assisted syntheses, a fivefold excess of amino acid and HBTU and a tenfold excess of DIEA were used, and the coupling and deprotection were carried out at 75°C using 25 W microwave power for 5 min and 60 W microwave power for 3 min, respectively.
The single chain disulfide-constrained peptides were synthesized as described previously (Shabanpoor et al. 2007). Analogues 30 and 31 (Table 1) with two inter-chain disulfide bonds were synthesized with two Cys(Trt)s and two Cys(Acm)s, one of each in either chain. The formation of a disulfide bond between the two Cys(Trt) was carried out by dissolving the A and B-chains in an equimolar ratio in 0.1 M NH4CO3, adding 300 μl of 100 mM DPDS and stirring the reaction mixture for 30 min. The second inter-chain disulfide bond was formed by first dissolving the peptide in acetic acid (2 mg/ml) followed by the addition of 60 mM HCl (0.1 ml/mg) and 20 mM I2 (42 eq/Acm). The reaction mixture was stirred at room temperature for 1 h and the progression of the reaction was monitored by HPLC.
The all-linear form of the lactam-constrained peptides were synthesized at the 0.1-mmol scale on PAL-PEG-PS resin (substitution 0.20 mmol/g) using a microwave-assisted peptide synthesizer and the conditions described above. The formation of an amide bond between the side chains of two residues, Lys or Dab and Glu or Asp, was carried out on-resin. The phenylisopropyl ester (OPip) of aspartic and glutamic acids and methyltrityl (Mtt) group of lysine and Dab were removed by treating the peptide resin with 3% TFA/5% TIPS in DCM (2 × 30 min) (Shepherd et al. 2006). The on-resin cyclization was carried out in three different ways. In the first instance, we attempted to cyclize the peptide on-resin using a standard protocol of coupling with 3 equivalents of HBTU and 3.5 equivalents of DIPEA in 3 ml of DMF overnight. Second, the resin-bound peptide was treated with PyBOP/HOAt/DIPEA (3:3:3.5) in 3 ml of DMF/DMSO/NMP (1:1:1) overnight. Finally, the cyclization was carried out in a microwave-assisted peptide synthesizer using HBTU (3 eq) DIPEA (3.5 eq) for 10 min at 75°C, 25 W.
The syntheses of peptides with helicogenic residues and N-caps were carried out in the same way as for the disulfide constrained mimetics. The N-terminus was either capped with acetic anhydride (10 eq) or 1,2,4,5-benzene-tetracarboxylic dianhydride (10 eq) in DMF in the presence of DIEA (10 eq).
The cleavage of peptides was carried out using a TFA:H2O:DODT:TIS (94:2.5:2.5:1, 20 ml) mixture for 90 min. Cleaved peptides were precipitated in ice-cold diethyl ether, centrifuged at 3,000 rpm for 3 min; the pellet was washed by resuspending it in ice-cold diethylether and centrifuging it again for three times. Peptides were analysed and purified by RP-HPLC on Waters XBridge™ columns (4.6 × 250 mm, C18, 5 μm) and (19 × 150 mm, C18, 5 μm), respectively, using H2O with 0.1% TFA as solvent A and acetonitrile with 0.1% TFA as solvent B, with a gradient of 1% change in buffer B per min over 30 min. Peptide 24 (Table 1) N-capped with 1,2,4,5-benzene-tetracarboxylic dianhydride was dissolved in 1 M (NH4)2CO3 and lyophilized before HPLC analysis and purification.
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/TOF MS, Bruker Daltonics, Germany) was used to characterize the peptides at each intermediate step using sinapinic acid, α-cyano-4-hydroxy-cinnamic acid and 2,5-dihydroxy benzoic acid (Bruker Daltonics, Germany) as matrices, based on the molecular size of a peptide. The matrices were made up in 50% acetonitrile containing 0.05% TFA. The peptide content was determined using vapour-phase acid hydrolysis in 6 M HCl containing 2% phenol at 110°C for 24 h. The individual amino acids were converted to stable, fluorescent derivatives using Waters AccQ.Tag kit (Waters, Sydney, Australia). The derivatized amino acids were separated using a Shim-Pak XR-ODS (3 × 75 mm, 2.2 μm) column on a Shimadzu RP-HPLC system (Shimadzu, VIC, Australia). The concentrations of individual amino acids were standardized against an internal standard (norvaline) at a concentration of 100 pmol/μl sample injected.
