
Abstract
Progressive microcephaly and neurodegeneration are genetically heterogenous conditions, largely associated with genes that are essential for the survival of neurons. In this study, we interrogate the genetic etiology of two siblings from a non-consanguineous family with severe early onset of neurological manifestations. Whole exome sequencing identified novel compound heterozygous mutations in VARS that segregated with the proband: a missense (c.3192G>A; p.Met1064Ile) and a splice site mutation (c.1577-2A>G). The VARS gene encodes cytoplasmic valyl-tRNA synthetase (ValRS), an enzyme that is essential during eukaryotic translation. cDNA analysis on patient derived fibroblasts revealed that the splice site acceptor variant allele led to nonsense mediated decay, thus resulting in a null allele. Three-dimensional modeling of ValRS predicts that the missense mutation lies in a highly conserved region and could alter side chain packing, thus affecting tRNA binding or destabilizing the interface between the catalytic and tRNA binding domains. Further quantitation of the expression of VARS showed remarkably reduced levels of mRNA and protein in skin derived fibroblasts. Aminoacylation experiments on patient derived cells showed markedly reduced enzyme activity of ValRS suggesting the mutations to be loss of function. Bi-allelic mutations in cytoplasmic amino acyl tRNA synthetases are well-known for their role in neurodegenerative disorders, yet human disorders associated with VARS mutations have not yet been clinically well characterized. Our study describes the phenotype associated with recessive VARS mutations and further functional delineation of the pathogenicity of novel variants identified, which widens the clinical and genetic spectrum of patients with progressive microcephaly.
Similar content being viewed by others


Introduction
Microcephaly is a genetically heterogeneous neurodevelopmental disorder characterized by marked reduction in brain size accompanied by varying degrees of intellectual disability, seizures and abnormal facial features that manifest either congenitally or progressively within 1 or 2 years of birth. Major cases of congenital primary microcephaly are due to defects in genes involved in transcription regulation, cell cycle progression, centrosome maturation, mitotic spindle formation, and migration of neuronal cells (Barkovich et al. 2012). In contrast to congenital microcephaly, progressive or post-migrational microcephaly is commonly accompanied by cerebral–cerebellar atrophy and associated neurodegenerative diseases. Mutations in genes involved in transcription and translation machinery largely account for the latter condition, one of which includes the amino acyl-tRNA synthetases (ARSs) family of genes (Antonellis and Green 2008). ARSs comprise an ancient group of housekeeping enzymes catalyzing the esterification of amino acids with their cognate tRNA bearing correct anticodon, to translate information to the protein level with high fidelity (Havrylenko and Mirande 2015). In human cells, there are 37 different ARSs encoded by different genes, 18 of which function exclusively in the cytoplasm, and 17 in the mitochondria, and the remaining two in both cell compartments. Mitochondrial ARS are implicated in autosomal recessive diseases affecting multiple organs, while genes encoding cytoplasmic ARS are generally inherited in both dominant and recessive fashion. Dominant mutations in cytoplasmic ARSs (cARSs) cause peripheral neuropathies such as Charcot Marie Tooth (CMT) diseases (Oprescu et al. 2017) and muscular dystrophy (Tsai et al. 2017). Most of the recessive cARSs mutations (AARS, DARS, IARS, LARS, RARS, SARS) are associated with severe neurological manifestations including epileptic encephalopathy, hypo myelination, microcephaly, neurodegeneration, growth retardation, and intellectual disability (Kopajtich et al. 2016; Latour et al. 2010; Musante et al. 2017; Taft et al. 2013; Wolf et al. 2014; Zhang et al. 2014). However, interstitial lung and liver diseases in LARS, MARS, and Usher syndrome type 3B in HARS have also been identified, suggesting diverse functions of individual ARSs (Casey et al. 2012; Gonzalez et al. 2013).
Here we describe two siblings presented with progressive microcephaly, brain atrophy and seizures. Through whole exome sequencing (WES), we identified bi-allelic mutations in VARS, encoding the cytoplasmic valyl-tRNA synthetase (ValRS) as the probable causative variants.
Materials and methods
Study subjects
Patients were clinically evaluated at the Amrita Institute of Medical Sciences, Kerala, India. Written informed consent for clinical details, photography and sample collection was obtained from parents under the protocol 76-HG-0238 (Diagnosis and treatment of patients with inborn errors of metabolism or other genetic disorders) approved by the Institutional Review Board of the National Human Genome Research Institute.
