
Abstract
Kobuviruses are small non-enveloped RNA viruses that probably cause diarrhea in cattle and swine. Since its discovery in 2003, few studies have addressed bovine kobuvirus (BKoV; a species of Aichivirus B) infections. BKoV has been reported in Europe, Asia, and South America, suggesting a worldwide distribution. To investigate the presence of BKoV in Egypt, 36 fecal specimens from diarrheic calves in two different Egyptian provinces (Cairo and Sharkia) were screened by RT-PCR and 24 (66.7%) were found positive for BKoV. RNA from one of the positive samples (BKoV/Egy-1/KY407744) was subjected to next-generation sequencing to determine the complete BKoV genome sequence. When compared to the only recorded BKoV genome sequence (BKoV/U-1/AB084788), the studied strain showed 94 amino acid (aa) substitutions through its entire polyprotein (2463 aa), one nucleotide (nt) insertion and one nt deletion in the 2B gene and 4-nt deletions in the UTRs (2 each). Additionally, five VP1 and seven 3D sequences were obtained from other samples by using RT-PCR and Sanger sequencing. A discrepancy in the phylogenetic topography of VP1 and 3D was observed, where the Egyptian VP1 sequences were classified as a distinct cluster within the proposed lineage 1 (genotype A), which also contained strains from the UK, Brazil, and Japan. While, the 3D sequences from Cairo were related to those of Chinese strains unlike Sharkia ones that were more closer to Korean strains. To the best of our knowledge, this is the first detection and genomic characterization of BKoV in Egypt or indeed Africa.
Similar content being viewed by others

Introduction
As a taxonomically classifiable member of the family Picornaviridae, kobuviruses are small, non-enveloped, and positive-sense single-stranded RNA viruses. Their genome consists of a single open reading frame (ORF) that encodes a large polyprotein [1]. After translation, the protein precursor is proteolytically processed into several structural and non-structural proteins. With a total size of 8.2–8.4 kb, the kobuvirus genome is organized sequentially as follows: VPg, 5′UTR, leader protein (L), three structural proteins (VP0, VP3, and VP1), seven non-structural proteins (2A–2C and 3A–3D), 3′UTR, and poly(A) tail [2]. The 3D gene encodes for the RNA-dependent RNA polymerase (RdRp). According to the function of the encoded proteins, the kobuvirus genome can be arranged into three distinct regions, namely P1 (encoding structural capsid proteins), P2, and P3 (encoding non-structural proteins) [3].
Initially, kobuviruses included Aichivirus (AiV), bovine kobuvirus (BKoV), and porcine kobuvirus (PKoV) based on the hosts infected e.g. humans, cattle, and swine, respectively. Later, the genus Kobuvirus was divided into three species; Aichivirus A, B and C (with strains abbreviated to AiV A, B, and C). AiV A includes human AiV and kobuviruses of dogs, cats, and mice. AiV B includes bovine, ovine, and ferret kobuviruses while AiV C includes only PKoV [4]. Kobuviruses have also been detected in black goats, rabbits, European roller, and bats (http://www.picornaviridae.com/). Based on VP1 sequence analysis, human AiV strains have been divided into three major lineages (1, 2, and 3) with nucleotide (nt) sequence identities < 84.8%. These three lineages were later renamed as genotypes A, B, and C [5]. Recently, Chang et al. [6] proposed four genotypes (A, B, C, and D) of BKoV-VP1 strains based on 85.1% nt identity as a recommended cutoff value for genotyping.
In 1989, AiV was linked to gastroenteritis outbreaks in the Aichi Prefecture of Japan after being isolated from stools of patients who consumed raw oysters [7]. Later, the virus was classified as a member of the family Picornaviridae [8]. PKoV was first detected in domestic pigs in Hungary in 2008 [9]. In 2003, BKoV/U-1/AB084788 was identified in Japan as a cytopathic contaminant of HeLa cells, which have been used around the world for more than 30 years. Subsequently, the virus was detected in serum and fecal samples of apparently healthy animals [10] and has been reported from Europe, Asia, and South America [11].
