
Abstract
Porcine epidemic diarrhea (PED) is a contagious viral disease in pigs, caused by the coronavirus porcine epidemic diarrhea virus (PEDV). PEDV infection results in significant mortality in piglets in unvaccinated herds. Like many others RNA viruses, PEDV has high evolutionary rate and is prone to genetic mutations. In this study, we analyzed the complete genome sequence of the recently sequenced isolate PEDV/Belgorod/dom/2008. A recombination event in S gene of PEDV/Belgorod/dom/2008 was detected. Pairwise identity analysis of the whole genome sequences revealed that PEDV/Belgorod/dom/2008 is an intermediate between PEDV and transmissible gastroenteritis virus (TGEV) strains. These results can be used for further analysis of the evolutionary variability, prevalence, and epidemiology of the porcine epidemic diarrhea virus.
Similar content being viewed by others


Enteropathogenic porcine coronaviruses affect pig herds around the world, leading to significant financial losses. Porcine epidemic diarrhea (PED) is a highly contagious viral disease in pigs, caused by an RNA-containing virus belonging to the family Coronaviridae. PED is characterized by debilitating diarrhea, dehydration, and high mortality. The disease affects pigs of all age groups, but the most susceptible are newborn piglets (up to two weeks old) among which mortality ranges between 50 and 100% [1, 2].
PED is common in the United States, Canada, China, Korea, Japan, Thailand and Vietnam, as well as European countries, with the exception of Ireland, Denmark and Sweden [3,4,5,6,7]. PED was first introduced into large pig farms in the Russian Federation in 2006. At present, the prevalence of PED is increasing within regions with a high pig concentration in Russia. However, limited information is available about the genetic characteristics of PEDV strains currently circulating in Russia. All porcine epidemic diarrhea virus (PEDV) isolates form one serotype but have different degrees of virulence in the field [8].
The spike (S) protein of PEDV is the viral protein that is subjected to the greatest immunological pressure and variability. Deletions (S-INDELs) or small insertions have been observed in the S gene nucleotide sequences of many PEDV isolates [9]. The PEDV strains that are currently circulating in the European Union are similar to the American S-INDEL strains [10,11,12]. The phylogenetic classification of the PEDV strains is based on the analysis of complete genomes sequences obtained worldwide [13] or individual genes such as S, M, N, or ORF3 [9, 11, 14].
In this study, we analyzed the genome sequence of recently sequenced PEDV isolate, PEDV/Belgorod/dom/2008 (GenBank accession number MF577027) [15].
Pathological samples from the intestine and stomach were taken from one-month-old sick piglets from the Belgorod region of Russia in 2008 [15]. Total RNA was extracted from a 10% organ suspension using TRIzol Reagent (ThermoFisher Scientific) according to the manufacturer’s instructions. Next-generation sequencing was done using an Illumina MiSeq instrument with a MiSeq reagent kit v3 in 2- × 300-bp PE mode (Illumina, San Diego, CA, USA) [15]. The PEDV/Belgorod/dom/2008 isolate was subsequently isolated from the small-intestine tissue in Vero cell culture.
Prediction of homologous recombination events was carried out using RDP4 (Recombination Detection Program) and SIMPLOT [16, 17]. Pairwise identity analysis was performed using SDT v1.2 software [18] and 18 whole genomic PEDV sequences, three transmissible gastroenteritis virus (TGEV) sequences, and one swine enteric coronavirus sequence from the GenBank database. Multiple alignment was performed using MUSCLE software [19]. Phylogenetic trees were constructed based on PEDV M and S gene sequences using the maximum-likelihood method in MEGA 6.0. [20]. Bootstrap values were estimated for 1000 replicates.
The complete coding sequence of the PEDV/Belgorod/dom/2008 is 28,315 nucleotides (nt) in length (GenBank accession number MF577027) [15]. Two putative recombination sites were detected in the genome of recombinant PEDV/Belgorod/dom/2008 at nt 20476 in ORF1B and nt 24403 in the S gene (Fig. 1). PEDV strain LZC (EF185992) and PEDV strain SLO/JH-11/2015 (KU297956) were identified as the major and minor parental viruses, respectively. The recombinant event was identified by six modules (RDP, MaxChi, Chimaera, Geneconv, Bootscan, SiScan) with high confidence (average p-value, 2.77 × 10−23).

