
Abstract
Feline herpesvirus type 1 (FHV-1) is a highly contagious pathogen of domestic cats and other members of the family Felidae. Point-of-care diagnosis of persistent infection in cats is essential for control of its spread. A recombinase polymerase amplification (RPA) assay (RPA-LFD-FHV) combined with a lateral flow dipstrip (LFD) was developed that uses human body heat for incubation. Sensitivity was evaluated by testing a serial dilution of a control plasmid, and specificity was evaluated by testing related viruses. Swab samples from cats with suspected infection were tested by RPA-LFD-FHV, and the results were compared to those obtained by PCR to evaluate its clinical performance. The RPA-FLD-FHV assay was carried out successfully within 20 min, using body heat for incubation. The RPA-FLD-FHV had a detection limit of 103 copies of the FHV-1 gD gene, which was lower than that of PCR, which was 104 copies. The assay could detect templates of FHV-1 but not those of other feline and canine viruses. Viruses in boiled samples could be efficiently detected by the RPA-FLD-FHV. Thirty-one out of the 80 samples were positive by the RPA-FLD-FHV assay, whereas only 27 were positive by PCR. DNA sequencing confirmed that the four samples that were positive by RPA-FLD-FHV but negative by PCR were indeed positive. These results indicate that RPA-FLD-FHV is applicable for clinical use. The RPA-FLD-FHV assay is a simple, rapid, and reliable method for point-of-care diagnosis of FHV-1 infection.
Similar content being viewed by others

Introduction
Feline herpesvirus type 1 (FHV-1) is a highly contagious pathogen of domestic cats and other members of the family Felidae, including South China tigers [7] and the cheetahs [9]. Young and adolescent cats are at risk of acute primary disease, and most of them develop a persistent infection. About half of persistently infected cats shed virus throughout their life [3]. These carriers are able to spread infection, making prevention and control of the disease more challenging. When buying or introducing potentially susceptible animals, FHV-1 detection in the field is the first measure for prevention and control infection. Field diagnosis is essential for identification of persistent infection in cats, and it is also important for timely treatment at the early stage of infection. At present, a gold immunochromatography strip assay is the only available assay for rapid FHV-1 detection, but it can only be used during the symptomatic period of disease. A more sensitive method for point-of-care detection of FHV-1 needs to be developed.
DNA-based methods for pathogen detection are widely used because they are precise and rapid. However, a limitation of these assays is that they require sophisticated equipment, which restricts their application under field conditions. Recombinase polymerase amplification (RPA) [6] combined with chromatographic lateral flow dipstrips (RPA-LFD) could meet the requirements for field detection. RPA-LFD has the advantages of high speed, portability of equipment, and accessibility, making it suitable for time-saving and accurate tests in situations with low levels of resources and skills [4]. In this study, we developed a simple RPA-LFD method for detection of FHV-1 that uses body heat for incubation and boiling for sample treatment.
Materials and methods
Virus
The Fel-O-Vax PCT feline panleukopenia–rhinotracheitis–calicivirus vaccine (Boehringer-Ingelheim), the Vanguard Plus 5/CV-L canine distemper virus, parvovirus, adenovirus type 1, adenovirus type 2, parainfluenza virus, coronavirus vaccine (Zoetis) and pseudorabies virus Bartha-K61 strain (Boehringer-Ingelheim) were purchased from the affiliated veterinary hospital of Shenyang Agriculture University.
Plasmid extraction
The recombinant plasmid pET-FHV-1-gD was constructed by cloning the full-length gD gene into the multiple cloning site of pET-28a using standard procedures. Plasmid pET-FHV-1-gD was extracted and purified using a MiniBEST Plasmid Purification Kit (Takara Biotechnology Co., Ltd., Dalian, China) according to the manufacturer’s instructions. Purified plasmid was quantified using a NanoDrop 2000 Spectrophotometer (ThermoFisher, US). Genome equivalents (GE) were calculated based on a plasmid size of 6.5 kb [1].