Kobuviruses are implicated in gastroenteritis of animals and humans. However, their role in the pathogenesis of clinical diarrhea is still unclear because they have been detected in both healthy and diarrheic animals. Most cases of diarrhea are not routinely tested for kobuvirus and hence the full impact of this virus has not been accurately determined. As a newly recognized virus, GenBank contains just a single BKoV complete genome. In this study, we report BKoV infections among cattle populations of Egypt, the full-length genome of one Egyptian strain (BKoV/Egy-1/ KY407744) as well as the phylogenetic analysis of BKoV strains from Cairo and Sharkia provinces.
Materials and methods
Samples and RNA extraction
Massive outbreaks of calf diarrhea were reported from the Sharkia and Cairo provinces of Egypt in late 2014 and early 2015. Clinically, calves (ages 2 weeks to 10 months) had severe diarrhea, fever, weakness, and dehydration. A total of 36 diarrheic fecal specimens were collected aseptically and transferred to the diagnostic laboratory. The total RNA was extracted from supernatants of 20% fecal homogenates using Blood/Liquid Sample Total RNA Rapid Extraction Kit (Bioteke Corporation, Beijing, China) and stored at -80 °C.
Screening of samples using reverse transcription-polymerase chain reaction (RT-PCR)
The RNA samples (n = 36) were subjected to RT-PCR for the detection of BKoV using degenerate primers (Univ-Kobu-F and Univ-Kobu-R), which amplify a conserved region in the 3D (RdRp) gene of all kobuviruses [12] (Table 1). The PCR reaction was performed using one step RT-PCR kit (TaKaRa PrimeScript OneStep RT-PCR kit Ver.2 Dye Plus; Cat. #. RR057A, TaKaRa Bio Inc, Japan). Briefly, each mixture contained 2 μl of one-step enzyme mix, 25 μl of one-step buffer, 1 μl (20 μM) each of upstream and downstream primers, 2 μl of test RNA and 19 μl of RNase-free water. The 50 μl-reaction mix was incubated in the thermocycler at 50 °C for 30 min for reverse transcription and at 95 °C for 15 min for initial denaturation. Then, 35 cycles of amplification (denaturation at 94 °C for 1 min, annealing at 50 °C for 30 sec and elongation at 72 °C for 1 min) were applied followed by a step of final extension at 72 °C for 10 min. Part of the amplified PCR products (8 μl) were analyzed by 2% agarose gel electrophoresis and ultraviolet illumination. To detect infection with bovine rotavirus (BRV), bovine coronavirus (BCV), and bovine viral diarrhea virus (BVDV), RT-PCR was carried out using specific primers for these respective viruses (data not shown).
Next-generation sequencing (Illumina sequencing)
A 20-μl aliquot of one RNA sample from Sharkia province (namely; BKoV/Egy-1) was selected for the next-generation sequencing (NGS) using the Illumina platform to obtain the complete genome of BKoV. The NGS workflow was performed at the University of Minnesota Genomic Center (UMGC). For quality control purposes, the total RNA was quantified using RiboGreen fluorometry and checked for purity. The library was created using Illumina’s Truseq RNA sample preparation kit (RS-122-2001). The flow cell was loaded on an Illumina MiSeq and sequenced by 250 cycles paired-end reads sequencing.