Whole exome sequencing
Whole exome sequencing was performed using the Agilent Sure Select Target Enrichment Kit and the Illumina Hiseq 2000/2500 sequencer (Illumina, San Diego, CA) according to manufacturer’s instructions. Identified reads were aligned to a human reference genome (hg19; NCBI build 37; Feb. 2009), variant calling files were generated (Genome Analysis Tool Kit, Broad Institute) and functionally annotated using ANNOVAR (Chang and Wang 2012). Variants were filtered based on allele frequency less than 0.01 with truncating nature of the variants and predicted pathogenicity according to an online prediction tool (Supplementary Table 1). Identified candidates were Sanger evaluated for confirmation and family segregation. The novel variants identified in this study have been deposited in the Leiden Open Variation Database (LOVD) and now publicly available online (https://databases.lovd.nl/shared/transcripts/00022556).
Cell culture
Patient fibroblasts were cultured from skin biopsy as described (Stephen et al. 2016). Human normal fibroblast cell lines purchased form ATCC (Manassas, VA, USA) and Coriell (Camden, NJ, USA) were used as controls for our experiments. Fibroblasts were cultured and maintained in high-glucose (4.5 g/L) DMEM with 10% fetal bovine serum (FBS; Gemini Bio-Products, West Sacramento, CA, USA), 1% MEM non-essential amino acid solution and 1% penicillin–streptomycin with glutamine. Mouse embryonic haploid stem cells were generated as previously described (Elling et al. 2011, 2017) and transferred to feeder-free conditions using Matrigel (BD Biosciences) and maintained with standard mouse embryonic cell culture media (Sigma). Both human and mouse cell lines were lysed using RIPA buffer (Sigma) and quantified as described previously (Stephen et al. 2017). VARS native overexpression lysate was purchased from Novus Biologicals (NBP2-07645) and used for western blotting.
Splicing analysis
Total RNA was extracted from the patient skin derived fibroblasts using the RNeasy Mini Kit (Qiagen, CA, USA). cDNA synthesis was performed using a High Capacity RNA to cDNA synthesis kit (Applied Biosystems) according to the manufacturer’s protocol. Primers specific to two predominant transcripts were designed (Supplementary Table 2), amplified by PCR and further evaluated by Sanger sequencing.
Quantitative RT-PCR and western blotting
Relative mRNA expression level of VARS was measured in patient skin fibroblasts using SYBR Green PCR master mix and analyzed as described elsewhere (Stephen et al. 2016). Primers probes used for qPCR are shown in supplementary Table 2. For western blotting, protein lysates derived from fibroblasts were electrophoresed on 3–8% Tris-Acetate gel, blotted on nitrocellulose membrane and treated with the primary Anti-VARS monoclonal antibody produced in rabbit that targets the N-terminal region of VARS (Abcam). Immunoblots were treated with appropriate secondary antibodies and imaged under IR Odyssey® Imaging System (LiCOR, Lincoln, NE).
Homology modeling
The crystal structure of a complex of Thermus thermophilus valyl-tRNA synthetase (ValRS) and tRNAVal (pdb code: 1gax) (Fukai et al. 2000) was used as the template to build a three-dimensional model of the human ValRS complex. The Prime software tools (Schrodinger, LLC) were used to build the model, which was subsequently refined to relieve strain in and around Phe 1063. Using CHARMM (Brooks et al. 2009), the phenylalanine side chain was rebuilt with ideal geometry, and the energy calculated with the SCPISM solvent description (Hassan and Steinbach 2011) was minimized while allowing only residues Cys 927, Phe 1007, Thr 1011, and Phe 1063 to move. The model was rendered using the programs MolScript (Kraulis 1991) and Raster3D (Merritt and Bacon 1997).