Circular dichroism spectroscopy
The peptides were made up to a concentration of 0.1 μM in phosphate buffered saline (PBS: 10 mM potassium phosphate buffer containing 137 mM NaCl pH 7.4). The far UV circular dichroism (CD) spectra of peptides were acquired using a JASCO model J815 spectropolarimeter between the wavelengths of 195–250 nm at room temperature with a resolution of 0.1 nm, bandwidth of 0.1 nm and a cell of 0.1 cm path length (P). The recorded spectra in millidegrees of ellipticity (θ) were converted to mean residue ellipticity (MRE) in degcm2dmol−1. The CD spectra data were first transformed from machine unit θ to delta epsilon (∆ε) using GraphPad PRISM 4 (GraphPad Inc., San Diego, USA) with a user defined formula (∆ε = θ × (0.1 × MRW)/(P × C) × 3,298) (C: Peptide concentration, MRW: Peptide molecular weight/number of residues). The converted values were then submitted to the DichroWeb server (Lees et al. 2006; Lobley and Wallace 2001; Lobley et al. 2002; Whitmore and Wallace 2004) for the calculation of secondary structure using the CDSSTR (Compton and Johnson 1986) and K2D (Andrade et al. 1993) analysis algorithms.
Ligand binding assay
Human embryonic kidney (HEK)-293T cells stably transfected with RXFP2 and europium-labelled INSL3 were used in whole cell binding assays. Cells were plated out at a density of 80,000 cells per 200 μl per well in 96 well Isoplate with white wall and clear bottom precoated with poly-l-lysine. Competition binding experiments were carried out as described previously (Shabanpoor et al. 2008) with 300 pM of Eu-DTPA-INSL3 (Kd: 0.892 nM) in presence of increasing concentrations of peptide analogues. Non-specific binding was determining with an excess (500 nM) of unlabelled INSL3. Each concentration point was performed in triplicate and the data expressed as the mean ± SEM (standard error of mean) of the percentage of total specific binding of triplicates from at least three independent experiments. Curves were fitted using a one-site binding model in GraphPad PRISM 4 (GraphPad Inc., San Diego, USA). The inhibition constants (Ki) were determined from IC50 values using the Cheng-Prusoff equation, and the statistical differences in pKi values were calculated using one-way ANOVA followed by Bonferroni’s multiple comparison test for multiple group comparisons.
Functional cAMP assay
A cAMP reporter gene assay was used to assess the receptor signalling of INSL3 and analogue 31 in HEK-293T cell line co-transfected with RXFP2 (LGR8) and a pCRE-β-galactosidase reporter plasmid. The assay was carried out as described previously (Scott et al. 2006). Briefly, co-transfected cells were incubated with increasing concentrations of INSL3 and analogue 31 for 6 h after which the medium was removed and cells frozen at −80°C overnight. The amount of cAMP-driven β-galactosidase expression was measured by lysing the cells. Each concentration point was performed in triplicate and the data expressed as the mean ± SEM of three independent experiments.
Results and discussion
INSL3 binds to its receptor principally using residues confined to the B-chain. Deletion of the INSL3 A-chain leads to the loss of B-chain α-helical structure and, therefore, loss of receptor binding affinity of the B-chain in isolation. The α-helix is the most abundant secondary structure which accounts for 30% of all protein residues (Barlow and Thornton 1988). The helix plays a crucial role in many protein-mediated biological processes such as receptor binding (Beck-Sickinger and Jung 1995) and thus is an attractive target for the design of mimetics. However, peptides derived from these regions may not be biologically active when in isolation. This is usually due to the loss of α-helicity where the peptides exhibit little or no secondary structure as a result of loss of stabilizing interactions within the parent protein. As part of our ongoing effort to design short mimetics of INSL3 B-chain with antagonistic action, we have utilized and evaluated various possible ways of increasing the α-helicity within the B-chain of INSL3. Towards this aim, we have designed three series of analogues that include disulfide (primarily helix to strand) constraints, i to i + 4 lactam constraints, and helix-inducing N-caps and residues (Table 1).
All analogues were synthesized on the solid-phase and subjected to analysis by RP-HPLC and mass spectrometry. The on-resin cyclization of lactam analogues was difficult. In the first instance, using standard coupling procedures, the reaction did not give the desired cyclic compound. A second attempt using different coupling reagents, PyBOP and HOAt, gave the lactam but also resulted in the formation of various major side products. Best results were obtained using HBTU as coupling reagent and heating to 75°C using microwave power, in that the reaction went to completion with minor side-products formation only and an overall yield of 10–15%.