Primer design, RNA retesting by RT-PCR, and Sanger sequencing of partial 3D, VP1, and 2BC regions
Based on the complete genome of BKoV/Egy-1, three primer sets were designed to partially span the 3D, VP1, and 2BC regions (Table 1). Two primer pairs were used to retest some of the study samples by RT-PCR to analyze the molecular phylogenetics of the 3D and VP1 genes. The last primer pair partially covered the 2BC region to confirm the specific mutations (one insertion and one deletion) observed in the 2B gene of BKoV/Egy-1 and to search for them in other study samples. The reaction mixtures and PCR thermal profiles were the same as above except for the annealing step that was performed at 50 °C for 1 min. After electrophoresis in a 1% agarose gel, products of expected sizes were visualized under a UV transilluminator. Amplicons for partial regions of 3D (n = 7), VP1 (n = 5) and 2BC (n = 4) were purified using QIAquick PCR purification kit (Qiagen, Valencia, CA, USA). The purified DNA was sent to the UMGC for Sanger sequencing of the three regions in both directions (using the same forward and reverse primers used in RT-PCR).
Sequence and phylogenetic analysis
The NGS reads were trimmed to remove adaptor sequences followed by testing for sequence quality. The host sequences were removed and the remaining reads were assembled into contigs (minimum length = 500 nt) using de novo default settings in the CLC Genomic Workbench 6.0 software (http://www.clcbio.com/products/clc-genomics-workbench/). To determine viral sequences in the sample, extracted contigs were subjected to BLASTn analysis against NCBI databases (cutoff E value = 0.001). The ORF finder tool of NCBI (https://www.ncbi.nlm.nih.gov/orffinder/) was used to find ORFs. To determine the completeness of the genome and to predict structural and non-structural proteins, nt and amino acid (aa) sequences were aligned with reference kobuviruses. The alignments and phylograms were made using MEGA 6.0 software [13].
The Sanger sequences of partially-amplified genes were imported to Sequencher software version 5.1 (http://genecodes.com/) for evaluating chromatograms, trimming of poor quality nucleotides, alignment of forward and reverse reads, and generation of final contigs. The partial 3D, VP1, and 2BC sequences were compared to previously reported ones by using the MEGA 6.0 software [13]. Sequences were edited and aligned using the Clustal-W method followed by constructing neighbor-joining phylogenetic trees using the Kimura 2-substitution model with a bootstrap value of 1000 replicates. The nt and aa identities were calculated using the p-distance method. Deduced aa analysis was performed on VP1 sequences to spot possible substitutions.
GenBank accession
Study sequences were submitted to GenBank under the following accession numbers: KY407744 for the complete genome of BKoV/Egy-1, and KY260608-KY260611, KY260612-KY260618, and KY260619-KY260623 for the partial 2BC, 3D, and VP1 regions, respectively.
Results
RT-PCR detection of BKoV in diarrheic fecal materials
Of the 36 fecal samples tested, 24 (66.7%) were positive for BKoV-RNA (95% CI: 49.03% to 81.44%). The rates of kobuvirus detection in the Cairo and Sharkia provinces were 75% and 60%, respectively. The infection rate was the highest in young calves and decreased with age; being 82.6% (19/23) for calves ≤ one-month-old and 38.5% (5/13) for older calves. Some samples (25%) were positive for BRV without showing BKoV infection. All samples were negative for BCV and BVDV. Based on RNA quality, one BKoV-positive sample (BKoV/Egy-1/ KY407744) was selected for Illumina sequencing.
Sequence analysis of the BKoV complete genome
The full genome sequence of BKoV/Egy-1/ KY407744 was obtained by Illumina MiSeq. The resulting genome, excluding the poly A tail, was 8319 nt in length and contained a single ORF encoding a polyprotein of 2463 aa, which started with a methionine (AUG) initiation codon (756-758). It was organized as a 5′UTR of 755 nt, a 7392 nt ORF (756-8147), and a 3′UTR of 172 nt.
BKoV/Egy-1/KY407744 starts with a leader (L) protein of 561 nt (187 aa). The genomic organization and potential polyprotein cleavage sites were verified by using several NCBI tools. The cleavage sites Q/C (1044/1045) and Q/G (1678/1679) were predicted to separate P1 from the P2 region and P2 from P3 region, respectively (Table 2). The polyprotein of BKoV/Egy-1/ KY407744 aligned well with that of BKoV/U-1/AB084788, including the highly conserved 1474GPPGTGKS1481 motif in the nt binding domain of the putative picornavirus helicase, within the 2C protein and the 2154KDELR2158, 2322YGDD2325 and 2371FLKR2374 motifs in the RdRp protein. A phylogenetic analysis, based on the complete coding region, clustered the BKoV/Egy-1/ KY407744 with other members of Aichivirus B (Fig. S1).
