
Abstract
The number of descriptions of emerging viruses has grown at an unprecedented rate since the beginning of the 21st century. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), is the third highly pathogenic coronavirus that has introduced itself into the human population in the current era, after SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV). Molecular and cellular studies of the pathogenesis of this novel coronavirus are still in the early stages of research; however, based on similarities of SARS-CoV-2 to other coronaviruses, it can be hypothesized that the NF-κB, cytokine regulation, ERK, and TNF-α signaling pathways are the likely causes of inflammation at the onset of COVID-19. Several drugs have been prescribed and used to alleviate the adverse effects of these inflammatory cellular signaling pathways, and these might be beneficial for developing novel therapeutic modalities against COVID-19. In this review, we briefly summarize alterations of cellular signaling pathways that are associated with coronavirus infection, particularly SARS-CoV and MERS-CoV, and tabulate the therapeutic agents that are currently approved for treating other human diseases.
Similar content being viewed by others


Introduction
The number of descriptions of emerging viruses has grown at an unprecedented rate since the beginning of the 21st century [1, 2]. More than 100 years after the start of the 1918 influenza pandemic, we are now experiencing another pandemic. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has emerged as the third highly pathogenic coronavirus to introduce itself into the human population in the current era, after SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) [2]. The advent of SARS-CoV in 2002-2003 in China [3] and of MERS-CoV in the Kingdom of Saudi Arabia in 2012 [4] provoked general concern about their possible threat to global health security. SARS-CoV spread to several countries, with more than 8000 cases and more than 750 deaths [5, 6]. MERS-CoV infections have resulted in more than 600 deaths and caused severe respiratory disease in more than 1600 people [7, 8]. Numerous similarities and differences in the epidemiology, clinical properties, and handling of SARS and MERS have been recognized [9].
Viruses, including coronaviruses, manipulate the host cell machinery in order to complete their replication cycle [10]. Interfering with signaling pathways that regulate processes such as DNA repair and replication, immune response, transcription, metabolism, cell cycle, and survival is one of the ways for the virus to take over cellular processes [11]. Alteration of various signaling pathways involved in the central physiological functions of the cell takes place after coronavirus infection [12]. According to recent research reports, the phosphatidylinositol 3‑kinase (PI3K)/protein kinase B (AKT), interferon, p38 mitogen-activated protein kinase (MAPK), epidermal growth factor receptor (EGFR), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways are altered following virus infection. These pathways are involved in antagonizing the host antiviral response and are vital for viral replication, entry, propagation, and apoptosis. Coronaviruses manipulate the molecular function of signaling pathways, and this kind of interaction between the host cell and the virus might be responsible for viral pathogenesis [13, 14].
Rapid intervention in the usual public-health behaviors and the development of antiviral compounds, antibodies, or vaccines are the keys to controlling the spread of a new virus and associated disease. MERS-CoV, SARS-CoV, and SARS-CoV-2 are betacoronaviruses share many characteristics but exhibit significant differences in their epidemiology, pathology, genetics, and protein composition. SARS-CoV-2 has around 79% genome sequence identity to SARS-CoV and nearly 50% to MERS-CoV [15, 16]. Because of these similarities, prior information about controlling SARS-CoV and MERS-CoV can serve as a guide to the pathogenesis and epidemiology of SARS-CoV-2 and to the improvement of therapeutic approaches to preventing viral infection.
This review provides a comparative overview of MERS-CoV, SARS-CoV, and the recently emerged SARS-CoV-2 in the interest of increasing understanding host-pathogen interactions, signaling pathways of the host, and immune evasion mechanisms by the pathogen that may be of use for the planning of new treatments for COVID-19.
Coronavirus structure and life cycle
Tyrrell and Bynoe reported the isolation of a virus from the respiratory tracts of adults with symptoms of the common cold. This virus was shown to have the ability to grow in human embryonic tracheal tissue culture and to induce cold symptoms when inoculated into healthy volunteers [17]. During the same period, Hamre and Procknown showed that an unknown virus obtained from samples taken from medical students complaining of cold symptoms could persist and infect tissue culture. These two unknown viruses were shown to be of the same type and named 229E [18]. These two novel respiratory viruses did not have the well-known properties of orthomyxoviruses or paramyxoviruses. They both had prominent crown-like extramembrane projections that were observed by electron microscopy and reported by Almeida and Tyrrell [19]. Subsequent comparison of these viruses to other contemporaneously discovered viruses, including infectious bronchitis virus, transmissible gastroenteritis virus of swine, and mouse hepatitis virus (MHV), led to the establishment of a new viral group named "coronavirus", in reference to their crown-like surface projections [20]. In the meantime, numerous coronaviruses have been identified, seven of which contribute to human diseases, with clinical expression ranging from asymptomatic or mild to lethal infections. Taxonomically, these viruses belong to the order Nidovirales, the family Coronaviridae, and the subfamily Coronavirinae, which is further divided into the genera Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus (International Committee on Taxonomy of Viruses [ICTV]). These genera include several viruses with the ability to infect human cells, and especially epithelial cells. Human coronaviruses (HCoV) belonging to the genera Alphacoronavirus and Betacoronavirus include HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1, which induce mild pathology in most cases, and SARS-CoV [21], MERS-CoV [22], and the causative agent of the new pandemic COVID-19, SARS-CoV-2 [23], which can induce severe syndromes. SARS-CoV-2 and SARS-CoV both belong to the subgenus Sarbecovirus of the genus Betacoronavirus, which also has four other subgenera: Merbecovirus (MERS-CoV), Nobecovirus, Embecovirus (HCoV-HKU1, MHV, HCoV-OC43, HCoV-B814), and Hibecovirus.
The coronavirus particle exhibits a pleomorphic spherical enveloped shape displaying a crown-like surface due to the presence of club-shaped spike (S) proteins (Fig. 1). In comparison to the other RNA viruses, this virus is large (approximately 120 nm in diameter) and possesses a single-stranded, positive-sense, helical RNA genome associated with nucleocapsid (N) proteins [24]. In addition to its role in assembly, the N protein can regulate coronavirus replication and the infected-cell response [25]. The next layer of the coronavirus particle contains the major structural proteins or so-called membrane (M) proteins, which function to stabilize and maintain the viral envelope. The last structural protein, the envelope (E) protein, is the smallest one and is abundantly expressed in coronavirus-infected cell, which contrasts with its low abundance in the virion itself [26]. The coronavirus genomic RNA, which ranges in size from 26.4 to 31.7 kb, contains genes coding for structural and non-structural proteins. The genome is organized as follows: 5’-leader-UTR-replicase/transcriptase-S-E-M-N-UTR-3’-poly(A) tail. The replicase gene encodes the non-structural proteins that are required for viral replication and completion of the viral life cycle [27].

The structure of a typical SARS-CoV-2 virion. The presence of the viral S protein gives a crown-like shape to the virion. The helical ssRNA is encapsidated with the N protein, and together they are covered by the E protein. There are several HE proteins on the surface of virion as well as spikes. The complete coronavirus virion has the capability to infect its target cells.
The coronavirus replication cycle starts with binding of the S protein to its receptor on the host cell via the receptor-binding domain (RBD). The interaction of S and the receptor determines not only which species can be infected but also which tissues can be involved in this infection [28]. The most abundant receptors used by coronaviruses are peptidases, and it has been shown that this binding is independent of the enzymatic domains of these peptidases. The receptor utilized by alphacoronaviruses such as HCoV-229E is usually aminopeptidase N (APN) [29], which is an enzyme that mostly functions during the immune response and oncogenesis and is involved in the antigen-presenting process [30], while angiotensin-converting enzyme 2 (ACE2), which could be involved in several pathological conditions, such as diabetes in pregnancy and related lung diseases [31], is the main receptor used by HCoV-NL63 [32], SARS-CoV [33], and the newly discovered SARS-CoV-2 [34]. CEACAM1 (carcinoembryonic antigen-related cell adhesion molecule 1) is employed during the entry stage of MHV infection [35], and MERS-CoV attaches to its host cell via binding to dipeptidyl peptidase 4 (DPP4) [36]. The next step in a coronavirus infection is the entry of the virus into the host cell, mediated by S protein cleavage via the activation of cathepsin, transmembrane serine protease 2 (TMPRSS2), or another protease in an acid-dependent manner, resulting in fusion of the viral membrane with the host cell membrane and the release of viral RNA into the cytosol (Fig. 2) [37]. The viral genomic RNA recruits the protein-production machinery of the host cell to translate the replicase-encoding region, which has two open reading frames (ORFs), rep1a (ORF1a), and rep1b (ORF1ab), to produce two polyproteins called pp1a and pp1ab. All of the non-structural proteins (nsps) are derived from these polyproteins. pp1a and pp1ab cleavage results in the production of nsp1-11 and nsp1-16, respectively [38]. The cleavage of polyproteins is mainly carried out by viral proteases encoded in the coronavirus RNA, including the papain-like protease (PLpro), produced from nsp3, and Mpro, produced from nsp5. MERS-CoV, SARS-CoV, and SARS-CoV-2 express only one type of PLpro, whereas the other expresses two types [39, 40]. In addition to the direct translation of the replicase gene and the production of nsps to initiate replication and transcription of structural proteins, the viral RNA is used as a template for the synthesis of genomic RNA for the formation of the complete virion and subgenomic RNA, which is translated into the coronavirus structural proteins E, S, and M, which are transmitted to endoplasmic reticulum (ER) and then extracted from the ER system into the ER-Golgi intermediate compartment (ERGIC) [41]. This kind of endosome is now ready for the incorporation of the encapsidated viral genomic RNA, producing and releasing a mature virion from the infected cell. For this purpose, the N protein is translated from a subgenomic RNA joining the newly synthesized viral genomic RNA and budding within the ERGIC [24]. Eventually, the mature virion is transported to the cell surface and released via exocytosis. Replication of the viral RNA produces double-stranded RNA intermediates, which are potent activators of host ??pattern recognition receptors?? (PRRs) especially since replication occurs in host-cell-membrane-containing replication complexes called double-membrane vesicles or viral replication organelles (ROs), which were previously shown to be recruited by SARS-CoV and MERS-CoV [42].

Replication and transcription of SARS-CoV-2. The life cycle of the virus is initiated when a virion binds to its receptor, and this event is followed by entry into the host cell and uncoating, translation of the replicase gene, and synthesis of the pp1a and pp1ab polyproteins. After production of the viral structural proteins S, M, and E, the genomic RNA is encapsulated by N protein budding into the ERGIC, which finally gives rise to the release of the complete virion from the host cell.
Coronavirus and host molecular modifications
NF-κB
NF-κB is an important family of transcription factors that can enhance the transcription of stress-response proteins and pro-inflammatory cytokines and chemokines. In the cytoplasm, inhibitor of κB (IκB) binds to NF-κB and causes its inactivation [43]. Cellular stimuli trigger the degradation, ubiquitination, and phosphorylation of IκB through the proteasome and induce NF-κB translocation. NF-κB activation plays a crucial role in the inflammatory response against respiratory viruses such as human coronaviruses [44]. It has been reported that several structural and non-structural proteins of SARS-CoV, including the N protein, S protein, nsp1, nsp3a, and nsp7a, can stimulate NF-κB activation [45]. NF-κB is activated following rSARS-CoVMA15- E and rSARS-CoV-MA15 infection in cell culture and in vivo and also regulates the expression of proinflammatory mediators such as tumor necrosis factor (TNF), CCL2, and CXCL2. Therefore, inhibition of NF-κB by CAPE and parthenolide increases the survival of mice after infection and reduces the expression of proinflammatory cytokines in the lungs [45]. As a response to SARS-CoV infection, which is a stress-inducing condition, enhanced secretion of interleukin (IL)-1β is needed, which is done through two pathways, cleavage of pro-IL-1β and stimulation of pro-IL-1β transcription. The elevated level of IL-1β, in turn, increases the expression of several pro-inflammatory cytokines such as TNF-α and IL-6, followed by the activation of inflammasomes [46].