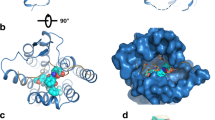
The introduction of a disulfide constraint between the strand and the helix of the B-chain had little impact on the level of helicity but improved the receptor binding affinity in the analogue 3 (pKi = 6.41 ± 0.11) compared to its linear counterpart analogue 1 (pKi = 5.31 ± 0.24). In order to determine the mode of receptor interaction of this analogue, the residues HB12, RB16, VB19, RB20 and WB27, which have been shown previously to be important for binding of native INSL3 to RXFP2, were each mutated in analogue 3. Alanine mutation of ArgB16 significantly reduced the receptor binding affinity of analogue 3 (Fig. 2). The replacement of HisB12 and ValB19 with Ala also caused 40–50 times reduction in the binding affinity of analogue 3 whereas replacement of ArgB20 caused only a slight drop in receptor binding affinity. Finally, replacement of TrpB27 in this analogue with Ala led to almost complete loss of receptor binding affinity. This trend of single replacement of binding residues in the B-chain disulfide constrained analogue and its loss of receptor binding affinity resembles that of native INSL3 (Rosengren et al. 2006), suggesting that, despite their lower affinity for RXFP2, the B-chain mimetics bind to the receptor in the same fashion as native INSL3.

Competition binding studies of native INSL3, disulfide constrained analogue 3 and its Ala-substituted analogues (Table 1). Europium-labelled-INSL3 (0.3 nM) was used as a labelled ligand in the competition binding assay in the presence of increasing concentration of INSL3 and analogues. Singly substituting RB12 (5) and WB27 (8) with Ala caused these peptides to almost completely lose their receptor binding affinity. Replacing HB12 (4) and V19 (6) with alanine also led to a major loss of receptor binding affinity. Mutation of RB20 had minor impact in the level of binding affinity
The small improvement in receptor binding of the linear B-chain with the disulfide constraint between the strand and the α-helix was probably not as a result of induction of further α-helical structure as there was little difference in the level of α-helicity between the linear compound 1 and its cyclic counterpart analogue 3. It is likely that the disulfide constraint between the strand and the helix of the B-chain pulls the helix and the strand—very flexible in native INSL3—closer together and this is possibly causing a conformational change in the spatial orientation of the TrpB27 at the C-terminus of the B-chain which might be better placed for interaction with the receptor. To further investigate this possibility, three residues (GB23,24 and PB25) toward the C-terminus of the B-chain were replaced with alanine. This change resulted in an analogue (11) with binding affinity (pKi = 5.74 ± 0.2, n = 3) similar to that of linear B-chain (analogue 1). The GGP residues appear to act as a hinge that provides flexibility for the B-chain C-terminal region and this flexibility may be required for proper interaction of TrpB27 with the receptor. Since TrpB27 is a crucial binding residue in the B-chain analogues, we further investigated the role of both the individual indole and benzene rings by replacing TrpB27 in analogue 3 with β-(benzothien-3-yl)alanine (Bal, analogue 9) and β-(naphtha-1-yl)alanine (1-Nal, analogue 10). Bal is an analogue of Trp that has sulfur instead of nitrogen in the indole ring and 1-Nal is another analogue in which the indole ring is replaced with a benzene ring. The drop in the level of receptor binding affinity of these two analogues was similar which shows both rings of TrpB27 are equally important in receptor interaction.
Further constraining analogue 3 by incorporating an i to i + 4 disulfide constraint along the helix at PheB14 and LeuB18 gave analogue 12 which exhibited low level of helicity even in the presence of 20% TFE. This peptide had a very low level of receptor affinity compared to analogue 3. The i to i + 4 disulfide constraint along the helix would cause analogue 12 to lose its flexibility around the helical region and prevent the peptide from adopting an active conformation for high affinity receptor binding. This was investigated by inserting only the i to i + 4 disulfide constraint at PheB14 and LeuB18 in analogue 13. This analogue did not show any affinity for receptor binding, which is likely due to the loss of flexibility and inability to adopt a conformation with the key receptor binding residues in a correct orientation to the binding pocket on the receptor.