The maximum comparative sequence identity for BKoV/Egy-1/ KY407744 was observed following comparisons with BKoV/U-1/AB084788 (89.5% at the nt level and 96.3% at the aa level) and then with sheep kobuvirus/TB3/GU245693 (81.5% at the nt level and 86.2% at the aa level). In comparison to BKoV/U-1/AB084788, the P1 region (capsid proteins) was less conserved (86.8% at the nt level and 96.4% at the aa level) with the VP1 gene being highly variant (sharing only 85.9% nt and 95.5% aa identities) among the three structural proteins. VP3 had the least variation; sharing 87.9% nt and 96.9% aa identities. The P3 region (3A-3D non-structural proteins) showed the highest identity (92.9% at the nt level and 98.2% at the aa level) followed by the P2 region (non-structural proteins (2A-2C), which showed identities of 89.6% at the nt level and 96.4% at the aa level (Fig. 1). Collectively, in relation to BKoV/U-1/AB084788, BKoV/Egy-1/ KY407744 showed total nt and aa substitutions of 852 and 94, respectively through its entire genome (Table S1).

The percentage nucleotide and amino acid identities between the studied strain (BKoV/Egy-1/KY407744) and the reference strain (BKoV/U-1/AB084788)
The 5′UTR of BKoV/Egy-1/ KY407744 was 755 nt in length (53 nt shorter than that of BKoV/U-1/AB084788 with 91.4% nt identity). As many as 51 nt were missing due to unresolved sequencing of the 5′-terminus. The lengths of the pyrimidine tract (Y), spacer sequence (X), and AUG initiation codon were expressed as a formula (Y7-X37-AUG) with two deletions being present (A307 and C706). Five AUG codons were detected in the 5′UTR as well as the AUG initiation codon (756-758). The 3′UTR was 172-nt long (2-nt deletions; T8337 and T8338) and had 94.8% nt identity when compared to the reference sequence BKoV/U-1/AB084788.
Sequence and phylogenetic analysis of partial 3D, VP1, and 2BC regions
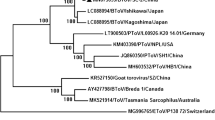
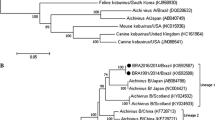
The partial 3D sequences from seven BKoV strains were highly conserved (overall identities of 93.7-99.9% at the nt level and 98.5-100% at the aa level). Based on phylogenetic analysis of 35 GenBank reference strains, BKoV sequences from Cairo (n = 5) clustered with Chinese strains from Xinjiang territory while those from Sharkia (n = 2) aligned with Korean strains. Cairo and Sharkia strains were obviously separated (Fig. 2). Between each other, the partial VP1 sequences identified in this study (n = 5) showed 94.3-99.6% nt and 98.9-100% aa identities. Closely related reference BKoV-VP1 strains were included in the phylogenetic analysis and all study sequences belonged to lineage 1 (genotype A), which contains strains from Brazil, Scotland (UK) and Japan (Fig. 3). The VP1 sequences from Sharkia (EGY-1, Sharkia-5 and Sharkia-6) and Cairo (Cairo-1, -4, and -7) clustered phylogenetically in lineage 1 but were further distinguishable into two subclades (Fig. 3). On contrast, the separation of the two Sharkia-Cairo subclades was more profound following analysis of the 3D gene, where the Sharkia and Cairo strains did not cluster with a significant bootstrap value in the phylogenetic tree.