Recombination breakpoints in the genome of the PEDV/Belgorod/dom/2008 isolate predicted by RDP4. The potential parental strains and the recombinant isolate are shown in teal (major), purple (minor) and yellow (recombinant), respectively. Arrows indicate recombinant breakpoints. UTR, untranslated region; ORF, open reading frame; S, spike; E, envelope; M, membrane; N, nucleocapsid
A similarity plot showed high overall sequence similarity between the PEDV/Belgorod/dom/2008 strain and the parental PEDV strains, but with a marked drop in the nucleotide sequence similarity in the S gene region (Fig. 1).
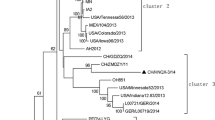
Phylogenetic analysis of the complete genomes showed that PEDV/Belgorod/dom/2008 has a distant relationship to known PEDV strains. The PEDV/Belgorod/dom/2008 isolate does not belong to any groups formed by the American or Chinese strains and forms a separate cluster together with the SeCoV-ITA09 recombinant strain isolated in Italy (Fig. 2).

Phylogenetic tree of the PEDV/Belgorod/dom/2008 isolate (highlighted in black) and other PEDV, TGEV and SeCoV strains of different geographical origin, based on an alignment of predicted amino acid sequences derived from complete genome sequences. The isolation year of LZC is unknown but should be before 2006 according to the GenBank submission date
Since only M gene sequences are available in the GenBank database for the Russian PEDV isolates, we rebuilt the phylogenetic tree to refine the analysis. Based on the phylogenetic analysis of the M gene, the PEDV/Belgorod/dom/2008 isolate belongs to the same clade as other virulent Russian PEDV strains, indicating a high degree of sequence homogeneity in the M gene (Fig. 3a). Interestingly, PEDV/Belgorod/dom/2008 carries a significant number of nucleotide substitutions with reference to the PEDV isolate Belgorod/05/07 (EU179730), which was isolated earlier from the same region.

Maximum-likelihood phylogenies of the PEDV isolates and closely related coronaviruses (TGEV and swine enteric coronavirus strain) based on predicted amino acid sequences. Phylogenetic trees based on the M gene (a) and the S gene (b) are presented. Bootstrap values of 60 or greater are shown at the nodes. The trees show robust incongruence for the PEDV/Belgorod/dom/2008 topology between the M and S genes. PEDV/Belgorod/dom/2008 is indicated by a black circle
The S gene phylogeny of PEDV and related coronaviruses demonstrates that the PEDV/Belgorod/dom/2008 isolate is genetically distinct and does not belong to any group (Fig. 3b). This robust incongruence between the M- and S-gene-based trees may be explained by a recombination event within the genome of the PEDV/Belgorod/dom/2008 isolate. Such variability can lead to dramatic changes in viral virulence, pathogenicity, and antigenicity.
Pairwise identity analysis based on the spike amino acid sequences revealed that PEDV/Belgorod/dom/2008 is an intermediate between PEDV and TGEV and is also distantly related to other PEDV strains (Fig. 4).

Genome-wide pairwise identity matrix of PEDV/Belgorod/dom/2008 and representative spike amino acid sequences of PEDV and TGEV. The PEDV/Belgorod/dom/2008 isolate is indicated by a black circle
PEDV/Belgorod/dom/2008 has a unique spike protein sequence and low similarity to other PEDV isolates. Changes in the S glycoprotein gene play an important role, since this protein is important for tissue tropism and virulence [5]. A preliminary animal study with PEDV/Belgorod/dom/2008 demonstrated that this recombinant is highly virulent in unvaccinated suckling piglets [21, 22].
Recombination events are possible and can sometimes be observed in cases where pigs have been vaccinated or infected with a mixture of TGEV and PEDV. Such recombination events can potentially result in a loss of vaccine efficacy.