Primer and probe design
The FHV-1 gD sequence D30767.1 was obtained from GenBank (http://www.ncbi.nlm.nih.gov/genbank) and aligned using the multiple sequence alignment tool ClustalW. After alignment, FHV-1 gD-specific PCR and RPA primers and probes were designed and analysed using Prime-Blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast) (Table 2). All oligonucleotide primers and probes were synthesised by Sangon Biotech Co. Ltd. (Shanghai, China).
RPA assay development
RPA reactions were performed using a TwistAmp nfo kit (TwistDx, UK). A 50-μL reagent mixture, which containing 1X rehydration buffer, 5 μM each RPA primer (F1+R1), and 106 copies of the recombined plasmid pET-FHV-1-gD was added to the dry enzyme pellet and thoroughly mixed. The reaction was then initiated by addition of 280 mM magnesium acetate. The RPA reaction was performed using human body heat by placing the tubes under the tester’s waistband for 10 to 60 minutes. The reactions were stopped by adding 50 μL of a mixture of chloroform and isoamyl alcohol (1:1). The mixture was then centrifuged at 12,000 g for 1 min, and 5 µl of the supernatant was electrophoresed in a 2.0% (w/v) agarose gel.
RPA-LFD assay development
To adapt them for the RPA-LFD assay, the gD LF-R1 primers were labeled at the 5’ end with biotin, and an internal RPA LF probe (LF probe) was designed (Table 1). Each primer (F1 + LF-R1) at a concentration of 5 μM, 0.5 μM LF probe, and 0.25 M betaine (Sigma Aldrich, UK) were added to the RPA reaction mixture. After incubation, 0.2 μl of the mixture was diluted in 100 μl of running buffer (Milenia Biotec, Germany), and a Milenia HybriDetect (MGHD) Dipstrip (Milenia Biotec, Germany) was placed vertically into the running buffer. The incubation was performed at room temperature for 5 min, and the final result was judged visually with the naked eye.
Sensitivity analysis
For evaluating the sensitivity of the RPA assay, a tenfold serial dilution of the pET-FHV-1-gD plasmid was made to obtain concentrations ranging from 106 to 102 copies/μL. One μL of each dilution was amplified by RPA as well as PCR using the same primers. One μL of template was mixed with 5 μL of Taq PCR MasterMix (Takara Biotechnology Co., Ltd., Dalian, China), 2 μL of distilled water and 2 μL of F2 and R2 primers. PCR was performed as follows: stage 1, 95 °C for 4 min; stage 2, 40 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 45 s; and stage 3, 72 °C for 10 min. The RPA product was detected by 2% agarose gel electrophoresis and LFD.
Specificity analysis
To evaluate the specificity of the RPA assay, nucleic acid was extracted from the Fel-O-Vax PCT feline panleukopenia–rhinotracheitis–calicivirus vaccine, Vanguard Plus 5/CV-L canine distemper virus, parvovirus, adenovirus type 1, adenovirus type 2, parainfluenza virus, coronavirus vaccine (Zoetis), and the pseudorabies virus Bartha-K61 strain (Boehringer-Ingelheim) and detected by RPA. The RPA product was detected by 2% agarose gel electrophoresis and LFD. The specific fragment from the RPA assay separated by 2% agarose gel was extracted and sequenced to confirm FHV-1 gD.
Simple sample treatment
To develop a simple sample treatment procedure that could be performed at the point of care, we tested the RPA assay with templates subjected to two different preparation procedures. For the simple treatment procedure, 100 μL of Fel-O-Vax PCT feline vaccine (Boehringer-Ingelheim) was boiled for 10 min and used directly as a template without centrifugation. For comparison, 100 μL was extracted using a MiniBEST Viral RNA/DNA Extraction Kit (Takara Biotechnology Co., Ltd., Dalian, China). The treated samples or templates were then tested by RPA assay and PCR.