A neighbor-joining phylogenetic tree representing the relationship between the detected BKoV-3D sequences and reference strains. Bootstrap values (> 50%) are shown above the branches. The study sequences are indicated by solid circles. BKoV, bovine kobuvirus; PKoV, porcine kobuvirus

A neighbor-joining phylogenetic tree of Egyptian VP1 sequences (801 bp) including representative BKoV-VP1 reference strains. Bootstrap values (> 50%) are shown above the branches. The study sequences are indicated by solid circles. BKoV, bovine kobuvirus; PKoV, porcine kobuvirus
Furthermore, a deduced aa analysis of 101 BKoV-VP1 sequences revealed that the study strain had six residue substitutions. All study strains had an aa substitution of A258 to S258. In addition, Cairo strains (BKoV-1, BKoV-4 and BKoV-7) had a unique aa change at position 70 (V70 to I70). Sharkia strains (BKoV-5 and BKoV-6) and BKoV/Egy-1/KY407744 had two aa substitutions T207 to A207 and I248 to V248. Except for BKoV-7, all study strains showed a residue change of A259 to V259. BKoV-7 strain had one aa exchange from A260 to T260.
Four sequences representing the 2BC region shared 92.1-100% and 98.1-100% nt and aa identities, respectively. When compared to the reference BKoV/U-1/AB084788 sequence, all sequences had one insertion (G4718) and one deletion (C4726) in the 2B gene, which were also seen in BKoV/Egy-1/ KY407744.
Discussion
Classifiable members of the family Picornaviridae are known to cause a wide range of important diseases in humans and animals. Two picornaviruses have been reported in Egyptian farm animals; the endemic foot and mouth disease virus [14] and the recently recognized bovine enterovirus [15]. Neonatal calf diarrhea (NCD) results in huge economic losses in both beef and dairy industries in Egypt. Recent studies have described the presence of different enteric viruses in Egyptian cattle herds [15, 16] and have raised questions about their actual contribution to NCD.
Kobuviruses have been incriminated in animal and human cases of gastroenteritis. They are transmitted through the fecal-oral route either directly from one animal to another or indirectly by consumption of contaminated food or water [4]. The present study reports, for the first time, the existence of BKoV (species Aichivirus B) in Egypt, in cattle showing clinical features of calf diarrhea. Using generic kobuvirus primers, 24 of 36 (66.7%) diarrheic fecal samples tested positive for BKoV. These results are somewhat different from those reported in prior surveys. For example, kobuvirus was detected in 16.7% of healthy cattle in Japan [10], 8.3% of diarrheic cattle in Thailand [17], and 18.2% of diarrheic cattle in Brazil [18, 19]. In other countries, the prevalence has been described as follows: 6.25% in Hungary [12], 1% in Belgium [20], 25.8 -34.6% in Korea [21, 22], 77.8% in the Netherlands [23], 4.9% in Italy [11], and 34.9% in China [6]. The highest rates of BKoV have been observed in Egypt (66.7% in this study) and the Netherlands (77.8) possibly due to the smaller sample size tested in these two studies (n = 36 and n = 9, respectively). Extensive epidemiologic surveillance should be planned to determine the endemicity of BKoV in Egypt.
The genome sequence we characterized (BKoV/Egy-1/KY407744) was compared to that of BKoV/U-1/AB084788, which, to date, is the only available BKoV complete genome sequence. The VP1 gene was highly divergent while the 3D gene was highly conserved (Fig. 1). The study strain had one nt insertion (G4718) and one nt deletion (C4726) in the 2B region and Sanger sequencing confirmed the presence of this insertion/deletion in BKoV/Egy-1/KY407744 as well as in four other study strains. The insertion-deletion resulted in three successive aa substitutions (L1304A, R1305S, and T1306H). Variant PKoV strains frequently express deletions in their 2B genes. The significance of these mutations is not yet known. It has been assumed that the 2B protein has multiple functions in different picornaviruses [24]. However, the 2B gene may not be responsible for kobuvirus pathogenesis [25].