Boniotti et al. reported a virus possessing a TGEV genome sequence in which the S protein sequence was identical to that of a PEDV isolate (SeCoV-ITA09) [23]. This chimeric virus was probably generated by recombination between TGEV and PEDV. Similar chimeric viruses have been found by other research groups in Germany [24] and Eastern Europe [25].
The genome sequence of one PEDV isolate (CH/HNQX-3/14) from China shows that this strain appeared due to naturally occurring recombination of the attenuated strains CV777 and DR13 with the circulating field strain CH/ZMDZY/11. The recombination events occurred in the S, ORF3, and N-structural protein-coding region and the replicase ORF1a region [26].
The results of phylogenetic and recombination analysis revealed a discrepancy between the S gene sequence of PEDV/Belgorod/dom/2008 and the sequences of other isolates available in the GenBank database. Our results indicate that PEDV/Belgorod/dom/2008 is a new recombinant strain. Interestingly, PEDV/Belgorod/dom/2008 and SeCoV-ITA09 (a recombinant strain from Italy) form a unique phylogenetic group.
Pairwise identity analysis demonstrated that the amino acid sequence of the S gene of PEDV/Belgorod/dom/2008 is 60% identical to the S gene of other PEDV strains and 50% identical to those of TGEV strains. These data argue that PEDV/Belgorod/dom/2008 occupies an intermediate position between TGEV and PEDV.
The identification of recombinant regions in PEDV/Belgorod/dom/2008 can be useful for further analysis of evolutionary variability, epidemiology, and development of a new diagnostic gene-based assay for porcine epidemic diarrhea virus.
References
Pensaert M (2006) Porcine epidemic diarrhea. In: Straw BE et al (eds) Diseases of swine, 9th edn. Blackwell, Ames, pp 367–372
Pan Y, Tian X, Li W, Zhou Q, Wang D, Bi Y, Chen F, Song Y (2012) Isolation and characterization of a variant porcine epidemic diarrhea virus in China. Virol J 9:195. https://doi.org/10.1186/1743-422X-9-195
Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q (2012) New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis 18:1350–1353. https://doi.org/10.3201/eid1808.120002
Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ (2013) Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Investig 25:649–654. https://doi.org/10.1177/1040638713501675
Chen JF, Sun DB, Wang CB, Shi HY, Cui XC, Liu SW, Qiu HJ, Feng L (2008) Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes 36:355–364. https://doi.org/10.1007/s11262-007-0196-7
Sun M, Ma J, Wang Y, Wang M, Song W, Zhang W, Lu C, Yao H (2015) Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia. J Clin Microbiol 53:1484–1492. https://doi.org/10.1128/JCM.02898-14
Park SJ, Moon HJ, Yang JS, Lee CS, Song DS, Kang BK, Park BK (2007) Sequence analysis of the partial spike glycoprotein gene of porcine epidemic diarrhea viruses isolated in Korea. Virus Genes 35:321–332
Kubota S, Sasaki O, Animoto K, Okada N, Kitazima T, Yasuhara H (1999) Detection of porcine epidemic diarrhea virus using polymerase chain reaction and comparison of the nucleocapsid protein genes among strains of the virus. J Vet Med Sci 61:827–830
Jung K, Saif LJ (2015) Porcine epidemic diarrhea virus infection: Etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet J 204:134–143. https://doi.org/10.1016/j.tvjl.2015.02.017
Oka T, Saif LJ, Marthaler D, Esseili MA, Meulia T, Lin CM, Vlasova AN, Jung K, Zhang Y, Wang Q (2014) Cell culture isolation and sequence analysis of genetically diverse US porcine epidemic diarrhea virus strains including a novel strain with a large deletion in the spike gene. Vet Microbiol 173:258–269. https://doi.org/10.1016/j.vetmic.2014.08.012