Aminoacylation assay
Bovine liver crude tRNA (Sigma) was re-suspended in 1 mL of 10 mM HEPES pH 7.5, 0.1 mM EDTA. Since mammalian tRNA often lacks CCA at the 3′ -end, a previously published method was employed to process the tRNA using E. coli CCA-adding enzyme, with the exception of utilizing a temperature of 37 °C (Pang and Martinis 2009). The full-length tRNA was phenol-extracted using 2 mL Phase Lock Gel tubes (Eppendorf) and subsequently ethanol-precipitated overnight. The tRNA was re-suspended in 10 mM HEPES buffer pH 7.5, 0.1 mM EDTA for use in aminoacylation reaction. Aminoacylation was carried out in the presence of 50 mM HEPES, pH 7.4, 15 mM MgCl2, 20 mM KCl, 1 mM dithiothreitol, 50 μM L-[14C]-valine (0.27 Ci/mmol), 1.5 mg/mL of full length bovine liver tRNA and mouse embryonic cell lysate containing 320 μg/mL of protein or human fibroblast cell lysate at 920 μg/mL. The human fibroblast lysate had reduced expression of ValRS when compared to the mouse embryonic stem cell lysates. Therefore, to carry out the aminoacylation assay, the lysates from human fibroblast cells were used at a higher protein concentration with equal overall protein concentration. Aliquots (5 μL) were taken at different time points and quenched on Whatman filter pads that were pre-soaked with 10% trichloroacetic acid (TCA). The pads were washed three times for 8 min each with ice-cold 5% TCA and then treated with ice-cold 95% ethanol for 8 min. The washed pads were then dried under a heat lamp. Radioactivity was quantified in a Beckman LS 6500 scintillation counter (Beckman Coulter, Fullerton, CA).
Results
Clinical features
The elder sibling of the proband (Fig. 1a-II.1) was born at 32 weeks of gestation, had a birth weight of 2160 g (50th–75th centile), birth length of 40 cm (< 5th centile) and head circumference of 28 cm (< 3rd centile, − 6SD); his mother had gestational diabetes during the pregnancy and was on diet control. The infant had a very small anterior fontanel and prominent sutural ridging and weak cry. He had progressive microcephaly and developed seizures by the first month of age (Fig. 1b). The head circumference at 2 months was only 30 cm (< 3rd centile, − 8SD). He did not acquire any motor milestones or speech and expired at 1 year of age due to a severe respiratory infection. He had an episode of status epilepticus by 30 days of life and had recurrent lower respiratory tract infections. There was no record of hepatosplenomegaly. The metabolic evaluation did not reveal any abnormality. Brain stem auditory evoked response showed bilateral conductive hearing loss. Ophthalmological evaluation showed a myopic astigmatism of 3D and visual evoked response revealed prolonged P100 latency. Plain CT of the brain in the new-born appeared normal and there was no intracranial calcification. Axial T2 and coronal T2 MRI of brain at 4 months of age showed diffuse cerebral atrophy with thinning of corpus callosum and normal cerebellum (Fig. 1c, d). There was no diffusion restriction.

Pedigree and clinical features of the family. a Pedigree of the family (arrow points to the proband). b Clinical photographs of the elder sibling of the proband (A-II.1) showing severe microcephaly, low set ears, upslant of eyes, sloping forehead, micrognathia, short nose and flat nasal bridge. c, d MRI of A-II.1 at the age of 4 months showing diffuse cerebral atrophy, thin corpus callosum and normal cerebellum. e, f T2 Axial and coronal MRI images of the proband (A-II.2) at 52 days of life showing minimal diffuse brain atrophy with prominent fissures and sulci and no significant cerebellar atrophy. g, h MRI repeated at 2 years of proband (A-II.2) showing progressive worsening of cerebral and cerebellar atrophy (arrow in H) and thinning of corpus callosum
The Proband (Fig. 1a-II.2) was a 32-month-old male born as the second child of a non-consanguineous couple from India, evaluated for global developmental delay. He was born as a full-term baby following caesarean section due to breech presentation. His mother (Fig. 1a-I.2) had gestational diabetes in the second pregnancy and was managed with insulin. At birth, he had a birth weight of 4070 g (75th–90th centile) and a head circumference of 33 cm (10th centile, − 2SD). At around 1.4 months of age (49 days) he had right focal seizures, of no obvious etiology, but was diagnosed to have anemia, which was corrected with a single blood transfusion. At 2 months, his head circumference was noticeable smaller for age (34.5 cm, < 3rd centile, − 4SD). During the most recent hospital visit at 32 months, he continued to have refractory seizures that were moderately controlled by five antiepileptic medications. On examination, he had severe microcephaly (40 cm, < 3rd centile, between − 8SD and − 9SD), bilaterally low set ears, long eyelashes, sloping forehead, micrognathia, and short nose. He did not attain head control and speech. He had generalized hypotonia; deep tendon reflexes were present in the upper extremity but were not elicited at the knees and ankles. He also had contracture of ankles and clawing of fingers. Because he had difficulty in swallowing, he was fed through a nasogastric tube. Axial T2 plain sequences of magnetic resonance imaging (MRI) of brain done at 52 days of life showed minimal diffuse brain atrophy with prominent fissures and sulci (Fig. 1e). Coronal T2 imaging did not show significant cerebellar atrophy (Fig. 1f). MRI brain imaging repeated at 2 years of life showed progressive worsening of cerebral and cerebellar atrophy, reduction in white matter volume, bilateral frontal and occipital subcortical white matter hyper intensities and thinning of corpus callosum in Axial T2 coronal T2 sequences (Fig. 1g, h). Unusually prominent vascular flow voids were noted along cerebrospinal fluid spaces especially over temporal polar and over convexities in coronal T2 sequence. Electroencephalogram showed bilateral occipital epileptiform abnormalities. Fundus evaluation was normal. Echocardiogram, visual evoked potential, TORCH screening, and immunoglobulin estimation in serum were normal. Flow cytometry revealed a normal level of CD3 and CD19 (CD3–65.8%, CD19–17.9%). A comprehensive clinical summary of both the patients is shown in Table 1.