The phylogenetic analysis of sequences representing partial 3D regions showed that BKoV strains from Cairo grouped with Chinese strains while those from Sharkia clustered with Korean strains. The Cairo strains shared identities of 97.8-99.9% at the nt level and 99.1-100% at the aa level while the two Sharkia strains shared identities of 99.9% and 100% at the nt and aa levels, respectively. Similarly, Chang et al. [6] and Candido et al. [19] stated that BKoV-3D sequences from the same epizootic areas grouped together and shared higher identities. Earlier studies have concentrated mostly on detection and sequencing of a smaller part of the BKoV-3D (RdRp) gene [21, 18, 19, 23]. The 3D genes are usually conserved. Thus, they are mainly used, phylogenetically, to discriminate BKoV from other kobuviruses but not to differentiate BKoV into distinct lineages [6, 26].
On the other hand, VP1 is the most exterior, immune-determinant capsid protein in BKoV and is used for serotyping as it carries several neutralization domains [1, 27, 28]. Molecular typing of the VP1 gene correlated with serotype specificity as strains from the same serotype tend to be monophyletic. This information is valuable in assigning taxonomic groups [29]. Until now, the observed evolutionary variations among different BKoV-VP1 strains have been studied based only on sequencing of 17 BKoV-VP1 gene sequences of Chinese origin. Four lineages (genotypes) were proposed based on phylogenetics and nt sequence identities (with an 85.1% cutoff value) [6]. BKoV VP1 sequences from Egypt belonged to lineage 1 (Genotype A), which includes BKoVs from Brazil, Japan (only U-1 strain), and Scotland. The members of genotype A shared nt identities higher than 85.1%, validating the genotyping of Chang et al. [6]. Other genotypes (B, C, and D) contained BKoV-VP1 strains from China only, so it appears that genotype A is the most prevalent cluster worldwide. Also, it was noticed that some strains from Scotland (SC13, n = 14) formed a new group (tentatively named genotype E/ lineage 5) within the phylogenetic tree (Fig. S2) with nt identities less than 85.1% (74.6-78% with genotype A, 75.7-77% with genotype B, 74.6-75.3% with group C, and 77.8-78.3% with genotype D), indicating the presence of other genotypes that have not yet been acknowledged. It was noticed that BKoV-3D sequences from this study showed different phylogenetic topography than that of VP1 sequences, suggesting different evolutionary histories for the structural and non-structural regions. Due to the limited number of BKoV-VP1 sequences in GenBank, the present study provides important data for future genotyping and classification of BKoV.
VP1 protein sequences from this study contained six non-synonymous substitutions at positions 70, 207, 248, 258, 259, and 260 compared to reference sequences (101 sequences in total); with one of them (V70 to I70) considered unique to Cairo strains (BKoV-1, BKoV-4, and BKoV-7). Between BKoV/Egy-1/KY407744 and BKoV/U-1/AB084788, twelve aa substitutions were observed within the VP1 protein (Table S1). Adaptive mutations may play an important role in inducing diarrheal outbreaks through the generation of new variants that may not be recognized by host immune defenses. Moreover, they may participate in the escape of kobuviruses from the gastrointestinal tract to the circulatory system [30]. The discrepancy in the phylogenetic topography of VP1 (Fig. 3) and 3D (Fig. 2) between the Cairo and Sharkia strains is likely due to differences in evolutionary pressure and substitution rates between structural (VP1) and non-structural regions (3D). However, recombination events in non-structural genes, including the 3D gene, cannot be ruled out as it is common for all picornavirus to recombine in such regions [31]. Whole genome sequencing and the continued surveillance of BKoV diversity are required to increase information about this virus in Egypt and to document genetic shift caused by recombination events.
BKoV detection and analysis of its complete genome, particularly the VP1 gene, are highly significant for improving our understanding of kobuvirus biology. Our findings further confirm that BKoV is distributed globally and that further epidemiological studies are needed in Egypt. The genetic diversity of kobuviruses and its role in NCD should be monitored carefully. RT-PCR and NGS approaches, applied in this study, should be useful in this regard.