The introduction of a lactam bridge in the B-chain was the second approach that was investigated for the restoration of helical structure in the isolated B-chain of INSL3. Side chain–side chain lactam bridges have long been used as a convenient and flexible method for introducing conformational constraints into a peptide structure. The points to consider in the design of peptides with a lactam bridge are the spacing of the two residues to be linked, the side-chain orientation and the position of the bridge. A Glu–Lys (E-K) lactam at the spacing of i and i + 4 in the middle of the peptide has been shown to be more effective at stabilizing helical structure than two Glu-Lys lactams positioned one at each end of the peptide (Houston et al. 1995). In the B-chain of INSL3, we introduced a lactam bridge (i, i + 4) in the mid-region of the B-chain α-helix by mutating PheB14 and LeuB18 to Glu, Asp, Lys or Dab. Various orientations of the lactam bridge were evaluated in the long form (analogues 14–16) (Fig. 3a) and truncated B-chain of INSL3 (analogues 17–22).

a An INSL3 B-chain analogue in which a lactam constraint was incorporated in the middle of the α-helix on the side opposite to where the binding residue is located. F14 and L18 were replaced with K, E, D or Dab in different orientations. b Truncated INSL3 B-chain until GB11 and the N-terminus capped with either acetic anhydride or 1,2,4,5-benzene-tetracarboxylic dianhydride
Analogues 14 and 15 had higher α-helicity in PBS (17 and 18%, respectively) compared to the linear analogue 1 (10%); this small improvement in α-helicity was accompanied by a small increase in receptor binding affinity. In analogue 15, LysB18 was replaced with diaminobutyric acid (Dab) (analogue 16). This modification was observed to not only reduce the level of α-helicity but also cause a drop in receptor binding affinity (Table 1). This may be due to the shorter side-chain of Dab which, on cyclization with the Glu side-chain, results in an unfavourable conformational change in the B-chain. The introduction of a lactam bond in the truncated INSL3 B-chain analogues had little effect on either α-helicity or receptor binding. Analogues 17 and 21 with K-D and E-K lactam orientation, respectively, were more effective in inducing helical structure compared to D-K and K-E lactams in analogues 18 and 20. Analogue 18 with D-K lactam orientation did not have higher α-helicity compared to its linear counterpart analogue 19. On the other hand, analogue 21 with E-K lactam orientation exhibited higher α-helicity compared to its linear analogue 22. Both the short and long forms of E-K orientated lactams (15 and 21) had the highest helical content both in PBS and 20% TFE compared to any other lactam orientation and this is in accordance with previous literature reports (Houston et al. 1995). Despite some of the lactam constrained analogues being able to induce α-helicity in the B-chain, this did not result in high affinity binding. The introduction of a lactam bridge in the mid-region of the B-chain backbone likely led to the loss of B-chain flexibility and a more rigid structure, which is no longer capable of adjusting binding residues to align with the binding pocket on the receptor.
Analogues 23–25 show the effect of “capping” the N-terminal of the truncated B-chain with acetyl and 2,4,5 benzenecarboxylate (Fig. 3B), the latter which has recently been reported to increase the α-helicity of a model peptide from 17 to 70% by providing carbonyl groups that act as surrogate H-bond acceptors (Mimna et al. 2007). The N-terminus capping of the B-chain α-helix was not effective in inducing α-helicity in PBS but in 20% TFE the level of α-helicity increased significantly, which was likely a reflection of the tendency of these peptides to adopt a helical structure.
The α-helix-inducing effect of residues such as Ala and Aib on the INSL3 B-chain helix was also investigated. The use of α-helicogenic residues in enhancing the activity of peptides has been demonstrated by substituting these at specific positions in the helical N-terminal fragment of parathyroid hormone PTH(1–11), which made the resulting peptide 3,500 times more potent (Barazza et al. 2005). The α-helix stabilization of alanine has been related to the hydrophobic interaction, steric effects and solvation of the polar groups of the α-helix backbone (Avbelj 2000; Avbelj and Fele 1998; Bai and Englander 1994; Bai et al. 1993; Blaber et al. 1993, 1994; Connelly et al. 1993). Aib-based peptides have a remarkable tendency to form α-helical conformations in solution due to the restricted rotation about (N-Cα) and (Cα-C1) bonds, which is caused by the presence of two methyl groups on the Cα atom (Ma et al. 2007; Marshall et al. 1990). Alanine and Aib residues were incorporated along the α-helical segment of INSL3 B-chain in place of valine in analogues 26–29. None of these analogues had high helicity in PBS, although the helicity increased upon addition of 20% TFE. These peptides also showed very low receptor binding affinity due to the lack of helical structure.