Karyotype analysis of the proband (II.2) showed 46 XY. Metabolic screening with tandem mass spectrometry and gas chromatography of urine sample did not suggest an underlying metabolic defect. A stress cytogenetics study using mitomycin C showed increased chromosomal breakages. In view of microcephaly, sloping forehead, diffuse brain atrophy and increased chromosomal breakages, and a family history a microcephaly and death in a sibling, the possibility of Nijmegen breakage syndrome was considered but no mutations in NBN were found. Clinical exome sequencing of a panel of genes (4829 genes), including known disease-causing genes of microcephaly, did not identify any pathogenic mutations. Hence, trio WES (parents and the proband) was performed to identify the underlying genetic cause. DNA samples from the elder sibling of the proband (II.1) were unavailable for the clinical genetic evaluation.
Identification of mutations in VARS, splice analysis and protein modeling
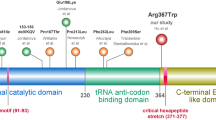
Exome sequencing followed by variant filtering identified compound heterozygous mutations in VARS: a splice site mutation, Chr6:31752087T>C (NM_006295.2:c.1577-2A>G;p.?) inherited from father and a missense mutation, Chr6:31747481C>T (NM_006295.2:c.3192G>A;p.Met1064Ile) inherited from mother (Fig. 2a). A summary of exome analysis and the final list of variants are given in supplementary Table 1. Both the mutations in VARS are novel (not found in any online databases ExAC, genomAD) and have high CADD Phred scores (17.55 and 23.7, respectively). The missense mutation is predicted to be damaging per various prediction tools: SIFT (score: 0), Mutation Taster (p value: 1), and PolyPhen-2 (score: 0.99), and located in the highly conserved area of the anticodon-binding domain of the protein (Fig. 2b). The splice site mutation is in the canonical acceptor splice site before exon 13 which encodes a part of the catalytic domain of the enzyme. Exon 13 resides in the intronic region of two predicted splice variants, XM_017011246.1 and XM_017011247.1 (Fig. 2c). Both control and patient’s cDNA amplification showed two bands: the upper band (383 bp) for the longest isoform; and the lower one (288 bp), which naturally lacks exon 13, for the other two isoforms (Fig. 2d). Skipping of exon 13 is predicted to lead to premature termination codon (NM_006295.2:c.1577_1671del; p.(Asp526Alafs*22) and result in the degradation of mutant mRNA through nonsense mediated decay. This was supported by the finding that amplification of the missense mutation at exon 27 on concentrated cDNA showed a single mutant peak (c.3192A) in the patient compared to the control, which indicates that the mRNA allele harboring the splice site variation is severely reduced or absent (Fig. 2e). Thus, both the missense and splice site mutations are predicted to affect the functional domains of the ValRS suggesting the possible pathogenicity (Fig. 2f).