References
Reuter G, Boros Á, Pankovics P (2011) Kobuviruses—a comprehensive review. Rev Med Virol 21:32–41
Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, Pallansch MA, Palmenberg AC, Simmonds P, Skern T, Stanway G, Yamashita T, Zell R (2012) Picornaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on taxonomy of viruses. Elsevier, San Diego, pp 855–880
Racaniello VR (2007) Picornaviridae: the viruses and their replication. In: Knipe DM, Howley PM (eds) Fields virology. Lippincott Williams & Wilkins, Philadelphia, pp 795–838
Khamrin P, Maneekarn N, Okitsu S, Ushijima H (2014) Epidemiology of human and animal kobuviruses. Virusdisease 25:195–200
Pham NTK, Trinh QD, Khamrin P, Nguyen TA, Dey SK, Phan TG, Hoang LP, Maneekarn N, Okitsu S, Mizuguchi M, Ushijima H (2008) Sequence analysis of the capsid gene of Aichi viruses detected from Japan, Bangladesh, Thailand, and Vietnam. J Med Virol 80:1222–1227
Chang J, Wang Q, Wang F, Jiang Z, Liu Y, Yu L (2014) Prevalence and genetic diversity of bovine kobuvirus in China. Arch Virol 159:1505–1510
Yamashita T, Kobayashi S, Sakac K, Nakata S, Chiba S, Ishihara Y, Isomura S (1991) Isolation of cytopathic small round viruses with BS-Cl cells from patients with gastroenteritis. J Infect Dis 164:954–957
Yamashita T, Sakae K, Tsuzuki H, Suzuki Y, Ishikawa N, Takeda N, Miyamura T, Yamazaki S (1998) Complete nucleotide sequence and genetic organization of Aichi virus, a distinct member of the Picornaviridae associated with acute gastroenteritis in humans. J Virol 72:8408–8412
Reuter G, Boldizsar A, Kiss I, Pankovics P (2008) Candidate new species of Kobuvirus in porcine hosts. Emerg Infect Dis 14:1968–1970
Yamashita T, Ito M, Kabashima Y, Tsuzuki H, Fujiura A, Sakae K (2003) Isolation and characterization of a new species of kobuvirus associated with cattle. J Gen Virol 84:3069–3077
Di Martino B, Di Profio F, Di Felice E, Ceci C, Pistilli MG, Marsilio F (2012) Molecular detection of bovine kobuviruses in Italy. Arch Virol 157:2393–2396
Reuter G, Egyed L (2009) Bovine kobuvirus in Europe. Emerg Infect Dis 15:822–823
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Kandeil A, El-Shesheny R, Kayali G, Moatasim Y, Bagato O, Darwish M, Gaffar A, Younes A, Farag T, Kutkat MA, Ali MA (2013) Characterization of the recent outbreak of foot-and-mouth disease virus serotype SAT2 in Egypt. Arch Virol 158:619–627
Sobhy NM, Mor SK, Mohammed MEM, Bastawecy IM, Fakhry HM, Youssef CRB, Abouzeid NZ, Goyal SM (2015) Isolation and molecular characterization of bovine enteroviruses in Egypt. Vet J 206:317–321
Mohamed FF, Mansour SM, El-Araby IE, Mor SK, Goyal SM (2017) Molecular detection of enteric viruses from diarrheic calves in Egypt. Arch Virol 162:129–137
Khamrin P, Maneekarn N, Peerakome S, Okitsu S, Mizuguchi M, Ushijima H (2008) Bovine kobuviruses from cattle with diarrhea. Emerg Infect Dis 14:985–986
Mauroy A, Scipioni A, Mathijs E, Thys C, Thiry E (2009) Molecular detection of kobuviruses and recombinant noroviruses in cattle in continental Europe. Arch Virol 154:1841–1845
Park SJ, Kim HK, Song DS, Moon HJ, Park BK (2011) Molecular detection and genetic characterization of kobuviruses in fecal samples collected from diarrheic cattle in Korea. Infect Genet Evol 11:1178–1182
Jeoung HY, Lim JA, Jeong W, Oem JK, An DJ (2011) Three clusters of bovine kobuvirus isolated in Korea, 2008–2010. Virus Genes 42:402–406