Wang L, Byrum B, Zhang Y (2014) New variant of porcine epidemic diarrhea virus, United States, 2014. Emerg Infect Dis 20:917–919. https://doi.org/10.3201/eid2005.140195
Hanke D, Jenckel M, Petrov A, Ritzmann M, Stadler J, Akimkin V, Blome S, Pohlmann A, Schirrmeier H, Beer M, Hoper D (2015) Comparison of porcine epidemic diarrhea viruses from Germany and the United States, 2014. Emerg Infect Dis 21:493–496. https://doi.org/10.3201/eid2103.141165
Jarvis MC, Lam HC, Zhang Y, Wang L, Hesse RA, Hause BM, Vlasova A, Wang Q, Zhang J, Nelson MI, Murtaugh MP, Marthaler D (2015) Genomic and evolutionary inferences between American and global strains of porcine epidemic diarrhea virus. Prev Vet Med 123:175–184. https://doi.org/10.1016/j.prevetmed.2015.10.020
Vlasova AN, Marthaler D, Wang Q, Culhane MR, Rossow KD, Rovira A, Collins J, Jung K (2014) Distinct characteristics and complex evolution of PEDV strains, North America, May 2013–February 2014. Emerg Infect Dis 20:1620–1628. https://doi.org/10.3201/eid2010.140491
Strizhakova O, Hanke D, Titov I, Blome S, Malogolovkin A (2017) Complete genome sequence of a porcine epidemic diarrhea virus isolated in Belgorod, Russia, in 2008. Genome Announc 5:e01026–17. https://doi.org/10.1128/genomeA.01026-17
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. https://doi.org/10.1093/ve/vev003
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9:e108277
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Strizhakova O (2013) Isolation and identification of porcine epidemic diarrhea virus in pigs under the outbreak at a large farm. Sel’skokhozyaistvennaya Biologia 4:65–69. https://doi.org/10.15389/agrobiology.2013.4.65eng
Strizhakova OM, Malogolovkin AS, Titov IA, Zhivoderov SP, Kurinnov VV, Strizhakov AA (2017) Biological characteristics of an epizootic isolate BS-08 of porcine epidemic diarrhea virus. Veterinariya 3:31–34
Boniotti MB, Papetti A, Lavazza A, Alborali G, Sozzi E, Chiapponi C, Faccini S, Bonilauri P, Cordioli P, Marthaler D (2016) Porcine epidemic diarrhea virus and discovery of a recombinant swine enteric coronavirus, Italy. Emerg Infect Dis 22:83–87. https://doi.org/10.3201/eid2201.150544
Akimkin V, Beer M, Blome S, Hanke D, Hoper D, Jenckel M, Pohlmann A (2016) New chimeric porcine coronavirus in swine feces, Germany, 2012. Emerg Infect Dis 22:1314–1315. https://doi.org/10.3201/eid2207.160179
Belsham GJ, Rasmussen TB, Normann P, Vaclavek P, Strandbygaard B, Botner A (2016) Characterization of a novel chimeric swine enteric coronavirus from diseased pigs in Central Eastern Europe in 2016. Transbound Emerg Dis 63:595–601. https://doi.org/10.1111/tbed.12579
Li R, Qiao S, Yang Y, Guo J, Xie S, Zhou E, Zhang G (2016) Genome sequencing and analysis of a novel recombinant porcine epidemic diarrhea virus strain from Henan, China. Virus Genes 52:91–98. https://doi.org/10.1007/s11262-015-1254-1
Acknowledgements
We thank Olga Strizhakova for providing PEDV/Belgorod/dom/2008. We also acknowledge Yegor Bazykin and Alexey Neverov for fruitful discussions of virus evolution and recombination analysis. We appreciate Sandra Blome and Dennis Hanke for PEDV/Belgorod/dom/2008 complete genome sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Our work was supported by the Ministry of Science and Higher Education of the Russian Federation.
Conflict of interest
The authors declare no competing interests.
Additional information
Handling Editor: Diego G. Diel.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nefedeva, M., Titov, I. & Malogolovkin, A. Molecular characteristics of a novel recombinant of porcine epidemic diarrhea virus. Arch Virol 164, 1199–1204 (2019). https://doi.org/10.1007/s00705-019-04166-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04166-4