Sanger validation of the variants, conservation and splice analysis. a Sanger validation of identified compound heterozygous variants in the proband: the splice site variation (c.1577-2A>G) inherited from the father and the missense variation (c.3192G>A) inherited from the mother. b Conservation of amino acids around the position of missense variation (c.3192G>A; p.Met1064Ile); methionine is highly conserved up to C.elegans and Baker’s yeast. c Diagrammatic representation of different splice variants of VARS and primer designing scheme for splice analysis and qPCR. Each box represents the exonic regions of the gene; the black boxes are coding parts and the boxes with dashed line denote untranslated regions, and ‘ATG’ marks the start of the coding frame. Two splice variants (XM_017011247.1 and XM_017011246.1) lack 5′ coding regions and exon 13 compared to the longest variants (NM_006295.2 and XM_005249362.2) and have their start codon in-frame downstream. Position of primers for splice defect analysis (F1–R1, F2–R2) and qPCR (Q1F–Q1R, Q2F–Q2R, Q3F–Q3R) are also depicted. d Gel images of the PCR products of F1–R1 flanking the splice site mutation. The lower band (288 bp) which lacks exon 13 is present in both control and patient, results from the amplification of shorter splice variants that lack exon 13. e Chromatogram showing the sequencing results of F2–R2 amplifying cDNA, depicting the presence of only one allele (c.3192A) in the patient (lower panel) compared to control (upper panel), showing the absence of G allele, which harbors the splice site variation. f Schematic representation of different functional domains in the full length VARS protein (Uniprot: P26640). The splice site variation is predicted to abolish both tRNA synthetase and anticodon-binding domains while the missense variation is in a highly conserved area of the anticodon-binding domain
To assess the possible functional impact of the missense mutation (M1064I), a model of human ValRS bound to tRNA was generated using the template crystal structure of Thermus thermophilus valyl-tRNA synthetase (ValRS) and tRNAVal complex (Fig. 3a). The highly conserved region surrounding Met 1064 (position of missense variation) is shown in Fig. 3b. Note that although Cys 950 corresponds to an alanine in the Thermus thermophilus template structure, cysteine is, in fact, highly conserved at this position (alignment not shown). Thus, the neighboring residues of Met 1064 are highly conserved, and the mutation M1064I presumably alters side chain packing, possibly displacing the nearby alpha helix that includes Cys 950 and conserved residues that are proximal to the bound tRNA, namely, Arg947, Asn951, Lys952, Asn955, Ala956, and Phe959 (Fig. 3c). The mutation may also destabilize the interface between the catalytic domain and the anticodon-binding domain. In the Conserved Domain Database (Marchler-Bauer et al. 2017), residues 335–938, including the conserved DALR motif, are assigned to the catalytic core domain, and the anticodon-binding domain ranges from residue 938–1075. It is possible that the M1064I variant perturbs the association of these domains, straining the ValRS structure and possibly shifting residues near the aminoacylation site, which could affect the amino acylation activity of the enzyme (Fig. 3c).

The model of human valyl-tRNA synthetase with tRNA bound. a, b The protein main chain (alpha-carbon atoms) is colored according to sequence identity with the template structure, yellow where identical (39%) and light blue where different. a Stereo view, with the tRNA backbone (phosphorus atoms) shown in red, a Val-AMP analog shown as ball and stick, bound in the aminoacylation site, and the side chain of Met 1064 shown as space filling. The location of an unmodeled region inserted in the human sequence between the KMSKS loop and the DALR motif is marked by the blue spheres representing Asp 875 and Cys 917. b Stereo view of residues neighboring Met 1064, with carbon atoms in side chains of residues identical in the model and template colored dark gray. The DALR motif and other conserved residues are labeled. For clarity, only the tip of Leu 953 is shown. c Protein-tRNA interface, with tRNA shown as ball and stick and the Val-AMP analogue shown as sticks. The possible impact of the M1064I mutation on ValRS-tRNA binding is illustrated by showing side chains as space filling for Met 1064 and Cys 950, as well as for conserved residues at or near the interface: Arg 947, Phe 949, Asn 951, Lys 952, Asn 955, Ala 956, and Phe 959. The M1064I mutation might also perturb the interface between the catalytic core domain (colored salmon) and the anticodon-binding domain (green)
Expression of VARS is severely reduced in patient skin fibroblasts
Valyl-tRNA synthetase is ubiquitously expressed in tissues, including skin, with a diverse array of cellular functions (Lo et al. 2014). We measured the level of mRNA in skin derived fibroblasts from the patient using three different primer probes specific to the N-terminal (Q1F–Q1R), middle (Q2F–Q2R) and C-terminal (Q3F–Q3R) regions of the transcripts (Fig. 2c). Supplementary Table 2 lists the sequence information for each primer probes. Realtime PCR amplification using the three primer probes showed significant reduction of transcripts (50–60%) in patient compared to control (Fig. 4a). Further examination of protein expression levels using immunoblotting showed severely reduced expression of VARS protein in the proband cells (Fig. 4b). Note that in control cell lines, there is some degree of variability in protein expression.