Barry AF, Ribeiro J, Alfieri AF, van der Poel WH, Alfieri AA (2011) First detection of kobuvirus in farm animals in Brazil and the Netherlands. Infect Genet Evol 11:1811–1814
Ribeiro J, Lorenzetti E, Alfieri AF, Alfieri AA (2014) Kobuvirus (Aichivirus B) infection in Brazilian cattle herds. Vet Res Commun 38:177–182
Candido M, Batinga MCA, Alencar ALF, de Almeida-Queiroz SR, da Glória Buzinaro M, Livonesi MC, Fernandes AM, de Sousa RLM (2017) Molecular characterization and genetic diversity of bovine kobuvirus, Brazil. Virus Genes 53:105–110
de Jong AS, de Mattia F, Van Dommelen MM, Lanke K, Melchers WJ, Willems PH, van Kuppeveld FJ (2008) Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J Virol 82:3782–3790
Okitsu S, Khamrin P, Thongprachum A, Kalesaran AF, Takanashi S, Shimizu H, Maneekarn N, Mizuguchi M, Hayakawa S, Ushijima H (2014) Molecular characterization and sequence analysis of the 2B region of Aichivirus C strains in Japan and Thailand. Infect Genet Evol 26:89–94
Oberste MS, Maher K, Pallansch MA (2003) Genomic evidence that simian virus 2 and six other simian picornaviruses represent a new genus in Picornaviridae. Virology 314:283–293
Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, Hecht HJ, Johnson JE, Kamer G, Luo M, Mosser AG (1985) Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 317:145–153
Mateu MG (1995) Antibody recognition of picornaviruses and escape from neutralization: a structural view. Virus Res 38:1–24
Oberste MS, Maher K, Kilpatrick DR, Pallansch MA (1999) Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol 73:1941–1948
Fan S, Sun H, Ying Y, Gao X, Wang Z, Yu Y, Li Y, Wang T, Yu Z, Yang S, Zhao Y (2013) Identification and characterization of porcine kobuvirus variant isolated from suckling piglet in Gansu province, China. Viruses 5:2548–2560
Lukashev AN (2010) Recombination among picornaviruses. Rev Med Virol 20:327–337
Acknowledgements
We thank Dr. Hany Abdalla (Department of Theriogenology, Faculty of Veterinary Medicine, Zagazig University) for help in collection of samples and for providing historical data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors confirm that this article content has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Zhenhai Chen.
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2018_3758_MOESM1_ESM.jpg
Supplementary material 1 Fig. S1 A neighbor-joining phylogenetic tree of the complete coding region of BKoV/Egy-1/KY407744 as compared to other picornaviruses. Bootstrap values (> 50%) are shown above the branches. The sequence of our strain is indicated by a solid circle. BKoV, bovine kobuvirus; PKoV, porcine kobuvirus (JPEG 65 kb)
705_2018_3758_MOESM2_ESM.jpg
Supplementary material 2 Fig. S2 A neighbor-joining phylogenetic tree of BKoV VP1 sequences (801 bp) showing the new lineage 5 (genotype E); marked in red. Bootstrap values of >50% are shown above the branches. BKoV- bovine kobuvirus; PKoV- porcine kobuvirus (JPEG 389 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Mohamed, F.F., Mansour, S.M.G., Orabi, A. et al. Detection and genetic characterization of bovine kobuvirus from calves in Egypt. Arch Virol 163, 1439–1447 (2018). https://doi.org/10.1007/s00705-018-3758-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-3758-1