Performance validation with clinical samples
Nasal and ocular conjunctival swab samples were collected from cats at veterinary clinics in Liaoning Province in 2017. These cats had conjunctivitis, rhinitis, or other symptoms of upper respiratory tract infections. The swab was placed and washed in a tube containing 200 μl of sterile water and stored in a -80°C freezer. The samples were boiled and then tested by RPA-LFD and PCR.
Results and discussion
FHV-1 and feline calicivirus are the most important viral agents associated with feline upper respiratory tract disease. A significant proportion of FHV-1 infected cats develop persistent infection and continuously shed virus [3]. This characteristic of persistent infection results in spreading of the virus, causing great difficulties in the prevention and control of the disease. Convenient detection of these viruses in homes and in catteries is essential for treatment and control of their spread. Despite the availability of a gold immunochromatography strip for convenient detection of FHV-1, it can only be used during symptomatic periods due to its low sensitivity. A more sensitive detection method needs to be developed. RPA assays have the advantage that they are suitable for molecular diagnosis of diverse pathogens under field conditions. In this study, an RPA-LFD assay was developed for rapid and specific detection of FHV-1.
RPA has the advantage that amplification can be carried out at a constant temperature, making it possible to apply it under field conditions. However, a constant temperature is difficult to achieve in homes and in catteries. Although the optimum RPA reaction temperature is around 37°C, all RPA reactions can amplify DNA to detectable levels at 31 °C [5]. We therefore tried to carry out the PRA assay using body heat for the incubation step. It has been reported that RPA assay can be carried out with excellent results by placing the samples under the axilla [2]. However, under field conditions, this would impair the operator’s normal activities. In this study, we chose to place the tubes under the operator’s waistband. This has the advantages of easy placement, convenience, even for a long time, and a large area for multiple tubes and tests. We measured the temperature under the waistband of six volunteers using thermometer at 8, 12 and 18 o’clock. The average temperature was 35.4 ± 0.4 °C (data not shown). This temperature is higher than that of the abdomen under clothing [2]. This temperature difference may depend on the measuring location, season, tools, and race. The results showed that the expected 307-bp band was clearly visible on an agarose gel when the reaction time was extended to 20 min (Fig. 1). At lower incubation temperatures, more time is required for the reaction to reach completion. At 37 °C, 10 minutes are enough for the amplification, but for the waistband incubation, at least 15 minutes are needed, and 20 minutes are enough for even the lowest concentration of templates (data not shown). Therefore, all of the remaining RPA reactions were performed for 20 min when using body heat.

RPA-FHV can be carried out using body heat. Reaction tubes were placed under the waistband for different times, and amplification products were detected by agarose gel electrophoresis. M, DNA marker; lanes 1, 3, 5, 7 and 9, incubation times of 20 min, 30 min, 40 min, 50 min and 60 min; 2, 4, 6, 8 and 10, negative controls
RPA products can be detected by several methods, including fluorescence detection and lateral flow (LF) dipstrips. For applications under field conditions, particularly in homes and in catteries, the lateral flow strip method is more feasible because it does not require any other equipment. Results of RPA assays can be read by means of lateral flow strips at the detection site. A red test line on the LF strip represents positive detection of gD RPA-LFD amplicons (Fig. 2). During the development of the assay, we found that when the products were detected directly using LF strips, a long run time can result in the generation of a confusing red line at the test line. This might be the result of a high concentration of the amplification products. To test this, the products were diluted and then detected using LF strips for different lengths of time. As expected, no false positive lines were observed when the products were diluted more than tenfold (data not shown). For field applications, increasing the amplicon dilution to 1:100 could eliminate these false positives.