VARS expression studies and aminoacylation activity in proband cells. a VARS mRNA expression studies using three different primer probes (see the primer designing scheme in Fig. 2c) showing 50–60% reduction of transcripts in proband, relative to control. b VARS immunoblotting using cell lysates derived from three controls and proband showing severe reduction in protein expression in proband cells. VARS overexpression lysate (OEL) produced in HEK293T cells was used as positive control and compared to empty vector lysate (EVL) (Novus Biologicals, NBP2-07645). Results were normalized to beta actin loading control. c, d Decreased aminoacylation activity in proband fibroblast cell lysate. Valylation was carried out using cell lysate containing 920 μg/mL of protein and 1.5 mg/mL full length bovine liver tRNA. c Enzyme activity of fibroblasts from control unaffected individual (closed squares) and proband II.2 (closed circles) was measured and normalized with total protein content. Mean values from three different control cell lines are shown. d The assay was optimized using mouse embryonic stem cells (wild type, closed squares; knockout, closed circles). All assays were done in triplicates and the error bars represent standard error of means
Aminoacylation is reduced in patient derived fibroblasts
ValRS recognizes tRNAVal via the C-terminal anticodon-binding domain’s interactions with A35 and C36 anticodon identity elements of tRNAVal (Tamura et al. 1991). X-ray crystallographic studies on T. thermophilus ValRS have confirmed these interactions (Fukai et al. 2003). Nucleotides A35 and C36 form base stacking interactions with a conserved Phe residue from the anticodon-binding domain of ValRS. The first and third helices of this domain of ValRS contact the tRNAVal and help hold it in position. Sequence alignment shows that Met1064 is a conserved residue of the anticodon-binding domain (Fig. 2b, f). Though this residue is not directly involved in the interaction between ValRS and tRNAVal, it is located near the turn of the second helix of the anticodon-binding domain. Mutation of this residue possibly results in structural changes that influence the activity of ValRS, and the other allele with the splice mutation is predicted to be null due to the splice defect. Aminoacylation activity of lysates from patient fibroblast cells was tested in the presence of bovine total tRNA. Compared to three different biological control fibroblast cells, lysate from the patient had about four times reduced aminoacylation activity of ValRS (Fig. 4c) which could possibly impact protein synthesis and affect cellular metabolism. Embryonic stem cells derived from wild type (Vars+/+) and knock out (Vars−/−) mice were used as experimental controls to optimize the aminoacylation experiment (Fig. 4D).
Discussion
The human brain is particularly sensitive to defects in genes involved in transcriptional, RNA processing and translational machinery. Bi-allelic loss of function mutations in genes encoding cytoplasmic aminoacyl tRNA synthetase are implicated for overlapping neurological abnormalities, peripheral neuropathy, liver dysfunction, lung fibrosis, visual and hearing impairment; for instance, mutations in AARS, RARS, DARS, and QARS account for core neurological manifestations including cerebral atrophy, seizures, hypo myelination and microcephaly (Supplementary Table 3). This study reports a family with recessive mutations in VARS causing such severe early onset neurological manifestations. The elder sibling of the proband (II.1) manifested severe phenotype and expired at 1 year of age. DNA samples from him were unavailable for analysis. However, similar clinical presentation compared to proband suggests that he probably harbored the same mutations.