Regarding the ability of NLRP3 (NLR family pyrin domain containing 3) to facilitate the oligomerization of apoptosis-associated speck-like protein containing CARD (ASC) and to activate inflammasomes, SARS-CoV E protein, ORF3a, and ORF8b have been shown to be activators of NLRP3 inflammasomes. ORF3a and the E protein can induce the NF-κB activation required for pro-IL-1β gene transcription. In addition, the E protein, through Ca2+ transport, causes activation of NLRP3 inflammasomes [47]. SARS-CoV ORF3a accomplishes these phenomena through TNF receptor-associated factor 3 (TRAF3)-dependent ubiquitination of p105. Moreover, different mechanisms allow SARS-CoV ORF3a to promote assembly of NLRP3 inflammasomes via TRAF3-dependent ku3 ubiquitination of ASC in HEK293T cells [48]. Therefore, the inhibition of NLRP3 inflammasomes through molecular inhibitors such as INF58 and MCC950 are most likely to be beneficial in COVID-19 treatment; however, further investigations are required to prove this [48]. Another study has shown that, among five accessory proteins of SARS-CoV tested using a reporter plasmid expressing luciferase (κB-Luc), 3a/X1, and 7a/X4 were capable of activating NF-κB in HEK293T cells [49]. Based on in vitro data, the spike protein of SARS CoV induced activation of NF-κB signaling, resulting in IL-6 and TNF-α secretion by murine macrophages [50].
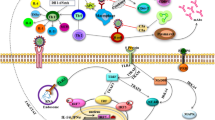
During MERS-CoV infection in Huh-7 and Calu-3 cells, the ORF4b protein binds to karyopherin-α4 (importin-α3), resulting in the inhibition of its interaction with NF-κB-p65 and preventing nuclear translocation of NF-κB. Accordingly, the expression of pro-inflammatory cytokines, as well as NF-κB-dependent cytokines, increases as a result of the latter event (Fig. 3) [44].

Cellular signaling pathways that are altered upon coronavirus infection. Once a coronavirus infects its host cell, several cellular signaling pathways are recruited to facilitate the replication of the virus. Despite this recruitment, the activation of such signaling pathways induces inflammation in the host. Most of these signaling pathways, such as EGFR, PI3K/AKT, JNK, can be overactivated by coronavirus proteins, which are mostly affected by the N protein. The viral proteins might also have an impact on P38 MAPK signaling and dysregulate the expression of BAX and BCL2, which are important proteins involved in the apoptosis process. The essential signaling pathway that is mainly inactivated by viral proteins is IFN signaling, and this leads to escape from the host immune response.
Recently, it was reported that SARS-CoV-2 nsp13 can interact with several transducin-like enhancer (TLE) family proteins regulating the inflammatory response of NF-κB. Physiological evidence shows that TLE 1 is a central negative regulator of inflammation. Also, ORF9c can moderate IκB kinase activity and the NF-κB signaling pathway via the interaction with NDFIP2, NLRX1, F2RL1 [51].
Transforming growth factor β (TGF-β)
TGF-β1 is a type of cytokine that binds to the TGFbRI and TGFbRII serine‐threonine kinase receptors through the TGF‐β signaling pathway, resulting in the activation of the SMAD‐dependent and SMAD‐independent pathways [52]. TGF-β1 is a moderator that is capable of enhancing Fas-mediated cell apoptosis, and it also can be the cause of thrombocytopenia and lymphopenia in SARS patients. Generally, viral pathogens such as coronaviruses can target TGF‐β signaling in several types of cells [53].
During SARS-CoV infection, through the ROS/p38 MAPK/STAT3 axis correlating with elevated pro-fibrotic responses, PLpro induces Egr-1-dependent TGF-β1 promoter activation in human lung epithelial cells and in mouse models [54]. Another study has indicated that SARS-CoV PLpro upregulates the expression of type I collagen via SMAD-independent TGF‐β1 signaling, leading to a pro-fibrotic responses in human lung epithelial cells and mouse pulmonary tissues [55]. The SARS-CoV N protein promotes the formation of the SMAD3-p300 complex; however, it can inhibit the activation of SMAD3 and SMAD4-mediated apoptosis in human peripheral lung epithelial HPL1 cells. These results reveal an interesting effect of the N protein of SARS-CoV: inhibiting apoptosis in host cells and promoting tissue fibrosis. They also suggest novel strategies for the treatment of SARS by targeting TGF-signaling compartments [56]. MERS-CoV, like other lethal human coronaviruses, can induce renal and lung cell apoptosis through the upregulation of SMAD7, an important protein in the TGF-β signaling pathway in vivo and in vitro (Fig. 3) [57]. Given the similarities between of SARS-CoV-2 and SARS-CoV, it could be predicted that the causative agent of COVID-19 also affects the TGF‐β signaling pathway in humans.
Cytokine regulation
Macrophages play critical roles in the immune system through cytokine regulation pathways and phagocytosis in response to coronaviruses infection. These innate immune cells are the main sources of cytokines [58]. An immunological and serological study of SARS patients showed that they had a low level of antiviral cytokines (IL-12p40 and IFN-α/β/ γ) and an increased level of pro-inflammatory cytokines (IL-6 and TNF-α) and inflammatory chemokines such as macrophage inflammatory protein 1α (MIP-1α). In addition to the extreme upregulation of inflammatory chemokine, the insufficiency of antiviral cytokines may represent a mechanism by which SARS-CoV escapes from immune responses [59]. A systematic analysis of autopsy specimens from SARS patients demonstrated that the expression of pro-inflammatory cytokines, including TNF-α, monocyte chemoattractant protein-1 (MCP-1), TGF-β1, IL-6, and IL-1β could induce the proliferation of ACE2+ cells, which are infected by SARS-CoV- [53]. Exposure to COVID-19 results in higher activated lymphocyte counts and minor monocyte counts than are observed in influenza patients, and in COVID-19 patients, IL-6 and IL-8 levels are increased. IL-6 upregulation in COVID-19 and influenza patients triggers acute respiratory failure. In contrast, granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-γ, and IL-9 levels are decreased [60]. IL-6 directly or indirectly stimulates glucocorticoid (GC) production, and very high levels indicate COVID-19 immune dysregulation and disease progression [61].
In addition, SARS-CoV PLpro can inhibit the production of type I interferons and pro-inflammatory cytokines through the Toll-like receptor (TLR) and retinoic-acid inducible gene I (RIG-I) pathways. This viral protein has been shown to impede the production of imiquimod-induced cytokine utilizing the suppression of NF-κB activator protein 1 (AP-1) and interferon regulatory factor 3 (IRF-3) in vitro [62]. As discussed above, NF-κB can be translocated to the nucleus, leading to increased pro-inflammatory cytokine expression [44]. It has been reported that SARS-CoV ORF8b might also play a critical role in inflammasome activation and cytokine storm and that it triggers NLRP3 activation as well as the release of IL-1β in vitro [63]. In a SARS-CoV murine model, IP-10, MIG, and its receptor were upregulated in the lungs, implicating the CXCR3 cascade in the development of ARDS in SARS CoV infection [64].
Increased expression of inflammatory cytokines and chemokines such CXCR3, IL-8 (CXCL8), SOCS5, IL-1β, CCR2, and IL-1α confirms the lung immunopathology in the lower respiratory tracts of patients infected with MERS-CoV. This, along with a decreased Th1 and Th2 response probably leads to severe infection, an increased risk of death, immunopathology, and lung inflammation (Fig. 3) [65]. Moreover, clinical findings have shown that increased levels of IP-10 are also associated with disease progression and poor prognosis in cases of MERS-CoV infection [66].
The modification of upstream cytokine regulation pathways might be a successful strategy for COVID-19 treatment. It has been suggested that more attention should be paid to the dysregulated production of IFN-I in COVID-19 patients, and ALK, cGAS, and STING should be considered potential targets for treatment against a cytokine storm, which occasionally occurs in acute cases of SARS-CoV-2 and SARS-CoV [67]. Increased levels of Th17 CD4 T cells seen in COVID-19 patients can be explained by high levels of IL-6, which is involved in T helper 17 (TH17) cell development [68].
Based on in vitro data, there is a significant distinguishable pattern in the cytokine storm seen in COVID-19 that resembles that observed in SARS-CoV and MERS-CoV infection. Only three cytokines, IL-6, IP-10, and IFN-γ, show markedly elevated levels in all three of these highly pathogenic HCoV infections [69]. Lianhuaqingwen (LH), a traditional Chinese medicine, significantly inhibits SARS-CoV-2 proliferation in Vero E6 cells and significantly reduces the production of proinflammatory cytokines, including TNF-α, IL-6, CCL-2/MCP-1, and CXCL-10/IP-10 [70]. In SARS-CoV-2-infected individuals, the proinflammatory cytokines IL-6, IL-10, and TNF-α increase during the disease and decrease during recovery. In addition, IL-6, IL-10, and TNF-α levels are inversely related to CD4 and CD8 T cell counts. [71]. Previous studies on animals showed that the cytokine storm plays an important role in reducing the adaptive immune response to SARS-CoV infection [72].
p53
One of the critical regulators of the cell cycle that is activated in response to different types of stress through phosphorylation and other post-translational modifications such as acetylation is transcription factor p53 [73, 74]. The upregulation of p53 increases the expression of genes involved in DNA repair (Gadd45α, P53r2, Ddb2, Mgmt), apoptosis (Bax, Noxa, Puma, Pig3), and cell-cycle arrest (14-3-3σ, p21, Btg2, Reprimo) when the damage becomes irreversible [74, 75]. Also, the level of p53 protein in the cell is controlled by the factors MDM2 and MDMX, which together reduce intracellular p53 levels by activating E3 ubiquitin ligase, ultimately leading to degradation of p53 by the proteasome [76, 77]. p53 acts as a tumor suppressor in cancer cells, but recent reports have suggested that p53 can also be involved in antiviral cellular immune responses, assisting in eliminating pathogens such as viruses [78]. Therefore, as a type of cellular stress, viral infections can stimulate the activation of p53, which eventually can induce apoptosis and suppress viral replication [79,80,81]. Extensive studies have shown that p53 is expressed in response to viral infections that enhance the expression of interferon-stimulated genes (ISG15) and genes encoding interferon regulatory factors (IRF9/5/7) and Toll-like receptor 3 (TLR3), which are involved in the IFN-I-dependent antiviral response [78, 80, 82]. Several studies have revealed that p53 can have both positive and negative effects on various viral infections. Recent observations indicated that p53-dependent apoptosis is detrimental in the early stages of replication of some viruses, but in the late stages of replication, some viruses use apoptosis to transmit completely formed viral particles to other, healthy cells [83]. Previous studies have shown that respiratory viruses, including respiratory syncytial virus (RSV) and influenza virus, cause p53-dependent apoptosis [84, 85].
Also, the murine coronavirus mouse hepatitis virus (MHV) induces cell cycle arrest by activating transcription factor p53, which favors viral infection [86]. Previous studies have also illustrated that the SARS-CoV 3a protein leads to cell cycle arrest by increasing phosphorylation of p53 in HEK 293 cells [87].
Yuan et al., based on in vitro and in vivo data, demonstrated that PLpro of HCoV-NL63 inhibits the p53-dependent antiviral response by reducing the stability of the p53 transcription factor via increasing MDM2-mediated ubiquitination, which ultimately leads to enhanced virus replication [88]. Furthermore, Ma-Lauer et al. revealed that in coronaviruses such as MERS-CoV and SARS-CoV, p53 acts as a negative regulator and reduces virus replication. These researchers proposed that nsp3 has the protective SARS-unique domain (SUD) and PLpro, which counteract the antiviral effect of p53. The in vitro and in vivo data indicate that SUD and PLpro together destroy p53 and inhibit the host cell's defense response by stabilizing E3 ubiquitin ligase RCHY1 [89]. Moreover, the 3a protein of SARS-CoV increases apoptosis in host cells by indirect activation of p53. The 3a protein increases the expression level of p53 and Bax by increasing the activity of the p38 MAPK signaling pathway and inhibiting the STAT signaling pathway, which directly inhibits p53 in vitro [90]. Recent clinical studies have shown that SARS-CoV-2 infection can cause apoptosis of lymphocytes. Interestingly, in patients with lymphopenia, p53, an important factor associated with apoptosis, is significantly increased [91]. Compared to other coronaviruses such as MERS-CoV and SARS-CoV, it has been shown that SARS-CoV-2 is more sensitive to the IFN-I-dependent antiviral response in the early stages of infection [92]. Since p53 plays a critical role in inducing this response, it is destroyed by MERS-CoV and SARS-CoV. SARS-CoV-2 might also inhibit antiviral responses by destroying p53. Moreover, since MERS-CoV and SARS-CoV use p53 signaling to induce apoptosis and cell cycle arrest, SARS-CoV-2 might also use the p53 pathway to induce cell cycle arrest and apoptosis (Fig. 3).