RPA-LFD-FHV detection results with 20-minute amplification. A 2% agarose gel. M, DNA marker; lane 1, positive; lane 2, negative. B Lateral flow strip detection. Lane 1, positive; lane 2, negative
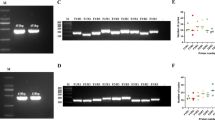
The sensitivity of the gD RPA-LFD assay was evaluated using a serial dilution of a plasmid containing the gD gene. As shown in Figure 3A, the detection limit of the assay was 103 copies/reaction, which was slightly higher than the published value for qPCR [8], but lower than the 104 copies/reaction observed for PCR (Fig. 3B). Since the virus titer in acute and persistent infection is relatively high, the sensitivity of the RPA-LFD-FHV is acceptable for point-of-care diagnosis. Therefore, the short time and small amount of resources required for RPA makes it an attractive tool for on-site diagnosis.

Sensitivity of the RPA-LFD-FHV assay. Different concentrations of pET-FHV-1-gD were amplified by RPA and detected by agarose gel or LF strips. For comparison, the plasmids were also amplified by PCR. The detection limit for RPA (103 copies/reaction) (A) and RPA-LFD (C) was tenfold higher than that of PCR (104 copies/reaction) (B). M, DNA marker; lane 1, 106 copies; lane 2, 105 copies; lane 3, 104 copies; lane 4, 103 copies; lane 5, 102 copies
Specificity analysis showed that a 307-bp band was successfully amplified from a feline vaccine (Fig. 4). Sequencing of products confirmed the expected sequence. In contrast, other common viruses, including FPV, BHV-1, PRV, CDV and CCV, were negative by the assay. This implies that the RPA assay for FHV-1 detection is specific.

Specificity of RPA. FHV-1 and other viruses were amplified by RPA and detected using agarose gel electrophoresis and lateral flow dipstrips. M, DNA marker; lane 1, feline panleukopenia-rhinotracheitis-calicivirus vaccine; lane 2, canine distemper virus-parvovirus-adenovirus type 1-adenovirus type 2-parainfluenza virus-coronavirus vaccine; lane 3, PRV Bartha-K61 strain
Sample treatment for nucleic acid detection methods is important for the whole diagnostic procedure. To be conveniently used, a simple sample treatment procedure for the RPA assay is necessary for the detection in homes or in the field. In this study, both boiling treatment and DNA extraction were used for template preparation for the RPA assay. Templates prepared by both methods yielded a clear red test line in strips (Fig. 5B), and the same size of bands in 2.0% (w/v) agarose gel (Fig. 5A). This indicated that swab samples can be treated by boiling for template preparation for the RPA-FHV and RPA-LFD-FHV assay. At the same time, to eliminate the centrifugation requirement, the boiled sample solution was used directly as the template without centrifugation. The template volume required was only 1 μl. This trace of template could be obtained by suction of fluid along the inner wall of the boiled sample tube. This further strengthens the field adaptation potential of RPA-LFD-FHV.

Comparison of sample treatment methods. Samples were treated by boiling or extracted using commercial kits. Templates were then amplified by RPA and detected by agarose gel electrophoresis (A) and LFD (B). Lane 1, boiling treatment; lane 2, DNA extraction with kit
To validate the performance of the newly developed RPA-LFD-FHV assay, a total of 80 clinical samples from cats with suspected infection were detected by RPA-LFD-FHV and PCR assay (Table 2). For PCR assay, 53 samples were negative and 27 samples were positive. For the RPA-LFD-FHV assay, 31 samples were positive. All 27 PCR positive samples were also positive by RPA-LFD assay. Four samples that were negative by PCR were positive by RPA-LFD. DNA sequencing of the products showed that these four samples were indeed positive. Positive accordance of the two detection assays was 87% (27/31) and negative accordance was 92.5% (49/53). The four samples that were negative by conventional PCR were positive in RPA-LFD-FHV, showing that RPA-LFD-FHV has higher sensitivity than PCR. All of these results indicate that RPA-LFD performed better than the PCR assay and is applicable for clinical use in the diagnosis of FHV-1.