Exome sequencing followed by variant filtering revealed bi-allelic variants in five candidate genes: HTRA4, LEXM, TLDC2, SPEM2, and VARS. HTRA4 is mainly expressed in placenta and is associated with endothelial dysfunction and capillary leakage (Singh et al. 2015). The TLDC2 gene has not yet been explored for any specific cellular function. In addition, healthy homozygotes have been reported for at least one of the variants of both HTRA4 and TLDC2, so we excluded them. Among the final three candidates (Supplementary Table 1), LEXM encodes an expansion molecule that may have a role in T-cell proliferation. SPEM2 is highly expressed in testes and knockout models have an abnormal reproductive system; so its role appears to be in reproduction. On the other hand, variants in the VARS gene had a comparatively higher CADD score (Supplementary Table 1) among our candidates, and the variants are predicted to affect important domains of the protein. Recently, in a large cohort of individuals with neurological malformations, Karaca et al. reported three affected individuals from two unrelated consanguineous families having homozygous missense mutations in VARS (Karaca et al. 2015); the affected individuals in the report had phenotypic overlap with our patients, presenting with severe microcephaly, developmental delay, intellectual disability and cortical atrophy. Considering the phenotype similarity and association of other cytoplasmic amino acyl tRNA synthetases (e.g., microcephaly and brain atrophy in patients with mutations in AARS, DARS, or QARS), we chose VARS as the best candidate.
ValRS belongs to the class I family of AARs and has high sequence similarity with mitochondrial ValRS, encoded by VARS2, the gene recently implicated for encephalomyopathies (Diodato et al. 2014). Like other ARSs, ValRS contains the catalytic domain and the anticodon-binding domain to catalyze the amino acylation of specific tRNAs; the catalytic domain catalyzes the binding of amino acid and ATP to form aminoacyl intermediate with the release of a pyrophosphate molecule. In a second step, the enzyme recognizes the appropriate tRNA with its anticodon-binding domain and covalently links the tRNA to amino acid to form amino acyl tRNA molecule (Datt and Sharma 2014). The truncating mutation in our patient is predicted to abolish both the catalytic domain and tRNA binding domain, while the missense mutation is in a highly conserved region of the tRNA binding domain (Fig. 2f), potentially affecting both the steps of enzymatic reaction. As per our protein modeling studies, it is possible that the Met1064Ile variant perturbs the association of these domains, straining the ValRS structure and possibly shifting residues near the aminoacylation site. In our mRNA expression studies, we observed 50–60% reduction in the transcripts due to nonsense mediated decay of the allele carrying splice site mutation. However, we observed severe reduction in protein levels, in contrast to mRNA expression, which could possibly due to a decreased protein stability of the protein with the missense mutation. The severe defect in aminoacylation we observed in our patient cells correlates with our expression and modeling data. Although the ValRS in-frame splice variants lack a catalytic domain, which has diverse cellular functions (Lo et al. 2014), we did not observe any defect other than neurological problems in our patients.
In conclusion, our study highlights the clinical characterization of individuals with bi-allelic mutations in VARS which encode an evolutionarily conserved enzyme essential for eukaryotic translation. VARS mutations produce severe neurological implications, illustrating the essential nature of this gene in the development and proper functioning of the nervous system. This study expands the molecular and clinical phenotyping of patients with bi-allelic VARS mutations. Mutations in VARS should be considered in patients with severe, early onset progressive microcephaly.
References
Antonellis A, Green ED (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genom Hum Genet 9:87–107. https://doi.org/10.1146/annurev.genom.9.081307.164204
Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB (2012) A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135:1348–1369. https://doi.org/10.1093/brain/aws019
Brooks BR, Brooks CL 3rd, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614. https://doi.org/10.1002/jcc.21287