EGFR
EGFR belongs to the family of receptor tyrosine kinases (RTKs). These are transmembrane proteins with an extracellular domain for ligand binding and an intracellular domain. They regulate many cellular processes by activating downstream signaling pathways such as the PI3K/AKT, Ras/Raf/MAPK, STAT, and Src kinase pathways via phosphorylation [93]. Upregulation of this pathway has been seen in many cancers that increase cell migration and proliferation. The over-activation of EGFR upon a viral infection has a major role in the entry of the virus into the host cell, producing more mucus and activating the inflammatory response [94, 95]. Recent reports have suggested that EGFR signaling stimulates the uptake of influenza A virus (IAV) and transmissible gastroenteritis virus (TGEV) into the host cell [96, 97]. In addition, other studies have shown that respiratory viral infections and PEV infections suppress the antiviral response by increasing the activity of the EGFR signaling pathway [98, 99]. Studies have also indicated that CoV-induced macropinocytosis is reliant on the activation of EGFR, which stimulates macropinocytosis pathways within the cell. SARS-CoV and MHV infections stimulate macropinocytosis, which occurs during late infection in vitro [100].
Extensive studies have shown that the EGFR signaling pathway plays a critical role in the progression of lung fibrosis. The role of EGFR in fibrosis progression has been investigated using in vivo models, primarily mice [101]. Using mouse models of SARS-CoV pathogenesis, it has been shown that the wound repair pathway, controlled by EGFR, is critical for recovery from SARS-CoV-induced tissue damage. SARS-CoV infection in the early stages causes acute lung injury, and in the middle stages, the symptoms of fibrosis appear in the lungs. Many survivors of SARS-CoV infection have a high risk of developing pulmonary fibrosis. EGFR signaling is an important regulator of SARS-CoV-induced lung damage. Hence, researchers have suggested that the upregulation of the EGFR pathway after SARS-CoV infection increases fibrosis. Therefore, the use of tyrosine kinase inhibitors such as erlotinib and related compounds is able to reverse or inhibit fibrosis development in a variety of animal models [102]. Activation of EGFR regulates downstream signaling pathways such as AKT, PI3K, and ERK, which are involved in inducing expression of wound-healing genes. It has also been seen that fibrosis caused by SARS-CoV is independent of the role of various types of interferon. Therefore, inhibition of EGFR could avert an excessive fibrotic response to SARS-CoV (Fig. 3) [102, 103]. SARS-CoV-2, like other emerging respiratory viruses, may induce pulmonary fibrosis in survivors of SARS-CoV-2 infection by activating the EGFR pathway. SARS-CoV-2 leads to pulmonary injury, and EGFR might facilitate its entry to the host cell and cause an inflammatory responses. Therefore, understanding how EGFR functions after a viral infection may lead to new treatments in the future.
JNK
C-Jun NH2-terminal kinase (JNK1/JNK2) signaling is an MAPK downstream pathway activated through phosphorylation by MKK7 and MKK4, which ultimately leads to numerous cellular processes, such as the inflammatory response, cell proliferation, survival, and death. The JNK signaling cascade is activated in response to various types of cellular stress, pathogens, and growth factors. One of the main targets of the JNK signaling pathway, which is involved in the pathogenicity of viruses by activating antiviral and pro-inflammatory cytokines, is the transcription factor activator protein 1 (AP-1) [104, 105]. Research shows that JNKs are important kinases that are activated in innate immune responses to viral infection and stimulate the activity of several significant cytokines, such as interleukins (IL-2, IL-4) and interferon gamma (IFN-γ) [106]. Recent reports have indicated that RSV and influenza A virus (IAV) increase the activity of the JNK/AP-1 signaling pathway [107, 108].
The coronavirus infectious bronchitis virus (IBV) stimulates the activity of the JNK signaling pathway by activating MKK7, which ultimately increases virus-induced apoptosis in H1299 cells. Paradoxically, inhibition of the JNK signaling pathway by SP600125 eliminates the inhibitory effect of JNK on the anti-apoptotic protein Bcl2 in Huh-7 cells, which ultimately reduces apoptosis during IBV infection [109]. Also, when Vero cells were transfected with a Bcl-2 expression plasmid and then infected with SARS-CoV, ??overexpression?? of Bcl-2 or other anti-apoptotic proteins protected cells against coronavirus-induced apoptosis [110, 111]. Another study found that the JNK signaling pathway is activated during HCoV-229E infection and plays an anti-apoptotic role through the modulation of Bcl2 family proteins. Activation of JNK also regulates innate immunity by inducing IFN-β and IL-8 [112]. Moreover, the JNK signaling pathway plays a crucial role in SARS-CoV infection [109]. Hence, the phosphorylation of JNK and its upstream pathways have been observed during this viral infection. JNK and PI3K/AKT signaling pathways could be phosphorylated by the N protein of SARS-CoV, causing the establishment of persistent SARS-CoV infection in Vero E6 cells [113, 114]. Moreover, the N protein of SARS-CoV is involved in the induction of apoptosis through the JNK activation and p38 MAPK signaling pathways in the absence of growth factors in COS-1 monkey kidney cells [115]. Also, Ye et al. observed that the protein encoded by ORF6 of SARS-CoV induces apoptosis by caspase-3-mediated ER stress and JNK-dependent signaling pathways in Vero E6 cells [116]. Upregulation of SARS-CoV 3a and 7a protein has also been demonstrated to induce JNK activation in HEK293T cells [49]. Furthermore, the SARS-CoV 3b protein stimulates the expression of the transcription factor AP-1 by activating JNK and ERK signaling pathways in Huh7 cells. Activation of AP-1 by the 3b protein stimulates the pro-inflammatory cytokines that are involved in cytokine storm production in SARS-CoV infection (Fig. 3) [117]. As with SARS-CoV, JNK and its upstream signaling pathways, such as MKK4 and MKK7, might play a major role in SARS-CoV-2-induced apoptosis. SARS-CoV-2, like SARS-CoV may induce the production of pro-inflammatory cytokines through AP-1 activation in response to the infection.
p38 MAPK
The p38 MAPK family comprises four groups of 38-kDa protein (p38) [118]. It is activated by phosphorylation at the sequence Thr180-Gly181-Tyr182, called the activation loop, when exposed to growth factors, environmental stresses such as viral infections, and inflammatory cytokines [118, 119]. The p38 MAPK signaling pathway plays various roles depending on the type of stimulation and also the type of tissue exposed to such stimulus. Hence, p38 MAPK signaling can increase both cell death and survival [120, 121]. Its key role has been observed in many viral infections that cause respiratory symptoms; in HCoV-229E, the activation of p38 MAPK is needed to induce CPE (cytopathic effect) and viral replication [122]. Mouse hepatitis virus (MHV, also known as murine coronavirus) requires p38 MAPK activation for its replication, and its activation leads to the phosphorylation of eIF4E to translate host proteins, such as IL-6 to synthesize a virus-specific protein [123]. Also, influenza A virus (H5N1) activates p38 MAPK to induce cytokines [124]. Avian infectious bronchitis virus (IBV) induces IL-6 and IL-8 production via phosphorylation of p38 MAPK, while dual-specificity phosphatase 1 (DUSP1), a protein phosphatase, in cells infected with IBV, is upregulated to dephosphorylate p38 MAPK in order to regulate pro-inflammatory cytokine [125].
In SARS-CoV-infected cells, at the in vitro stage, p38 MAPK is phosphorylated through downstream effectors such as STAT3, MAPK-activated protein kinase 2 (MAPKAPK2), eIF4E (eukaryotic translation initiation factor 4E) and activating transcription factor 1 (ATF-1) [126,127,128]. SARS-CoV-infected Vero E6 cells exhibit an elevated level of phosphorylated p38 MAPK [128]. Moreover, the 3a protein of SARS-CoV increases the p53 level, leading to p38 MAPK activation and the upregulation of Bax expression, which consequently triggers the intrinsic apoptosis pathway in vitro [90]. In vitro data have indicated that SARS-CoV induces apoptosis to cause intense damage in lung tissue, which is one of the prominent symptoms of this disease [129]. Besides the 3a protein, the 7a protein not only activates apoptosis by activating p38 MAPK but also inhibits translation in vitro [130]. Moreover, the N protein of SARS-CoV in COS-1 monkey kidney cells stimulates apoptosis via p38 MAPK upregulation [115]. In SARS-CoV, TGF-β1 is upregulated through the Egr-1-mediated pathway, which is induced by the papain-like protease (PLpro). TGF-β1 prompts lung fibrosis both in mouse and human lung tissues through the ROS/p38 MAPK/STAT3 pathway, triggered by SARS-CoV PLpro [54]. During SARS-CoV infection, syntenin activates p38 MAPK to increase inflammatory cytokines [131]. Cells infected with recombinant SARS-CoV lacking the envelope protein (E) PDZ-binding motif (PBM, a domain involved in protein-protein interactions) have lower levels of p38 MAPK, suggesting that E protein is involved in the severity of the infection in vivo [131]. An increased level of phosphorylated p38 MAPK in SARS patients results in high levels of IL-8 and aberrant cytokine levels [132]. Moreover, an in vitro experiment showed that the p38 MAPK inhibitor had an antiviral effect against MERS-CoV (Fig. 3) [133]. SARS-CoV-2 proteins may directly stimulate p38 activity to promote replication. The upregulation of p38 MAPK may also facilitate viral entry by ACE2 endocytosis and cause inflammation, thrombosis, and vasoconstriction in COVID-19 patients [134].
As mentioned above, SARS-CoV proteins such as 3a, 7a, N, and E are involved in activating p38 MAPK, suggesting that SARS-CoV-2 proteins may also be involved in triggering the p38 MAPK signaling pathway. One of the well-known symptoms of coronavirus infection is pulmonary injury. In SARS-CoV cases, the p38 MAPK pathway, via p53, TGF-β1, and syntenin, leads to apoptosis via interaction with different molecules, resulting in lung injury. SARS-CoV-2 is therefore likely to use p38 MAPK signaling to induce apoptosis and lung damage.
Extracellular signal-regulated kinase (ERK)
ERK is one of the main members of the MAPK signaling pathways [135]. ERK1 and ERK2, also referred to p44/42 MAPK, are involved in cell proliferation, survival, motility, and differentiation when exposed to mitogens and extracellular stimulants [136, 137]. Activation of the ERK pathway depends on binding of Ras to the cell membrane to initiate the cascade, after which ERK is phosphorylated at the sequence Thr183-Glu184-Tyr185 and is then translocated to the nucleus [138,139,140].
It has been shown that ERK1/2 and MEK1/2 knockdown prevents the replication of the murine coronavirus genome but does not affect protein translation or virus entry [141]. Endothelin 1 (ET-1) is upregulated in pulmonary fibrosis, induces the MEK/ERK MAP kinase pathway, and increases CCN2 mRNA and protein levels [142]. In idiopathic pulmonary fibrosis (IPF), TGFβ-1, through the ERK1/2 pathway, stimulates the transformation of human lung fibroblasts into myofibroblasts [143, 144]. It also increases α-SMA and collagen expression in the lung by activating ERK1/2 and inhibiting GSK-3 and β-catenin translocation to the nucleus [145]. Activation of the ERK signaling pathway has been observed during infections with respiratory viruses, including influenza virus (SIV, H1N1pdm), avian H9N2 influenza virus, and RSV [146, 147]. This suggests the participation of the ERK signaling pathway during respiratory virus infection. It has been demonstrated that coronaviruses can also recruit this pathway to facilitate their replication and pathogenesis.
Both p38 MAPK and ERK1/2 are phosphorylated and activated in Vero E6 cells infected with SARS-CoV [128]. P90RSK, a downstream element of ERK, is phosphorylated at Ser380 by p38 MAPK and plays a major role in SARS-CoV-infected cells when its Thr573 phosphorylation is decreased [148]. p90RSK controls apoptosis, which has been shown by treatment of SARS-CoV-infected Vero E6 cells with PD98059 (an ERK inhibitor) and SB203580 (a p38 inhibitor), which reduced p90RSK phosphorylation. However, the ERK inhibitor did not prevent major damage to the infected cell [148]. Thus, ERK phosphorylation is not sufficient to prevent apoptosis induced by SARS-CoV infection due to Akt activation [149].
Surjit et al. demonstrated that the SARS-CoV N protein induces apoptosis in COS-1 cells via downregulation of ERK in cells that are free from growth factors [115]. AP-1 controls cytokine transcription due to SARS-CoV infection, and ERK activation upregulates AP-1 activity via c-Fos in vitro [117]. In addition, 3b, one of the accessory proteins of SARS-CoV [150], stimulates AP-1-related genes [117]. ORF3b induces AP-1 transcriptional activity by activating ERK pathways [117]. SARS-CoV, via the S, PLpro, 3b proteins, and MERS-CoV, via Raf/MEKK2,3/Mos and then MKK1,2,5, induces ERK signaling [112]. Furthermore, the ERK/MAPK pathway, along with the PI3K/AKT/mTOR pathway, plays a crucial role in the pathogenesis of MERS-CoV, and inhibition of this pathway prevents MERS-CoV proliferation in vitro [151]. Taken together, the ERK signaling pathway is essential for pathogenicity, preventing host cell apoptosis and causing lung injury and viral spread. COVID-19 leads to the release of pro-inflammatory cytokines such as IL-1β and IL-6 to provoke lung inflammation, fever, and fibrosis [152]. As mentioned above, ERK stimulates AP-1 activation through 3b and ORF3b viral proteins to increase cytokine expression and via the N protein to increase survival of host cells infected with SARS-CoV (Fig. 3). It is therefore likely that ERK could activate AP-1 and cytokine expression in SARS-CoV-2 infection.