In conclusion, with the aim of detecting FHV-1 under field conditions such as in a cattery or family home, an RPA-LFD-FHV assay was developed. This assay can be effectively performed in 20 min by using body heat for incubation and boiling as the sample treatment procedure, avoiding the need for any other professional equipment. The RPA assay showed not only higher sensitivity (about 103 copies) than PCR (about 104 copies), but also high specificity. Thus, the goal of equipment-free detection almost has been realized. The boiling procedure can be carried out in a microwave oven or induction cooker in a family home. The RPA-LFD-FHV assay has great potential for applications in the rapid point-of-care diagnosis of FHV-1.
References
Clancy E, Higgins O, Forrest MS, Boo TW, Cormican M, Barry T, Piepenburg O, Smith TJ (2015) Development of a rapid recombinase polymerase amplification assay for the detection of Streptococcus pneumoniae in whole blood. BMC Infect Dis 15:481
Crannell ZA, Rohrman B, Richards-Kortum R (2014) Equipment-free incubation of recombinase polymerase amplification reactions using body heat. PLoS One 9:e112146
Gould D (2011) Feline herpesvirus-1: ocular manifestations, diagnosis and treatment options. J Feline Med Surg 13:333–346
James A, Macdonald J (2015) Recombinase polymerase amplification: emergence as a critical molecular technology for rapid, low-resource diagnostics. Expert Rev Mol Diagn 15:1475–1489
Lillis L, Lehman D, Singhal MC, Cantera J, Singleton J, Labarre P, Toyama A, Piepenburg O, Parker M, Wood R, Overbaugh J, Boyle DS (2014) Non-instrumented incubation of a recombinase polymerase amplification assay for the rapid and sensitive detection of proviral HIV-1 DNA. PLoS One 9:e108189
Piepenburg O, Williams CH, Stemple DL, Armes NA (2006) DNA detection using recombination proteins. PLoS Biol 4:e204
Sun H, Li Y, Jiao W, Liu C, Liu X, Wang H, Hua F, Dong J, Fan S, Yu Z, Gao Y, Xia X (2014) Isolation and identification of feline herpesvirus type 1 from a South China tiger in China. Viruses 6:1004–1014
Wang J, Liu L, Wang J, Sun X, Yuan W (2017) Recombinase polymerase amplification assay—a simple, fast and cost-effective alternative to real time PCR for specific detection of feline herpesvirus-1. PLoS One 12:e166903
Witte CL, Lamberski N, Rideout BA, Fields V, Teare CS, Barrie M, Haefele H, Junge R, Murray S, Hungerford LL (2013) Development of a case definition for clinical feline herpesvirus infection in cheetahs (Acinonyx jubatus) housed in zoos. J Zoo Wildl Med 44:634–644
Acknowledgements
This work was supported by National Special Project on Research and Development of Key Biosafety Technology (2017YFD0500901, 2016YFC1200100, 2017YFD0500305) and Shenyang Key Research and Development Plan (17-161-3-00). The authors would like to acknowledge friends for collecting swab samples suspected of containing FHV-1.
Funding
This work was supported by National Special Project on Research and Development of Key Biosafety Technology (2017YFD0500901, 2016YFC1200100, 2017YFD0500305) and Shenyang Key Research and Development Plan (17-161-3-00).
Author information
Authors and Affiliations
Contributions
ML and LY performed the experiments. WZ participated in sample collection. BL and ML analyzed the data. BL and ZC drafted the manuscript. BL conceived and designed the study.
Corresponding authors
Ethics declarations
Conflict of interest
The authors state that they have no competing interests.
Additional information
Handling Editor: Scott Schmid.
Rights and permissions
About this article

Cite this article
Liu, Mz., Han, Xh., Yao, Lq. et al. Development and application of a simple recombinase polymerase amplification assay for rapid point-of-care detection of feline herpesvirus type 1. Arch Virol 164, 195–200 (2019). https://doi.org/10.1007/s00705-018-4064-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-4064-7