The disulfide constrained analogue 30, where the B-chain helix was stabilized using the C-terminal α-helical region of the A-chain and analogue 31, which is a B-chain dimer, exhibited 23 and 32% α-helicity in PBS, which was higher than any other single B-chain analogues. The higher level of α-helicity was accompanied by an increase in receptor binding affinity of these two analogues (Fig. 4a). To determine if 31 was able to activate the receptor and induce intracellular cAMP production, the analogue was tested in HEK-293T cells expressing RXFP2 (LGR8). Native INSL3 induced cAMP production (pEC50 = 10.35 ± 0.12) whereas analogue 31 was unable to activate the receptor as intracellular cAMP accumulation was not induced (Fig. 4b), which suggests that this peptide is an antagonist of INSL3.

a Comparison of competition binding curves of INSL3 (pKi = 9.27 ± 0.06) with analogues 30 (pKi = 7.14 ± 0.02) and 31 (pKi = 8.43 ± 0.06). As the level of α-helicity increased (Table 1) so does the receptor binding affinity. b Functional cAMP studies of nINSL3 analogue 31. Native INSL3 can induce cAMP production (pEC50 = 10.35 ± 0.12) in HEK-293T cells co-transfected with RXFP2 (LGR8) and pCRE-β-galactosidase whereas treatment of these cells with analogue 31 does not induce cAMP production, i.e. it is an antagonist. Each concentration point was performed in triplicate and the data are expressed as the mean ± SEM of three independent experiments
In summary, a systematic approach was taken to induce α-helicity in the isolated INSL3 B-chain and study the binding mode of interaction with the receptor. Isolated INSL3 B-chain that was constrained with a disulfide bond was found to bind to the receptor in the same way as native INSL3. Attempting to induce an α-helical conformation by constraining the B-chain of INSL3 did not fully compensate for the stabilizing interactions of the missing A-chain as constraining the B-chain by either a disulfide or lactam bond did not increase α-helicity and receptor binding affinity simultaneously. However, constraining the B-chain with a short region of A-chain or dimerizing the B-chain not only increased the α-helicity but also the receptor binding affinity. On this basis, we have identified two novel INSL3 antagonists which can be used as lead compounds to be further minimized and optimized for development as clinically useful INSL3 antagonists which may be used as potential male and female contraceptives.
References
Adham IM, Burkhardt E, Benahmed M et al (1993) Cloning of a cDNA for a novel insulin-like peptide of the testicular Leydig cells. J Biol Chem 268:26668–26672
Andrade MA, Chacon P, Merelo JJ et al (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng 6:383–390. doi:10.1093/protein/6.4.383
Avbelj F (2000) Amino acid conformational preferences and solvation of polar backbone atoms in peptides and proteins. J Mol Biol 300:1335–1359. doi:10.1006/jmbi.2000.3901
Avbelj F, Fele L (1998) Role of main-chain electrostatics, hydrophobic effect and side-chain conformational entropy in determining the secondary structure of proteins. J Mol Biol 279:665–684. doi:10.1006/jmbi.1998.1792
Bachelot T, Ray-Coquard I, Catimel G et al (2000) Multivariable analysis of prognostic factors for toxicity and survival for patients enrolled in phase I clinical trials. Ann Oncol 11:151–4773. doi:10.1023/A:1008368319526
Bai Y, Englander SW (1994) Hydrogen bond strength and beta-sheet propensities: the role of a side chain blocking effect. Proteins 18:262–266. doi:10.1002/prot.340180307
Bai Y, Milne JS, Mayne L et al (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17:75–86. doi:10.1002/prot.340170110
Barazza A, Wittelsberger A, Fiori N et al (2005) Bioactive N-terminal undecapeptides derived from parathyroid hormone: the role of alpha-helicity. J Pept Res 65:23–35. doi:10.1111/j.1399-3011.2005.00207.x