TNF-α
TNF-α belongs to the TNF superfamily of transmembrane proteins and is a proinflammatory cytokine that is mainly secreted from macrophages [153]. Its essential roles in defense against pathogens are immunoregulation and inhibition of infection factors and tumor formation [154, 155], and it is associated with many diseases [156]. This cytokine mediates the inflammatory response and is important for the host cell defense against pathogens. Excessive expression of TNF-α can exacerbate disease [156]. Each person has a different ability to produce cytokines that is related to that individual's genetic background [157]. TNF-α is associated with autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, psoriasis, and refractory asthma [158]. It has various effects. For instance, it increases the release of pro-inflammatory factors, enhances the production of adhesion molecules, and increases migration of eosinophils and neutrophils. Moreover, its levels are elevated in mice infected with human adenovirus B1[159] and in humans infected with H1N1 influenza virus [160]. The capsid of hepatitis B virus strongly induces TNF-α production in human THP-1 macrophages [161]. Activation of Toll-like receptor 2 (TLR2) by binding to viruses such as hepatitis C virus (HCV) [162], measles virus [163], and various herpesviruses, including herpes simplex virus (HSV) [164] and cytomegalovirus [165], is followed by TNF-α production. TNF-α is associated with lung fibrosis [166] and pulmonary fibrosis [167], which, together with IL-1, is overexpressed in regenerating type 2 pneumocytes in the lung [166]. In IPF progression, epithelial cells release cytokines such as TNF-α, which induce the transformation of fibroblasts into myofibroblasts and the production of extracellular matrix molecules, leading to respiratory insufficiency [168].
In SARS patients, analysis of serum cytokine levels has shown large amounts of TNF-α, which suggests that TNF-α is involved in SARS pathogenesis [169] by causing excessive inflammation [170]. The TNF-α level is significantly increased in the acute stage and reduced in the convalescent stage of illness in SARS patients [171], suggesting that TNF-α plays a pivotal role in the pulmonary injury triggered by SARS coronavirus, and TNF-α inhibitors can be a potent factor in treatment of acute respiratory disease syndrome caused by coronavirus infection [172]. The SARS-CoV S protein attaches to the host cell receptor to initiate infection [33]. The S protein induces IL-6 and TNF-α production via the NF-κB pathway activation in RAW264.7 cells, which are murine macrophages [50]. Moreover, p38 MAPK activation gives rise to TNF-α and IL-6 production [173, 174]. It is thought that incubation of RAW264.7 cells with the S protein activates the p38 pathway, thus increasing TNF-α and IL-6 levels [50]. Additionally, SARS-CoV infection induces TNF-α and IL-6 production in dendritic cells and human macrophages [59, 175]. Haga et al., demonstrated in vitro that SARS-CoV S prompts TNF-α converting enzyme (TACE, also called ADAM17) to cause ACE-2 ectodomain shedding, which is followed by the production of TNF-α, facilitating viral entry and increasing tissue injury [176]. TACE continuously sheds ACE-2 in the epithelium of the human airway to release soluble ACE2 (sACE2), which is active [177]. There is no evidence that TNF-α obstruction is dangerous for COVID-19 patients [158]. TNF-α is present at high levels in immune response cells, such as dendritic cells and macrophages in the case of SARS-CoV infection. TNF-α also intensifies the harmful effects of SARS-CoV infection, such as lung tissue injury, and facilitates virus entry (Fig. 3). SARS-CoV-2 is known to cause severe pulmonary damage, which is consistent with the theory that TNF-α worsens the symptoms of SARS-CoV-2 infection and promotes overproduction of cytokines via the S protein and that TACE may also be involved in the entry of SARS-CoV-2.
Interferon signaling pathway
The innate immune response is the first step in protection against pathogens [178]. In viral infections, it stimulates the interferon signaling pathway and, ultimately, the expression of IFN-I, which leads to an antiviral response in cells [179]. IFN-I binds to cell-surface co-receptors and stimulates the Janus kinase (JAK) signal transducer and STAT signaling network, which upregulates several IFN-stimulated genes (ISGs). Numerous proteins encoded by these ISGs are responsible for antiviral functions [180]. However, viruses have developed various mechanisms to interfere with IFN expression, and this appears to apply to coronaviruses. In SARS-CoV, MERS-CoV, and other CoVs, the response to viral infection through IFN-I is hampered. These CoVs use different mechanisms to reduce the production of IFN-I. This suppression strategy is closely correlated with disease severity and mortality [181].
TGEV, a member of the genus Alphacoronavirus, escapes the effect of IFN-I through IRE1α-based manipulation of the miR-30a-5p/SOCS1/3 axis. Recent studies have indicated that the interaction between the C-terminus of the SARS-CoV (and also MERS-CoV) N protein and the SPRY domain of tripartite motif protein 25 (TRIM25) prevents TRIM25-mediated RIG-I ubiquitination, leading to the suppression of IFN production in vitro [182, 183]. Lui et al. have proposed a strategy through which MERS-CoV and SARS-CoV utilize their M proteins to inhibit IFN-I expression at the level of TANK-binding kinase 1 (TBK1)-associated phosphorylation and stimulation of interferon regulatory factor 3 (IRF3), leading to evasion of the innate antiviral response in HEK-293 cells [184]. A recent study revealed that the SARS-CoV proteins 8ab and 8b are IFN antagonists. This in vitro study demonstrated the direct association of these two proteins with IRF3. It was also found that they moderately inhibit IFN stimulation by restricting IRF3 activation and by increasing the proteasome-dependent destruction of IRF3 [185]. Moreover, of the SARS-CoV viral proteins, ORF3b, M, ORF6, N, PLP, nsp1, nsp7, and nsp15 have been identified as interferon antagonists [186,187,188]. Lee et al. have reported that ORF8b of MERS-CoV is an effective antagonist of both MDA5 (melanoma differentiation-associated protein 5)- and RIG-I (retinoic acid-inducible gene I)-mediated stimulation of IFN signaling in vitro [189].
In vitro data have shown that the expression of the membrane-anchored PLpro domain (PLpro-TM) of SARS-CoV prevents STING/TBK1/IKKε-based stimulation of IFN-I and disrupts the phosphorylation and dimerization of IRF3, which are triggered via STING (stimulator of interferon genes) and TBK1. Additionally, PLpro-TM physically interacts with TRAF3, TBK1, IκB kinase ε (IKKε), STING, and IRF3, the significant factors that form the STING-TRAF3-TBK1 complex for stimulation of IFN expression [190]. Yang and colleagues have proposed a model of the IFN antagonism strategy of MERS-CoV ORF4b in HeLa and 293T cells in which ORF4b binding to TBK1 and IKKε stops the molecular interplay between mitochondrial antiviral signaling protein (MAVS) and IKKε and prevents IRF3 phosphorylation and stimulation of IFN-β production (Fig. 3) [191]. Despite the well-known effects of IFN-I as an antiviral and immunomodulatory factor that contributes to protection against viral infections, it has become clear that IFN-I also supports adverse inflammatory responses in several bacterial, fungal, and chronic virus infections. Using BALB/c mice infected with SARS-CoV, Channappanavar et al. revealed that rapid virus replication and delayed IFN-I signaling results in inflammatory responses and lung immunopathology that threaten survival. This delayed IFN-I signaling stimulates the accumulation of inflammatory monocyte-macrophages, leading to increased cytokine/chemokine levels in the lung, vascular leakage, and reduced virus-specific T cell responses [72]. Thus, dysregulated IFN-I and inflammation pathways converge to cause lung damage in a SARS-CoV model. By screening the SARS-CoV-2 viral proteins, researchers have found that the viral proteins ORF6, ORF8, and nucleocapsid are potent inhibitors of the interferon type I signaling pathway. These proteins have been shown to inhibit interferon-stimulated genes (ISGs) such as ISG54 and ISG56 in HEK-293 T cells [192]. Moreover, by screening 23 viral proteins, it has been shown that the SARS-CoV-2 nsp, nsp3, nsp12, nsp13, nsp14, ORF3, ORF6, and M proteins inhibit Sendai-virus-induced IFN-β promoter activation [193]. Also, Xia et al. have shown that SARS-CoV-2 proteins suppress the IFN-I response in vitro. They found that nsp6, nsp13, and ORF6 suppress phosphorylation of TBK1 and IRF3, which is essential for the induction of the IFN-β pathway. Compared with SARS-CoV and MERS-CoV, IFN-I signaling is more efficiently inhibited by nsp1 and nsp6 of SARS-CoV-2 [194]. Mantlo et al. assessed the sensitivity of SARS-CoV-2 to both IFN-α and IFN-β in vitro. They found that IFN-I has potent anti-SARS-CoV-2 activity in cultured cells – a finding that could inform future treatment options for COVID-19 [195]. The progress of disease and viral transmission depend on immune antagonism and the level of viral replication. Using ex vivo human lung tissue explants, it has been shown that SARS-CoV-2 infection of lung tissues, despite producing a large amount of virus, induces a relatively small amount of IFN and pro-inflammatory cytokines/chemokines in comparison to SARS-CoV infection, which might explain why the virus is transmitted by individuals without symptoms and with delayed onset of COVID-19 disease [70]. The rapid appearance of SARS-CoV-2 has prompted increased investigation of innate immune responses to CoVs. Recent research has shown that SARS-CoV-2 stimulates insignificant expression of IFNs (type I, II, or III) in infected cells, even when compared to other coronaviruses. This lack of IFN production probably weakens the primary innate immune response to SARS-CoV-2 infection and suggests that exogenous IFN treatment can be successful against SARS-CoV-2 [196]. Most COVID-19 patients with acute respiratory failure, instead of a cytokine storm, have IFN-I and -II immunosuppression. The IFN-γ and IFN-α response pathways are seriously depressed in B cells, CD8+ T cells, ??mixed cytolytic lymphocyte populations?? (MCLPs), Tregs, ??plasmacytoid dendritic cells??, and monocyte/macrophage subsets of COVID-19-infected subjects [197].
Lokugamage et al. have shown that SARS-CoV-2 is much more sensitive to IFN-I than SARS-CoV is in Vero and in Calu3 cells. This increased sensitivity to IFN-I is probably due to differences in the viral proteins of these two CoVs [198]. Different studies have shown that SARS-CoV-2 nsp13 (helicase), nsp14 (exonuclease), nsp15 (endoribonuclease), and accessory protein ORF6 are the strongest viral interferon antagonists [193, 199]. Unlike other respiratory viruses, the IFN-I and IFN-III levels are reduced in SARS-CoV-2 infections, and it should be noted that increasing IFN leads to a significant reduction in virus replication, and even SARS-CoV is limited by IFN-α [200]. Moreover, high levels of circulating neutrophils caused by the induction of CXCL2 and CXCL8 contribute to disease severity in COVID-19 patients [201].
We therefore conclude that antagonism of IFN by SARS-CoV-2 and the ability to block other pathways of innate antiviral signaling might determine the severity of the disease. This prediction is based on the hypothesis that the immune response against SARS-CoV-2 is similar to that against other CoVs, which needs to be confirmed through future studies on SARS-CoV-2.
PI3K/AKT
The PI3K/AKT pathway controls various cellular processes, such as antiviral immunity, cell proliferation, protein translation, RNA processing, apoptosis, and autophagy [202]. Several host cellular signaling pathways are stimulated and exploited by viruses to allow successful cycles of replication. The PI3K/AKT pathway has recently received significant attention because of its function in controlling virus replication. This pathway has been shown to be essential not only for viral cell entry but also for subsequent intracellular trafficking and replication of some viruses [203]. Apoptosis functions as an operative antiviral mechanism in an infected organism that is simple and usually successful. Thus, to protect its replication, the virus must inhibit or postpone apoptosis. One of the mechanisms used by viruses to prolong viral replication and slow down apoptosis in both persistent and acute infections is altering PI3K/AKT signaling [204]. PI3K/AKT signaling is also involved in upregulating the interferon response, and increasing PI3K/AKT activity can stop viral spread due to the initiation of cellular defenses [205]. Stimulation of this pathway by influenza virus decreases IRF-3-based promoter functions and dimerization of IRF-3, resulting in a reduction in host antiviral activity [206]. Kindrachuk et al. demonstrated that in vitro MERS-CoV replication could be substantially inhibited by treatment with a group of licensed kinase inhibitors targeting the PI3K/AKT/mTOR pathway. This suggests that PI3K/AKT/mTOR signaling plays a central role in MERS-CoV infection and may indicate new drug targets for treatment [151]. Also, it is found that the PI3K/AKT signaling pathway is vital for the initiation of SARS-CoV infection in Vero E6 cells, and no viable cells were detected after treatment with the PI3K/AKT inhibitor LY294002 [114, 207]. Moreover, Chan et al. found that the M protein of SARS-CoV modulates the cellular AKT pro-survival pathway and mitochondrial cytochrome c secretion and stimulates apoptosis in both HEK293T cells and transgenic drosophila (Fig. 3) [208].
Based on information obtained from studies of other CoVs, we consider it likely that infection with SARS-CoV-2 stimulates PI3K/AKT signaling and that preventing P13K activation can lessen the severity of viral infections.
Signaling pathways that are likely to be dysregulated upon infection with SARS-CoV-2
The SARS-CoV-2 global crisis has reminded everyone that viruses are constantly changing genetically and functionally to better infect different species, including humans [209]. Coronaviruses, on the one hand, functionally paralyze their host cells and, on the other hand, force them to produce their essential proteins. In the meantime, many of the host cell's signaling pathways undergo alterations that can worsen the disease condition caused by these viruses. In the current review, we have summarized information about several signaling pathways that are affected by coronavirus infections and illustrated the mechanisms by which this occurs. The main purpose of a virus upon its entry into the host cell is to replicate its RNA/DNA, maintain or over-activate the machinery for protein production in the host cell, and evade immune responses by the host. Considering the ability of coronavirus infections, particularly SARS-CoV-2 infections, to induce inflammation and lung injury and also the similarity of this novel virus to MERS-CoV and SARS-CoV in its ability to infect lung epithelial cells, it is expected that this emerging life-threatening coronavirus utilizes some of the same cellular signaling pathways as MERS-CoV and SARS-CoV. As a therapeutic approach, the use of anti-inflammatory agents during COVID-19 to affect inflammatory signaling pathways might be beneficial for reducing the severity of the disease (Table 1). The NF-κB, cytokine regulation, ERK, and TNF-α signaling pathways have been shown to be likely causes of inflammation in MERS-CoV and SARS-CoV infections, with neutrophilia and basophilia exacerbating the disease in SARS patients [210]. The increased levels of inflammatory cytokines in serum of COVID-19 patients have shed light on the involvement of such signaling pathways in the pathogenesis infection of SARS-CoV-2. Further studies are needed to clarify the exact roles of cellular signaling pathways once the SARS-CoV-2 initiates infection in its host cell. Also, researchers in molecular medicine should consider the roles of the most strongly up- and downregulated components of cellular signaling pathways during COVID-19 to identify and design better molecular drugs that will decrease the fatality rate of this novel pandemic coronavirus.
References
Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P (2008) Global trends in emerging infectious diseases. Nature 451(7181):990–993
Brunetti O, Derakhshani A, Baradaran B, Galvano A, Russo A, Silvestris N (2020) COVID-19 infection in cancer patients: how can oncologists deal with these patients? Front Oncol 10:734
Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, Ahuja A, Yung MY, Leung C, To K (2003) A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 348(20):1986–1994
Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367(19):1814–1820
Sampathkumar P, Temesgen Z, Smith TF, Thompson RL (2003) SARS: epidemiology, clinical presentation, management, and infection control measures. In: Mayo Clinic Proceedings, 2003, vol 7. Elsevier, pp 882–890
Organization WH (2004) SARS (Severe Acute Respiratory Syndrome). https://www.who.int/ith/diseases/sars/en/
Modjarrad K (2016) MERS-CoV vaccine candidates in development: the current landscape. Vaccine 34(26):2982–2987
Organization WH (2019,) Middle East respiratory syndrome coronavirus (MERS-CoV) https://www.who.int/emergencies/mers-cov/en/
Hui DS, Memish ZA, Zumla A (2014) Severe acute respiratory syndrome vs. the Middle East respiratory syndrome. Curr Opin Pulmon Med 20(3):233–241
Kaminskyy V, Zhivotovsky B (2010) To kill or be killed: how viruses interact with the cell death machinery. J Intern Med 267(5):473–482
Grandvaux N, Servant MJ, Hiscott J (2002) The interferon antiviral response: from viral invasion to evasion. Curr Opin Infect Dis 15(3):259–267
de Wit E, van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14(8):523
Kindler E, Thiel V, Weber F (2016) Interaction of SARS and MERS coronaviruses with the antiviral interferon response. In: Advances in virus research, vol 96. Elsevier, pp 219–243
Yin Y, Wunderink RG (2018) MERS, SARS and other coronaviruses as causes of pneumonia. Respirology 23(2):130–137
Prompetchara E, Ketloy C, Palaga T (2020) Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol 38(1):1–9
Rabaan AA, Al-Ahmed SH, Haque S, Sah R, Tiwari R, Malik YS, Dhama K, Yatoo MI, Bonilla-Aldana DK, Rodriguez-Morales AJ (2020) SARS-CoV-2, SARS-CoV, and MERSCOV: a comparative overview. Infez Med 28:174–184
Tyrrell D, Bynoe M (1966) Cultivation of viruses from a high proportion of patients with colds. Lancet 76–77
Hamre D, Procknow JJ (1966) A new virus isolated from the human respiratory tract. Proc Soc Exp Biol Med 121(1):190–193
Almeida JD, Tyrrell D (1967) The morphology of three previously uncharacterized human respiratory viruses that grow in organ culture. J Gen Virol 1(2):175–178
Tyrrell D, Almeida J, Cunningham C, Dowdle W, Hofstad M, McIntosh K, Tajima M, Zakstelskaya LY, Easterday B, Kapikian A (1975) Coronaviridae. Intervirology 5(1–2):76–82
Kahn JS, McIntosh K (2005) History and recent advances in coronavirus discovery. Pediatr Infect Dis J 24(11):S223–S227
de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish ZA (2013) Commentary: Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J Virol 87(14):7790–7792
Of the International CSG (2020) The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 1:536–544
de Haan CA, Rottier PJ (2005) Molecular interactions in the assembly of coronaviruses. Adv Virus Res 64:165–230
McBride R, Van Zyl M, Fielding BC (2014) The coronavirus nucleocapsid is a multifunctional protein. Viruses 6(8):2991–3018
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16(1):69
Fehr AR, Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. In: Coronaviruses. Springer, pp 1–23
Cheng PK, Wong DA, Tong LK, Ip S-M, Lo AC, Lau C-S, Yeung EY, Lim WW (2004) Viral shedding patterns of coronavirus in patients with probable severe acute respiratory syndrome. Lancet 363(9422):1699–1700
Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV (1992) Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357(6377):420–422
Luan Y, Xu W (2007) The structure and main functions of aminopeptidase N. Curr Med Chem 14(6):639–647
Hamming I, Cooper ME, Haagmans BL, Hooper NM, Korstanje R, Osterhaus AD, Timens W, Turner A, Navis G, van Goor H (2007) The emerging role of ACE2 in physiology and disease. J Pathol 212(1):1–11
Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pöhlmann S (2005) Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci 102(22):7988–7993
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426(6965):450–454
Letko M, Marzi A, Munster V (2020) Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 5(4):562–569
Nedellec P, Dveksler GS, Daniels E, Turbide C, Chow B, Basile AA, Holmes KV, Beauchemin N (1994) Bgp2, a new member of the carcinoembryonic antigen-related gene family, encodes an alternative receptor for mouse hepatitis viruses. J Virol 68(7):4525–4537
Raj VS, Mou H, Smits SL, Dekkers DH, Müller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA (2013) Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495(7440):251–254
Belouzard S, Chu VC, Whittaker GR (2009) Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci 106(14):5871–5876
Ziebuhr J, Snijder EJ, Gorbalenya AE (2000) Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81(4):853–879
Mielech AM, Chen Y, Mesecar AD, Baker SC (2014) Nidovirus papain-like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res 194:184–190
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368(6489):409–412
Krijnse-Locker J, Ericsson M, Rottier P, Griffiths G (1994) Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J Cell Biol 124(1):55–70
Wolff G, Melia CE, Snijder EJ, Bárcena M (2020) Double-membrane vesicles as platforms for viral replication. Trends Microbiol 28:1022–1033
Lee JY, Bae S, Myoung J (2019) Middle East Respiratory Syndrome coronavirus-encoded accessory proteins impair MDA5-and TBK1-mediated activation of NF-kappaB. J Microbiol Biotechnol 29(8):1316–1323
Canton J, Fehr AR, Fernandez-Delgado R, Gutierrez-Alvarez FJ, Sanchez-Aparicio MT, Garcia-Sastre A, Perlman S, Enjuanes L, Sola I (2018) MERS-CoV 4b protein interferes with the NF-kappaB-dependent innate immune response during infection. PLoS Pathog 14(1):e1006838
DeDiego ML, Nieto-Torres JL, Regla-Nava JA, Jimenez-Guardeno JM, Fernandez-Delgado R, Fett C, Castano-Rodriguez C, Perlman S, Enjuanes L (2014) Inhibition of NF-kappaB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J Virol 88(2):913–924
Siu KL, Yuen KS, Castano-Rodriguez C, Ye ZW, Yeung ML, Fung SY, Yuan S, Chan CP, Yuen KY, Enjuanes L (2019) Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J 33(8):8865–8877
Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, Torres J, Aguilella VM, Enjuanes L (2015) Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485:330–339
Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY (2020) A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect 9(1):558–570
Kanzawa N, Nishigaki K, Hayashi T, Ishii Y, Furukawa S, Niiro A, Yasui F, Kohara M, Morita K, Matsushima K (2006) Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett 580(30):6807–6812
Wang W, Ye L, Ye L, Li B, Gao B, Zeng Y, Kong L, Fang X, Zheng H, Wu Z (2007) Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res 128(1–2):1–8
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O’Meara MJ, Guo JZ, Swaney DL, Tummino TA, Huettenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Naing ZZC, Zhou Y, Peng S, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Shen W, Shi Y, Zhang Z, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Ramachandran R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Wankowicz SA, Bohn M, Sharp PP, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Roesch F, Vallet T, Meyer B, White KM, Miorin L, Rosenberg OS, Verba KA, Agard D, Ott M, Emerman M, Ruggero D, García-Sastre A, Jura N, von Zastrow M, Taunton J, Ashworth A, Schwartz O, Vignuzzi M, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor S, Fraser JS, Gross J, Sali A, Kortemme T, Beltrao P, Shokat K, Shoichet BK, Krogan NJ (2020) A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. bioRxiv 2020.2003.2022.002386
Mirzaei H, Faghihloo E (2018) Viruses as key modulators of the TGF-beta pathway; a double-edged sword involved in cancer. Rev Med Virol 28(2):e1967
He L, Ding Y, Zhang Q, Che X, He Y, Shen H, Wang H, Li Z, Zhao L, Geng J (2006) Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J Pathol 210(3):288–297
Li SW, Wang CY, Jou YJ, Yang TC, Huang SH, Wan L, Lin YJ, Lin CW (2016) SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-beta1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep 6:25754
Wang CY, Lu CY, Li SW, Lai CC, Hua CH, Huang SH, Lin YJ, Hour MJ, Lin CW (2017) SARS coronavirus papain-like protease up-regulates the collagen expression through non-Samd TGF-beta1 signaling. Virus Res 235:58–66
Zhao X, Nicholls JM, Chen YG (2008) Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J Biol Chem 283(6):3272–3280
Yeung ML, Yao Y, Jia L, Chan JF, Chan KH, Cheung KF, Chen H, Poon VK, Tsang AK, To KK, Yiu MK, Teng JL, Chu H, Zhou J, Zhang Q, Deng W, Lau SK, Lau JY, Woo PC, Chan TM, Yung S, Zheng BJ, Jin DY, Mathieson PW, Qin C, Yuen KY (2016) MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat Microbiol 1:16004
Cheung CY, Poon LL, Ng IH, Luk W, Sia SF, Wu MH, Chan KH, Yuen KY, Gordon S, Guan Y, Peiris JS (2005) Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol 79(12):7819–7826
Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, Nicholls JM, Peiris JM, Lau YL (2005) Chemokine up-regulation in sars-coronavirus–infected, monocyte-derived human dendritic cells. Blood 106(7):2366–2374
Mudd PA, Crawford JC, Turner JS, Souquette A, Reynolds D, Bender D, Bosanquet JP, Anand NJ, Striker DA, Martin RS (2020) Targeted immunosuppression distinguishes COVID-19 from influenza in moderate and severe disease. medRxiv
Weber M, Michl P, Auernhammer C, Engelhardt D (1997) Interleukin-3 and interleukin-6 stimulate cortisol secretion from adult human adrenocortical cells. Endocrinology 138(5):2207–2210
Li S-W, Wang C-Y, Jou Y-J, Huang S-H, Hsiao L-H, Wan L, Lin Y-J, Kung S-H, Lin C-W (2016) SARS coronavirus papain-like protease inhibits the TLR7 signaling pathway through removing Lys63-linked polyubiquitination of TRAF3 and TRAF6. Int J Mol Sci 17(5):678
Shi CS, Nabar NR, Huang NN, Kehrl JH (2019) SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov 5:101
Glass WG, Subbarao K, Murphy B, Murphy PM (2004) Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J Immunol 173(6):4030–4039
Alosaimi B, Hamed M, Naeem A, Alsharef A, Alqahtani S, Aldosari K, Alamri A, Al-Eisa K, Khojah T, Assiri A, Enani M (2019) MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract. Cytokine 126
Kim ES, Choe PG, Park WB, Oh HS, Kim EJ, Nam EY, Na SH, Kim M, Song K-H, Bang JH (2016) Clinical progression and cytokine profiles of Middle East respiratory syndrome coronavirus infection. J Korean Med Sci 31(11):1717–1725
Deng X, Yu X, Pei J (2020) Regulation of interferon production as a potential strategy for COVID-19 treatment
Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8(4):420–422
Johnson BS, Laloraya M (2020) Cytokine storm in COVID-19 patients transforms to a cytokine super cyclone in patients with risk factors. Cytokine Growth Fact Rev 54:32–42
Runfeng L, Yunlong H, Jicheng H, Weiqi P, Qinhai M, Yongxia S, Chufang L, Jin Z, Zhenhua J, Haiming J (2020) Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2). Pharmacol Res 104761
Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, Chen L, Li M, Liu Y, Wang G (2020) Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol 11:827
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S (2016) Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19(2):181–193
Lane D, Levine A (2010) p53 research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2(12):a000893
Joerger AC, Fersht AR (2016) The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem 85:375–404
Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137(3):413–431
Wade M, Li Y-C, Wahl GM (2013) MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13(2):83–96
Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387(6630):296–299
Rivas C, Aaronson SA, Munoz-Fontela C (2010) Dual role of p53 in innate antiviral immunity. Viruses 2(1):298–313
Sato Y, Tsurumi T (2013) Genome guardian p53 and viral infections. Rev Med Virol 23(4):213–220
Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K (2003) Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 424(6948):516–523
Muñoz-Fontela C, Macip S, Martínez-Sobrido L, Brown L, Ashour J, García-Sastre A, Lee SW, Aaronson SA (2008) Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med 205(8):1929–1938
Nakamura H, Li M, Zarycki J, Jung JU (2001) Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J Virol 75(16):7572–7582
Teodoro JG, Branton PE (1997) Regulation of apoptosis by viral gene products. J Virol 71(3):1739
Bian T, Gibbs JD, Örvell C, Imani F (2012) Respiratory syncytial virus matrix protein induces lung epithelial cell cycle arrest through a p53 dependent pathway. PLoS ONE 7(5):e38052
Turpin E, Luke K, Jones J, Tumpey T, Konan K, Schultz-Cherry S (2005) Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. J Virol 79(14):8802–8811
Chen C-J, Sugiyama K, Kubo H, Huang C, Makino S (2004) Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J Virol 78(19):10410–10419
Yuan X, Yao Z, Wu J, Zhou Y, Shan Y, Dong B, Zhao Z, Hua P, Chen J, Cong Y (2007) G1 phase cell cycle arrest induced by SARS-CoV 3a protein via the cyclin D3/pRb pathway. Am J Respir Cell Mol Biol 37(1):9–19
Yuan L, Chen Z, Song S, Wang S, Tian C, Xing G, Chen X, Xiao Z-X, He F, Zhang L (2015) p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J Biol Chem 290(5):3172–3182
Ma-Lauer Y, Carbajo-Lozoya J, Hein MY, Müller MA, Deng W, Lei J, Meyer B, Kusov Y, von Brunn B, Bairad DR (2016) p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc Natl Acad Sci 113(35):E5192–E5201
Padhan K, Minakshi R, Towheed MAB, Jameel S (2008) Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J Gen Virol 89(8):1960–1969
Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, Guo D, Hu W, Yang J, Tang Z (2020) Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect 9(1):761–770
Sallard E, Lescure F-X, Yazdanpanah Y, Mentre F, Peiffer-Smadja N, Florence A, Yazdanpanah Y, Mentre F, Lescure F-X, Peiffer-Smadja N (2020) Type 1 interferons as a potential treatment against COVID-19. Antivir Res 178:104791
Lurje G, Lenz H-J (2009) EGFR signaling and drug discovery. Oncology 77(6):400–410
Sebastian S, Settleman J, Reshkin SJ, Azzariti A, Bellizzi A, Paradiso A (2006) The complexity of targeting EGFR signalling in cancer: from expression to turnover. Biochimica et Biophysica Acta (BBA) Rev Cancer 1766(1):120–139
Samani AA, Fallavollita L, Jaalouk DE, Galipeau J, Brodt P (2001) Inhibition of carcinoma cell growth and metastasis by a vesicular stomatitis virus G-pseudotyped retrovector expressing type I insulin-like growth factor receptor antisense. Hum Gene Ther 12(16):1969–1977
Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C (2010) The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog 6(9):e1001099
Hu W, Zhang S, Shen Y, Yang Q (2018) Epidermal growth factor receptor is a co-factor for transmissible gastroenteritis virus entry. Virology 521:33–43
Yang L, Xu J, Guo L, Guo T, Zhang L, Feng L, Chen H, Wang Y (2018) Porcine epidemic diarrhea virus-induced epidermal growth factor receptor activation impairs the antiviral activity of type I interferon. J Virol 92(8)
Kalinowski A, Galen BT, Ueki IF, Sun Y, Mulenos A, Osafo-Addo A, Clark B, Joerns J, Liu W, Nadel JA (2018) Respiratory syncytial virus activates epidermal growth factor receptor to suppress interferon regulatory factor 1-dependent interferon-lambda and antiviral defense in airway epithelium. Mucosal Immunol 11(3):958–967
Freeman MC, Peek CT, Becker MM, Smith EC, Denison MR (2014) Coronaviruses induce entry-independent, continuous macropinocytosis. MBio 5(4):e01340-e11314
Vallath S, Hynds RE, Succony L, Janes SM, Giangreco A (2014) Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur Respir Soc 44:513–522
Venkataraman T, Frieman MB (2017) The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antivir Res 143:142–150
Venkataraman T, Coleman CM, Frieman MB (2017) Overactive epidermal growth factor receptor signaling leads to increased fibrosis after severe acute respiratory syndrome coronavirus infection. J Virol 91(12):e00182-e1117
Bubici C, Papa S (2014) JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol 171(1):24–37
Weston CR, Davis RJ (2007) The JNK signal transduction pathway. Curr Opin Cell Biol 19(2):142–149
Chu W-M, Ostertag D, Li Z-W, Chang L, Chen Y, Hu Y, Williams B, Perrault J, Karin M (1999) JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity 11(6):721–731
Ludwig S, Ehrhardt C, Neumeier ER, Kracht M, Rapp UR, Pleschka S (2001) Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J Biol Chem 276(14):10990–10998
Li X, Sun S, Wu F, Shi T, Fan H, Li D (2016) Study on JNK/AP-1 signaling pathway of airway mucus hypersecretion of severe pneumonia under RSV infection. Eur Rev Med Pharmacol Sci 20(5):853–857
Fung TS, Liu DX (2017) Activation of the c-Jun NH 2-terminal kinase pathway by coronavirus infectious bronchitis virus promotes apoptosis independently of c-Jun. Cell Death Dis 8(12):1–13
Zhong Y, Liao Y, Fang S, Tam JP, Liu DX (2012) Up-regulation of Mcl-1 and Bak by coronavirus infection of human, avian and animal cells modulates apoptosis and viral replication. PLoS One 7(1):e30191
Bordi L, Castilletti C, Falasca L, Ciccosanti F, Calcaterra S, Rozera G, Di Caro A, Zaniratti S, Rinaldi A, Ippolito G (2006) Bcl-2 inhibits the caspase-dependent apoptosis induced by SARS-CoV without affecting virus replication kinetics. Adv Virol 151(2):369–377
Lim YX, Ng YL, Tam JP, Liu DX (2016) Human coronaviruses: a review of virus–host interactions. Diseases 4(3):26
Mizutani T, Fukushi S, Ishii K, Sasaki Y, Kenri T, Saijo M, Kanaji Y, Shirota K, Kurane I, Morikawa S (2006) Mechanisms of establishment of persistent SARS-CoV-infected cells. Biochem Biophys Res Commun 347(1):261–265
Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S (2005) JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochimica et Biophysica Acta (BBA) Mol Basis Dis 1741(1–2):4–10
Surjit M, Liu B, Jameel S, Chow VT, Lal SK (2004) The SARS coronavirus nucleocapsid protein induces actin reorganization and apoptosis in COS-1 cells in the absence of growth factors. Biochem J 383(1):13–18
Ye Z, Wong CK, Li P, Xie Y (2008) A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochimica et Biophysica Acta (BBA) Gen Subj 1780(12):1383–1387
Varshney B, Lal SK (2011) SARS-CoV accessory protein 3b induces AP-1 transcriptional activity through activation of JNK and ERK pathways. Biochemistry 50(24):5419–5425
Cuadrado A, Nebreda AR (2010) Mechanisms and functions of p38 MAPK signalling. Biochem J 429(3):403–417
Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410(6824):37–40
Juretic N, Santibáñez JF, Hurtado C, Martínez J (2001) ERK 1, 2 and p38 pathways are involved in the proliferative stimuli mediated by urokinase in osteoblastic SaOS-2 cell line. J Cell Biochem 83(1):92–98
Yosimichi G, Nakanishi T, Nishida T, Hattori T, Takano-Yamamoto T, Takigawa M (2001) CTGF/Hcs24 induces chondrocyte differentiation through a p38 mitogen-activated protein kinase (p38MAPK), and proliferation through a p44/42 MAPK/extracellular-signal regulated kinase (ERK). Eur J Biochem 268(23):6058–6065
Kono M, Tatsumi K, Imai AM, Saito K, Kuriyama T, Shirasawa H (2008) Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: involvement of p38 MAPK and ERK. Antivir Res 77(2):150–152
Banerjee S, Narayanan K, Mizutani T, Makino S (2002) Murine coronavirus replication-induced p38 mitogen-activated protein kinase activation promotes interleukin-6 production and virus replication in cultured cells. J Virol 76(12):5937–5948
Lee DC, Cheung C-Y, Law AH, Mok CK, Peiris M, Lau AS (2005) p38 mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factor alpha expression in response to avian influenza virus H5N1. J Virol 79(16):10147–10154
Liao Y, Wang X, Huang M, Tam JP, Liu DX (2011) Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology 420(2):106–116
Mizutani T (2007) Signal Transduction in SARS-CoV-Infected Cells. Ann N Y Acad Sci 1102(1):86–95
Mizutani T, Fukushi S, Murakami M, Hirano T, Saijo M, Kurane I, Morikawa S (2004) Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells. FEBS Lett 577(1–2):187–192
Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S (2004) Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem Biophys Res Commun 319(4):1228–1234
Yan H, Xiao G, Zhang J, Hu Y, Yuan F, Cole DK, Zheng C, Gao GF (2004) SARS coronavirus induces apoptosis in Vero E6 cells. J Med Virol 73(3):323–331
Kopecky-Bromberg SA, Martinez-Sobrido L, Palese P (2006) 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J Virol 80(2):785–793
Jimenez-Guardeno JM, Nieto-Torres JL, DeDiego ML, Regla-Nava JA, Fernandez-Delgado R, Castaño-Rodriguez C, Enjuanes L (2014) The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog 10(8):e1004320