Barlow DJ, Thornton JM (1988) Helix geometry in proteins. J Mol Biol 201:601–619. doi:10.1016/0022-2836(88)90641-9
Beck-Sickinger AG, Jung G (1995) Structure-activity relationships of neuropeptide Y analogues with respect to Y1 and Y2 receptors. Biopolymers 37:123–142. doi:10.1002/bip.360370207
Blaber M, Zhang XJ, Matthews BW (1993) Structural basis of amino acid alpha helix propensity. Science 260:1637–1640. doi:10.1126/science.8503008
Blaber M, Zhang XJ, Lindstrom JD et al (1994) Determination of alpha-helix propensity within the context of a folded protein. Sites 44 and 131 in bacteriophage T4 lysozyme. J Mol Biol 235:600–624. doi:10.1006/jmbi.1994.1016
Bogatcheva NV, Truong A, Feng S et al (2003) GREAT/LGR8 is the only receptor for insulin-like 3 peptide. Mol Endocrinol 17:2639–2646. doi:10.1210/me.2003-0096
Büllesbach EE, Schwabe C (1995) A novel Leydig cell cDNA-derived protein is a relaxin-like factor. J Biol Chem 270:16011–16015. doi:10.1074/jbc.270.27.16011
Büllesbach EE, Schwabe C (1999) Tryptophan B27 in the relaxin-like factor (RLF) is crucial for RLF receptor-binding. Biochemistry 38:3073–3078. doi:10.1021/bi982687u
Büllesbach EE, Schwabe C (2005) LGR8 signal activation by the relaxin-like factor. J Biol Chem 280:14586–14590. doi:10.1074/jbc.M414443200
Büllesbach EE, Schwabe C (2007) Structure of the transmembrane signal initiation site of the relaxin-like factor (RLF/INSL3). Biochemistry 46:9722–9727. doi:10.1021/bi700708s
Burkhardt E, Adham IM, Hobohm U et al (1994) A human cDNA coding for the Leydig insulin-like peptide (Ley I-L). Hum Genet 94:91–94. doi:10.1007/BF02272850
Compton LA, Johnson WC Jr (1986) Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal Biochem 155:155–167. doi:10.1016/0003-2697(86)90241-1
Connelly GP, Bai Y, Jeng MF et al (1993) Isotope effects in peptide group hydrogen exchange. Proteins 17:87–92. doi:10.1002/prot.340170111
Del Borgo MP, Hughes RA, Bathgate RA et al (2006) Analogs of insulin-like peptide 3 (INSL3) B-chain are LGR8 antagonists in vitro and in vivo. J Biol Chem 281:13068–13074. doi:10.1074/jbc.M600472200
Feng S, Cortessis VK, Hwang A et al (2004) Mutation analysis of INSL3 and GREAT/LGR8 genes in familial cryptorchidism. Urology 64:1032–1036. doi:10.1016/j.urology.2004.06.051
Ferlin A, Pepe A, Gianesello L et al (2008) Mutations in the insulin-like factor 3 receptor are associated with osteoporosis. J Bone Miner Res 23:683–693. doi:10.1359/jbmr.080204
Foresta C, Ferlin A (2004) Role of INSL3 and LGR8 in cryptorchidism and testicular functions. Reprod Biomed Online 9:294–298
Hossain MA, Rosengren KJ, Haugaard-Jonsson LM et al (2008) The A-chain of human relaxin family peptides has distinct roles in the binding and activation of the different relaxin family peptide receptors. J Biol Chem 283:17287–17297. doi:10.1074/jbc.M801911200
Houston ME Jr, Gannon CL, Kay CM et al (1995) Lactam bridge stabilization of alpha-helical peptides: ring size, orientation and positional effects. J Pept Sci 1:274–282. doi:10.1002/psc.310010408
Hsu SY (2003) New insights into the evolution of the relaxin-LGR signaling system. Trends Endocrinol Metab 14:303–309. doi:10.1016/S1043-2760(03)00106-1
Ivell R, Einspanier A (2002) Relaxin peptides are new global players. Trends Endocrinol Metab 13:343–348. doi:10.1016/S1043-2760(02)00664-1
Kawamura K, Kumagai J, Sudo S et al (2004) Paracrine regulation of mammalian oocyte maturation and male germ cell survival. Proc Natl Acad Sci USA 101:7323–7328. doi:10.1073/pnas.0307061101
Klonisch T, Mustafa T, Bialek J et al (2005) Human medullary thyroid carcinoma: a source and potential target for relaxin-like hormones. Ann N Y Acad Sci 1041:449–461. doi:10.1196/annals.1282.069
Lees JG, Miles AJ, Wien F et al (2006) A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 22:1955–1962. doi:10.1093/bioinformatics/btl327