Lee C-H, Chen R-F, Liu J-W, Yeh W-T, Chang J-C, Liu P-M, Eng H-L, Lin M-C, Yang KD (2004) Altered p38 mitogen-activated protein kinase expression in different leukocytes with increment of immunosuppressive mediators in patients with severe acute respiratory syndrome. J Immunol 172(12):7841–7847
Josset L, Menachery VD, Gralinski LE, Agnihothram S, Sova P, Carter VS, Yount BL, Graham RL, Baric RS, Katze MG (2013) Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio 4(3):e00165-e1113
Grimes JM, Grimes KV (2020) p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J Mol Cell Cardiol 144:63–65
Yang X, Gabuzda D (1999) Regulation of human immunodeficiency virus type 1 infectivity by the ERK mitogen-activated protein kinase signaling pathway. J Virol 73(4):3460–3466
Pelech SL, Sanghera JS (1992) Mitogen-activated protein kinases: versatile transducers for cell signaling. Trends Biochem Sci 17(6):233–238
Meloche S, Seuwen K, Pages G, Pouyssegur J (1992) Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol Endocrinol 6(5):845–854
Anderson NG, Maller JL, Tonks NK, Sturgill TW (1990) Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 343(6259):651–653
Payne D, Rossomando A, Martino P, Erickson A, Her J, Shabanowitz J, Hunt D, Weber M, Sturgill T (1991) Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J 10(4):885–892
Nguyen DH, Hussaini IM, Gonias SL (1998) Binding of urokinase-type plasminogen activator to its receptor in MCF-7 cells activates extracellular signal-regulated kinase 1 and 2 which is required for increased cellular motility. J Biol Chem 273(14):8502–8507
Cai Y, Liu Y, Zhang X (2007) Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J Virol 81(2):446–456
Shi-wen X, Howat SL, Renzoni EA, Holmes A, Pearson JD, Dashwood MR, Bou-Gharios G, Denton CP, du Bois RM, Black CM (2004) Endothelin-1 induces expression of matrix-associated genes in lung fibroblasts through MEK/ERK. J Biol Chem 279(22):23098–23103
Hu Y, Peng J, Feng D, Chu L, Li X, Jin Z, Lin Z, Zeng Q (2006) Role of extracellular signal-regulated kinase, p38 kinase, and activator protein-1 in transforming growth factor-β1-induced alpha smooth muscle actin expression in human fetal lung fibroblasts in vitro. Lung 184(1):33–42
Vancheri C, Gili E, Failla M, Mastruzzo C, Salinaro ET, LoFurno D, Pistorio MP, La Rosa C, Caruso M, Crimi N (2005) Bradykinin differentiates human lung fibroblasts to a myofibroblast phenotype via the B2 receptor. J Allergy Clin Immunol 116(6):1242–1248
Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A, Vancheri C (2008) TGF-β1 targets the GSK-3β/β-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res 57(4):274–282
Gao W, Sun W, Qu B, Cardona CJ, Powell K, Wegner M, Shi Y, Xing Z (2012) Distinct regulation of host responses by ERK and JNK MAP kinases in swine macrophages infected with pandemic (H1N1) 2009 influenza virus. PLoS ONE 7(1):e30328
Chen W, Monick MM, Carter AB, Hunninghake GW (2000) Activation of ERK2 by respiratory syncytial virus in A549 cells is linked to the production of interleukin 8. Exp Lung Res 26(1):13–26
Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S (2006) Regulation of p90RSK phosphorylation by SARS-CoV infection in Vero E6 cells. FEBS Lett 580(5):1417–1424
Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S (2004) Importance of Akt signaling pathway for apoptosis in SARS-CoV-infected Vero E6 cells. Virology 327(2):169–174
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen M-H (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300(5624):1394–1399
Kindrachuk J, Ork B, Hart BJ, Mazur S, Holbrook MR, Frieman MB, Traynor D, Johnson RF, Dyall J, Kuhn JH (2015) Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother 59(2):1088–1099
Conti P, Ronconi G, Caraffa A, Gallenga C, Ross R, Frydas I, Kritas S (2020) Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents 34(2)
Idriss HT, Naismith JH (2000) TNFα and the TNF receptor superfamily: Structure-function relationship (s). Microsc Res Technol 50(3):184–195
Aggarwal BB, Vilček J (1992) Tumor necrosis factors: Structure, function, and mechanism of action, vol 56. Marcel Dekker Incorporated
Vilcek J, Lee TH (1991) Tumor necrosis factor. New insights into the molecular mechanisms of its multiple actions. J Biol Chem 266(12):7313–7316
Sariban E, Imamura K, Luebbers R, Kufe D (1988) Transcriptional and posttranscriptional regulation of tumor necrosis factor gene expression in human monocytes. J Clin Investig 81(5):1506–1510
Wang S, Wei M, Han Y, Zhang K, He L, Yang Z, Su B, Zhang Z, Hu Y, Hui W (2008) Roles of TNF-α gene polymorphisms in the occurrence and progress of SARS-Cov infection: a case-control study. BMC Infect Dis 8(1):27
Russell B, Moss C, George G, Santaolalla A, Cope A, Papa S, Van Hemelrijck M (2020) Associations between immune-suppressive and stimulating drugs and novel COVID-19—a systematic review of current evidence. Ecancermedicalscience 14
Kajon AE, Gigliotti AP, Harrod KS (2003) Acute inflammatory response and remodeling of airway epithelium after subspecies B1 human adenovirus infection of the mouse lower respiratory tract. J Med Virol 71(2):233–244
Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE (1998) Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Investig 101(3):643–649
Cooper A, Tal G, Lider O, Shaul Y (2005) Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J Immunol 175(5):3165–3176
Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G (2004) Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127(5):1513–1524
Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, ter Meulen V, Schneider-Schaulies S (2002) Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol 76(17):8729–8736
Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW (2004) Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci 101(5):1315–1320
Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW (2003) Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol 77(8):4588–4596
Pan LH, Ohtani H, Yamauchi K, Nagura H (1996) Co-expression of TNFα and IL-1β in human acute pulmonary fibrotic diseases: an immunohistochemical analysis. Pathol Int 46(2):91–99
Whyte M, Hubbard R, Meliconi R, Whidborne M, Eaton V, Bingle C, Timms J, Duff G, Facchini A, Pacilli A (2000) Increased risk of fibrosing alveolitis associated with interleukin-1 receptor antagonist and tumor necrosis factor-α gene polymorphisms. Am J Respir Crit Care Med 162(2):755–758
Verma S, Slutsky AS (2007) Idiopathic pulmonary fibrosis—new insights. N Engl J Med 356(13):1370–1372
Zhang Y, Li J, Zhan Y, Wu L, Yu X, Zhang W, Ye L, Xu S, Sun R, Wang Y (2004) Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect Immun 72(8):4410–4415
McDermott JE, Mitchell HD, Gralinski LE, Eisfeld AJ, Josset L, Bankhead A, Neumann G, Tilton SC, Schäfer A, Li C (2016) The effect of inhibition of PP1 and TNFα signaling on pathogenesis of SARS coronavirus. BMC Syst Biol 10(1):93
Hsueh P-R, Chen P-J, Hsiao C-H, Yeh S-H, Cheng W-C, Wang J-L, Chiang B-L, Chang S-C, Chang F-Y, Wong W-W (2004) Patient data, early SARS epidemic, Taiwan
Folz RJ, Elkordy MA (1999) Coronavirus pneumonia following autologous bone marrow transplantation for breast cancer. Chest 115(3):901–905
He Y, Zhou Y, Liu S, Kou Z, Li W, Farzan M, Jiang S (2004) Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem Biophys Res Commun 324(2):773–781
Eliopoulos AG, Gallagher NJ, Blake SM, Dawson CW, Young LS (1999) Activation of the p38 mitogen-activated protein kinase pathway by Epstein–Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J Biol Chem 274(23):16085–16096
Tseng C-TK, Perrone LA, Zhu H, Makino S, Peters CJ (2005) Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol 174(12):7977–7985
Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y (2008) Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc Natl Acad Sci 105(22):7809–7814
Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, Hooper NM, Turner AJ (2005) Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem 280(34):30113–30119
McDonald DR, Levy O (2019) Innate immunity. In: Clinical immunology. Elsevier, pp 39–53 (e31)
Schulz KS, Mossman KL (2016) Viral evasion strategies in type I IFN signalling—a summary of recent developments. Front Immunol 7:498
Kane M, Zang TM, Rihn SJ, Zhang F, Kueck T, Alim M, Schoggins J, Rice CM, Wilson SJ, Bieniasz PD (2016) Identification of interferon-stimulated genes with antiretroviral activity. Cell Host Microbe 20(3):392–405
Dandekar AA, Perlman S (2005) Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol 5(12):917–927
Hu Y, Li W, Gao T, Cui Y, Jin Y, Li P, Ma Q, Liu X, Cao C (2017) The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J Virol 91(8):e02143-e12116
Chang C-Y, Liu HM, Chang M-F, Chang SC (2020) Middle East respiratory syndrome coronavirus nucleocapsid protein suppresses type I and type III interferon induction by targeting RIG-I signaling. J Virol
Lui P-Y, Wong L-YR, Fung C-L, Siu K-L, Yeung M-L, Yuen K-S, Chan C-P, Woo PC-Y, Yuen K-Y, Jin D-Y (2016) Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg Microbes Infect 5(1):1–9
Wong HH, Fung TS, Fang S, Huang M, Le MT, Liu DX (2018) Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology 515:165–175
Totura AL, Baric RS (2012) SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2(3):264–275
Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez-Santos YM, Wang J, Takayama J, Ghosh AK (2010) Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol 84(9):4619–4629
Kindler E, Gil-Cruz C, Spanier J, Li Y, Wilhelm J, Rabouw HH, Züst R, Hwang M (2017) V’kovski P, Stalder H, Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog 13(2):e1006195
Lee JY, Bae S, Myoung J (2019) Middle East respiratory syndrome coronavirus-encoded ORF8b strongly antagonizes IFN-β promoter activation: its implication for vaccine design. J Microbiol 57(9):803–811
Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z (2014) SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 5(5):369–381
Yang Y, Ye F, Zhu N, Wang W, Deng Y, Zhao Z, Tan W (2015) Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets. Sci Rep 5:17554
Li J-Y, Liao C-H, Wang Q, Tan Y-J, Luo R, Qiu Y, Ge X-Y (2020) The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res 286:198074
Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L (2020) Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11(1):1–12
Xia H, Cao Z, Xie X, Zhang X, Chen JY-C, Wang H, Menachery VD, Rajsbaum R, Shi P-Y (2020) Evasion of type I Interferon by SARS-CoV-2. Cell Rep 33(1):108234
Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C (2020) Antiviral activities of type I interferons to SARS-CoV-2 infection. Antivir Res 179:104811
O’Brien TR, Thomas DL, Jackson SS, Prokunina-Olsson L, Donnelly RP, Hartmann R (2020) Weak induction of interferon expression by SARS-CoV-2 supports clinical trials of interferon lambda to treat early COVID-19. Clin Infect Dis 71:1410–1412
Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D (2020) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–1045
Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R, Menachery VD (2020) SARS-CoV-2 sensitive to type I interferon pretreatment. BioRxiv
Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H, Cai J-P, Jin D-Y, To KK-W, Chan JF-W (2020) SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect 199:1–29
Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R, Menachery VD (2020) SARS-CoV-2 is sensitive to type I interferon pretreatment. BioRxiv
Felgenhauer U, Schoen A, Gad HH, Hartmann R, Schaubmar AR, Failing K, Drosten C, Weber F (2020) Inhibition of SARS–CoV-2 by type I and type III interferons. J Biol Chem 295(41):13958–13964
Dunn EF, Connor JH (2012) HijAkt: the PI3K/Akt pathway in virus replication and pathogenesis. In: Progress in molecular biology and translational science, vol 106. Elsevier, pp 223–250
Wang L, Yang L, Fikrig E, Wang P (2017) An essential role of PI3K in the control of West Nile virus infection. Sci Rep 7(1):1–9
Diehl N, Schaal H (2013) Make yourself at home: viral hijacking of the PI3K/Akt signaling pathway. Viruses 5(12):3192–3212
Ji W-T, Liu HJ (2008) PI3K-Akt signaling and viral infection. Recent Pat Biotechnol 2(3):218–226
Ehrhardt C, Marjuki H, Wolff T, Nürnberg B, Planz O, Pleschka S, Ludwig S (2006) Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol 8(8):1336–1348
Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S (2006) Characterization of persistent SARS-CoV. Nidoviruses 324, vol. 581
Chan C-M, Ma C-W, Chan W-Y, Chan HYE (2007) The SARS-Coronavirus Membrane protein induces apoptosis through modulating the Akt survival pathway. Arch Biochem Biophys 459(2):197–207
Sarkesh A, Sorkhabi AD, Sheykhsaran E, Alinezhad F, Mohammadzadeh N, Hemmat N, Baghi HB (2020) Extrapulmonary Clinical Manifestations in COVID-19 Patients. Am J Trop Med Hyg 103(5):1783–1796
Hemmat N, Derakhshani A, Bannazadeh Baghi H, Silvestris N, Baradaran B, De Summa S (2020) Neutrophils, crucial, or harmful immune cells involved in coronavirus infection: a bioinformatics study. Front Genet 11:641
Cho HK, Kim JR, Kim SY, Kyaw YY, Win AA, Cheong JH (2015) Sorafenib suppresses hepatitis B virus gene expression via inhibiting JNK pathway. Hepatoma Res 1:97–103
Dyall J, Gross R, Kindrachuk J, Johnson RF, Olinger GG, Hensley LE, Frieman MB, Jahrling PB (2017) Middle East respiratory syndrome and severe acute respiratory syndrome: current therapeutic options and potential targets for novel therapies. Drugs 77(18):1935–1966
Dai Q, Di Zhang HY, Xie W, Xin R, Wang L, Xu X, He X, Xiong J, Sheng H, Le Zhang KZ (2017) Berberine restricts coxsackievirus B type 3 replication via inhibition of c-Jun N-Terminal Kinase (JNK) and p38 MAPK activation in vitro. Med Sci Monit 23:1448
Warowicka A, Nawrot R, Goździcka-Józefiak A (2020) Antiviral activity of berberine. Adv Virol 165(9):1935–1945
Pizzorno A, Padey B, Dubois J, Julien T, Traversier A, Dulière V, Brun P, Lina B, Rosa-Calatrava M, Terrier O (2020) In vitro evaluation of antiviral activity of single and combined repurposable drugs against SARS-CoV-2. Antivir Res 181:104878
Zhang H, Niu X, Qian Z, Qian J, Xuan B (2015) The c-Jun N-terminal kinase inhibitor SP600125 inhibits human cytomegalovirus replication. J Med Virol 87(12):2135–2144
Cicenas J, Zalyte E, Rimkus A, Dapkus D, Noreika R, Urbonavicius S (2018) JNK, p38, ERK, and SGK1 inhibitors in cancer. Multidisciplinary Digital Publishing Institute
Cerbone A, Toaldo C, Pizzimenti S, Pettazzoni P, Dianzani C, Minelli R, Ciamporcero E, Roma G, Dianzani MU, Canaparo R (2012) AS601245, an anti-inflammatory JNK inhibitor, and clofibrate have a synergistic effect in inducing cell responses and in affecting the gene expression profile in CaCo-2 colon cancer cells. PPAR Res 2012
Jiang W, Wang Z, Jiang Y, Lu M, Li X (2015) Ginsenoside Rg1 ameliorates motor function in an animal model of Parkinson’s disease. Pharmacology 96(1–2):25–31
De Boer JP, van Egmond PW, Helder MN, de Menezes RX, Cleton-Jansen A-M, Beliën JA, Verheul HM, van Royen BJ (2012) Kaspers G-JJ, van Beusechem VW, Targeting JNK-interacting protein 1 (JIP1) sensitises osteosarcoma to doxorubicin. Oncotarget 3(10):1169
Su A-r (2017) Qiu M, Li Y-l, Xu W-t, Song S-w, Wang X-h, Song H-y, Zheng N, Wu Z-w, BX-795 inhibits HSV-1 and HSV-2 replication by blocking the JNK/p38 pathways without interfering with PDK1 activity in host cells. Acta Pharmacol Sin 38(3):402–414
Koff J, Min-Oo G, Ballon-Landa E, Kalinowski A, Ueki I, Lanier L, Nadel JA (2012) EGFR inhibition suppresses respiratory viral infection in vitro and in vivo. In: D73. receptor input of airway networking. American Thoracic Society, p A6274
Zhu X-F, Liu Z-C, Xie B-F, Li Z-M, Feng G-K, Yang D, Zeng Y-X (2001) EGFR tyrosine kinase inhibitor AG1478 inhibits cell proliferation and arrests cell cycle in nasopharyngeal carcinoma cells. Cancer Lett 169(1):27–32
Gan CJ, Li WF, Li CN, Li LL, Zhou WY, Peng XM (2020) EGF receptor inhibitors comprehensively suppress hepatitis B virus by downregulation of STAT3 phosphorylation. Biochem Biophys Rep 22:100763
Metro G, Finocchiaro G, Cappuzzo F (2006) Anti-cancer therapy with EGFR inhibitors: factors of prognostic and predictive significance. Ann Oncol 17:ii42–ii45
Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K (2004) A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Can Res 64(18):6652–6659
Weisberg E, Parent A, Yang PL, Sattler M, Liu Q, Liu Q, Wang J, Meng C, Buhrlage SJ, Gray N (2020) Repurposing of kinase inhibitors for treatment of COVID-19. Pharm Res 37(9):1–29
Tan C-S, Cho B-C, Soo RA (2016) Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor-mutant non-small cell lung cancer. Lung cancer 93:59–68
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV (1999) A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285(5434):1733–1737
Synnott NC, Bauer MR, Madden S, Murray A, Klinger R, O’Donovan N, O’Connor D, Gallagher WM, Crown J, Fersht AR (2018) Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett 414:99–106
Zhang Y, Xu L, Chang Y, Li Y, Butler W, Jin E, Wang A, Tao Y, Chen X, Liang C (2019) Therapeutic potential of ReACp53 targeting mutant p53 protein in CRPC. Prostate Cancer Prostat Dis 23:1–12
Underwood DC, Osborn RR, Bochnowicz S, Webb EF, Rieman DJ, Lee JC, Romanic AM, Adams JL, Hay DW, Griswold DE (2000) SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am J Physiol Lung Cell Mol Physiol 279(5):L895–L902
Prakash J, Sandovici M, Saluja V, Lacombe M, Schaapveld RQ, De Borst MH, Van Goor H, Henning RH, Proost JH, Moolenaar F (2006) Intracellular delivery of the p38 mitogen-activated protein kinase inhibitor SB202190 [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl) 1H-imidazole] in renal tubular cells: a novel strategy to treat renal fibrosis. J Pharmacol Exp Ther 319(1):8–19
Herman AP, Krawczyńska A, Bochenek J, Antushevich H, Herman A, Tomaszewska-Zaremba D (2014) Peripheral injection of SB203580 inhibits the inflammatory-dependent synthesis of proinflammatory cytokines in the hypothalamus. BioMed Res Int 2014
Jin X, Mo Q, Zhang Y, Gao Y, Wu Y, Li J, Hao X, Ma D, Gao Q, Chen P (2016) The p38 MAPK inhibitor BIRB796 enhances the antitumor effects of VX680 in cervical cancer. Cancer Biol Ther 17(5):566–576
Asaduzzaman M, Wang Y, Thorlacius H (2008) Critical role of p38 mitogen-activated protein kinase signaling in septic lung injury. Crit Care Med 36(2):482–488
Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M (2005) Identification of a selective ERK inhibitor and structural determination of the inhibitor–ERK2 complex. Biochem Biophys Res Commun 336(1):357–363
Wang F, Li D, Zheng Z, To KKW, Chen Z, Zhong M, Su X, Chen L, Fu L (2020) Reversal of ABCB1-related multidrug resistance by ERK5-IN-1. J Exp Clin Cancer Res 39(1):1–13
Lis K, Kuzawińska O, Bałkowiec-Iskra E (2014) Tumor necrosis factor inhibitors–state of knowledge. Arch Med Sci 10(6):1175
Tobinick E (2004) TNF-alpha inhibition for potential therapeutic modulation of SARS coronavirus infection. Curr Med Res Opin 20(1):39–40
Chen G, Guo Y, Yutian W (2016) Celastrol inhibits tumor necrosis factor-alpha induced proliferation and inflammatory responses in RAW264. 7 cells. Chin J Tissue Eng Res 20(37):5552–5559
Law S, Leung AW, Xu C (2020) Is the traditional Chinese herb, "Celastrol" effective to combat COVID-19? J Mater Environ Sci 11(8):1205–1208
Habtemariam S, Nabavi SF, Berindan‐Neagoe I, Cismaru CA, Izadi M, Sureda A, Nabavi SM (2020) Should we try the antiinflammatory natural product, celastrol, for COVID‐19? Phytother Res
Liu SP, Wang GD, Du XJ, Wan G, Wu JT, Miao LB, Liang QD (2017) Triptolide inhibits the function of TNF-α in osteoblast differentiation by inhibiting the NF-κB signaling pathway. Exp Ther Med 14(3):2235–2240
Chaparala S, Iwema CL, Chattopadhyay A (2020) SARS-CoV-2 infections-gene expression omnibus (GEO) data mining, pathway enrichment analysis, and prediction of repurposable drugs/compounds
Miller SC, Huang R, Sakamuru S, Shukla SJ, Attene-Ramos MS, Shinn P, Van Leer D, Leister W, Austin CP, Xia M (2010) Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanism of action. Biochem Pharmacol 79(9):1272–1280
Raaben M, Posthuma CC, Verheije MH, Te Lintelo EG, Kikkert M, Drijfhout JW, Snijder EJ, Rottier PJ, De Haan CA (2010) The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J Virol 84(15):7869–7879
Jiménez-Alberto A, Ribas-Aparicio RM, Aparicio-Ozores G, Castelán-Vega JA (2020) Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors. Comput Biol Chem 88:107325
Cho J, Lee YJ, Kim JH, Il Kim S, Kim SS, Choi B-S, Choi J-H (2020) Antiviral activity of digoxin and ouabain against SARS-CoV-2 infection and its implication for COVID-19. Sci Rep 10(1):1–8
Sauvat A, Ciccosanti F, Colavita F, Di Rienzo M, Castilletti C, Capobianchi MR, Kepp O, Zitvogel L, Fimia GM, Piacentini M (2020) On-target versus off-target effects of drugs inhibiting the replication of SARS-CoV-2. Cell Death Dis 11(8):1–11
Ghahremanpour MM, Tirado-Rives J, Deshmukh M, Ippolito JA, Zhang C-H, de Vaca IC, Liosi M-E, Anderson KS, Jorgensen WL (2020) Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. bioRxiv
Guo K, Wang Z, Gao P, Pu Q, Wu M, Huang C, Hur J (2020) Identification of repurposal drugs and adverse drug reactions for various courses of coronavirus disease 2019 (COVID-19) based on single-cell RNA sequencing data. arXiv:200507856
Klann K, Bojkova D, Tascher G, Ciesek S, Münch C, Cinatl J (2020) Growth factor receptor signaling inhibition prevents SARS-CoV-2 replication. bioRxiv
Xiao H, Zhang J, Xu Z, Feng Y, Zhang M, Liu J, Chen R, Shen J, Wu J, Lu Z (2016) Metformin is a novel suppressor for transforming growth factor (TGF)-β1. Sci Rep 6:28597
Sharma S, Ray A, Sadasivam B (2020) Metformin in COVID-19: a possible role beyond diabetes. Diabetes Res Clin Pract 164:108–183
de Gramont A, Faivre S, Raymond E (2016) Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology 6(1):e1257453–e1257453
Huynh LK, Hipolito CJ, Ten Dijke P (2019) A perspective on the development of tgf-β inhibitors for cancer treatment. Biomolecules 9(11):743
Suzuki E, Umezawa K, Bonavida B (2007) Rituximab inhibits the constitutively activated PI3K-Akt pathway in B-NHL cell lines: involvement in chemosensitization to drug-induced apoptosis. Oncogene 26(42):6184–6193
Wang F, Meng F, Wong SCC, Cho WC, Yang S, Chan LW (2020) Combination therapy of gefitinib and miR-30a-5p may overcome acquired drug resistance through regulating the PI3K/AKT pathway in non-small cell lung cancer. Ther Adv Respir Dis 14:1753466620915156
Lan W, Zhao J, Shang H, Peng J, Chen W, Lin J (2019) Anlotinib overcomes multiple drug resistant of the colorectal cancer cells via inactivating PI3K/AKT pathway. bioRxiv 821801
Bhatt R, Zhang L, Schor-Bardach R, Libermann T, Atkins M, Goldberg N, Kumar M, Mier J (2008) Effects of sorafenib administration on the IFN signaling pathway and on the expression of IFN-inducible genes in RCC tumor cells. AACR 68:1128
Acknowledgements
This project was supported by the Immunology Research Center, Tabriz University of Medical Sciences. A previous version of this article is also available at Preprints (https://www.preprints.org/manuscript/202009.0489/v1).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Data collection and categorization were performed by ZA, NKA, HA, and BN. The design of figures was done by AB, AD, and SN. The English editing also was performed by Dr. HBB and DJR. The first draft of the manuscript was written by NH, and all authors commented on previous versions of the manuscript. The corresponding authors of the manuscript are Dr. MAG and Dr. BB. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Zhenhai Chen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hemmat, N., Asadzadeh, Z., Ahangar, N.K. et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch Virol 166, 675–696 (2021). https://doi.org/10.1007/s00705-021-04958-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-04958-7