Lobley A, Wallace BA (2001) DICHROWEB: a website for the analysis of protein secondary structure from circular dichroism spectra. Biophys J 80:373
Lobley A, Whitmore L, Wallace BA (2002) DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 18:211–212. doi:10.1093/bioinformatics/18.1.211
Ma S, Bonaventure P, Ferraro T et al (2007) Relaxin-3 in GABA projection neurons of nucleus incertus suggests widespread influence on forebrain circuits via G-protein-coupled receptor-135 in the rat. Neuroscience 144:165–190. doi:10.1016/j.neuroscience.2006.08.072
Marshall GR, Hodgkin EE, Langs DA et al (1990) Factors governing helical preference of peptides containing multiple alpha, alpha-dialkyl amino acids. Proc Natl Acad Sci USA 87:487–491. doi:10.1073/pnas.87.1.487
Mimna R, Tuchscherer G, Mutter M (2007) Toward the design of highly efficient, readily accessible peptide N-caps for the induction of helical conformations. Int J Pept Res Ther 13:237–244. doi:10.1007/s10989-006-9073-9
Nef S, Parada LF (1999) Cryptorchidism in mice mutant for Insl3. Nat Genet 22:295–299. doi:10.1038/10364
Park JI, Semyonov J, Yi W (2008) Regulation of receptor signaling by relaxin A chain motifs: derivation of pan-specific and LGR7-specific human relaxin analogs. J Biol Chem 283(46):32099–32109
Roche PJ, Butkus A, Wintour EM et al (1996) Structure and expression of Leydig insulin-like peptide mRNA in the sheep. Mol Cell Endocrinol 121:171–177. doi:10.1016/0303-7207(96)03861-0
Rosengren KJ, Zhang S, Lin F et al (2006) Solution structure and characterization of the LGR8 receptor binding surface of insulin-like peptide 3. J Biol Chem 281:28287–28295. doi:10.1074/jbc.M603829200
Scott DJ, Layfield S, Yan Y et al (2006) Characterization of novel splice variants of LGR7 and LGR8 reveals that receptor signaling is mediated by their unique low density lipoprotein class A modules. J Biol Chem 281:34942–34954. doi:10.1074/jbc.M602728200
Shabanpoor F, Bathgate RA, Hossain MA et al (2007) Design, synthesis and pharmacological evaluation of cyclic mimetics of the insulin-like peptide 3 (INSL3) B-chain. J Pept Sci 13:113–120. doi:10.1002/psc.807
Shabanpoor F, Hughes RA, Bathgate RA et al (2008) Solid-phase synthesis of europium-labeled human INSL3 as a novel probe for the study of ligand-receptor interactions. Bioconjug Chem 19:1456–1463. doi:10.1021/bc800127p
Shepherd NE, Hoang HN, Desai VS et al (2006) Modular alpha-helical mimetics with antiviral activity against respiratory syncitial virus. J Am Chem Soc 128:13284–13289. doi:10.1021/ja064058a
Spiess AN, Balvers M, Tena-Sempere M et al (1999) Structure and expression of the rat relaxin-like factor (RLF) gene. Mol Reprod Dev 54:319–325. doi:10.1002/(SICI)1098-2795(199912)54:4<319::AID-MRD1>3.0.CO;2-Z
Tashima LS, Hieber AD, Greenwood FC et al (1995) The human Leydig insulin-like (hLEY I-L) gene is expressed in the corpus luteum and trophoblast. J Clin Endocrinol Metab 80:707–710. doi:10.1210/jc.80.2.707
Werle M, Bernkop-Schnürch A (2006) Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 30:351–367. doi:10.1007/s00726-005-0289-3
Whitmore L, Wallace BA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32:W668–W673
Zimmermann S, Steding G, Emmen JM et al (1999) Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol Endocrinol 13:681–691. doi:10.1210/me.13.5.681
Acknowledgments
We thank Tania Ferraro for help with binding assays. This work was funded by National Health and Medical Research Council of Australia Project grants #350245 and 509048 to JDW, RADB and RAH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shabanpoor, F., Hughes, R.A., Zhang, S. et al. Effect of helix-promoting strategies on the biological activity of novel analogues of the B-chain of INSL3. Amino Acids 38, 121–131 (2010). https://doi.org/10.1007/s00726-008-0219-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-008-0219-2