Casey JP, McGettigan P, Lynam-Lennon N, McDermott M, Regan R, Conroy J, Bourke B, O’Sullivan J, Crushell E, Lynch S, Ennis S (2012) Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol Genet Metab 106:351–358. https://doi.org/10.1016/j.ymgme.2012.04.017
Chang X, Wang K (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 49:433–436. https://doi.org/10.1136/jmedgenet-2012-100918
Datt M, Sharma A (2014) Evolutionary and structural annotation of disease-associated mutations in human aminoacyl-tRNA synthetases. BMC Genom 15:1063. https://doi.org/10.1186/1471-2164-15-1063
Diodato D, Melchionda L, Haack TB, Dallabona C, Baruffini E, Donnini C, Granata T, Ragona F, Balestri P, Margollicci M, Lamantea E, Nasca A, Powell CA, Minczuk M, Strom TM, Meitinger T, Prokisch H, Lamperti C, Zeviani M, Ghezzi D (2014) VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum Mutat 35:983–989. https://doi.org/10.1002/humu.22590
Elling U, Taubenschmid J, Wirnsberger G, O’Malley R, Demers SP, Vanhaelen Q, Shukalyuk AI, Schmauss G, Schramek D, Schnuetgen F, von Melchner H, Ecker JR, Stanford WL, Zuber J, Stark A, Penninger JM (2011) Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell 9:563–574. https://doi.org/10.1016/j.stem.2011.10.012
Elling U, Wimmer RA, Leibbrandt A, Burkard T, Michlits G, Leopoldi A, Micheler T, Abdeen D, Zhuk S, Aspalter IM, Handl C, Liebergesell J, Hubmann M, Husa AM, Kinzer M, Schuller N, Wetzel E, van de Loo N, Martinez JAZ, Estoppey D, Riedl R, Yang F, Fu B, Dechat T, Ivics Z, Agu CA, Bell O, Blaas D, Gerhardt H, Hoepfner D, Stark A, Penninger JM (2017) A reversible haploid mouse embryonic stem cell biobank resource for functional genomics. Nature 550:114–118. https://doi.org/10.1038/nature24027
Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S (2000) Structural basis for double-sieve discrimination of l-valine from l-isoleucine and l-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell 103:793–803
Fukai S, Nureki O, Sekine S-I, Shimada A, Vassylyev DG, Yokoyama S (2003) Mechanism of molecular interactions for tRNA-Val recognition by valyl-tRNA synthetase. RNA 9:100–111. https://doi.org/10.1261/rna.2760703
Gonzalez M, McLaughlin H, Houlden H, Guo M, Yo-Tsen L, Hadjivassilious M, Speziani F, Yang XL, Antonellis A, Reilly MM, Zuchner S (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry 84:1247–1249. https://doi.org/10.1136/jnnp-2013-305049
Hassan SA, Steinbach PJ (2011) Water-exclusion and liquid-structure forces in implicit solvation. J Phys Chem B 115:14668–14682. https://doi.org/10.1021/jp208184e
Havrylenko S, Mirande M (2015) Aminoacyl-tRNA synthetase complexes in evolution. Int J Mol Sci 16:6571–6594. https://doi.org/10.3390/ijms16036571
Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, Gonzaga-Jauregui C, Erdin S, Bayram Y, Campbell IM, Hunter JV, Atik MM, Van Esch H, Yuan B, Wiszniewski W, Isikay S, Yesil G, Yuregir OO, Tug Bozdogan S, Aslan H, Aydin H, Tos T, Aksoy A, De Vivo DC, Jain P, Geckinli BB, Sezer O, Gul D, Durmaz B, Cogulu O, Ozkinay F, Topcu V, Candan S, Cebi AH, Ikbal M, Yilmaz Gulec E, Gezdirici A, Koparir E, Ekici F, Coskun S, Cicek S, Karaer K, Koparir A, Duz MB, Kirat E, Fenercioglu E, Ulucan H, Seven M, Guran T, Elcioglu N, Yildirim MS, Aktas D, Alikasifoglu M, Ture M, Yakut T, Overton JD, Yuksel A, Ozen M, Muzny DM, Adams DR, Boerwinkle E, Chung WK, Gibbs RA, Lupski JR (2015) Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron 88:499–513. https://doi.org/10.1016/j.neuron.2015.09.048
Kopajtich R, Murayama K, Janecke AR, Haack TB, Breuer M, Knisely AS, Harting I, Ohashi T, Okazaki Y, Watanabe D, Tokuzawa Y, Kotzaeridou U, Kolker S, Sauer S, Carl M, Straub S, Entenmann A, Gizewski E, Feichtinger RG, Mayr JA, Lackner K, Strom TM, Meitinger T, Muller T, Ohtake A, Hoffmann GF, Prokisch H, Staufner C (2016) Biallelic IARS mutations cause growth retardation with prenatal onset, intellectual disability, muscular hypotonia, and infantile hepatopathy. Am J Hum Genet 99:414–422. https://doi.org/10.1016/j.ajhg.2016.05.027
Kraulis P (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24:946–950. https://doi.org/10.1107/s0021889891004399
Latour P, Thauvin-Robinet C, Baudelet-Mery C, Soichot P, Cusin V, Faivre L, Locatelli MC, Mayencon M, Sarcey A, Broussolle E, Camu W, David A, Rousson R (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 86:77–82. https://doi.org/10.1016/j.ajhg.2009.12.005
Lo WS, Gardiner E, Xu Z, Lau CF, Wang F, Zhou JJ, Mendlein JD, Nangle LA, Chiang KP, Yang XL, Au KF, Wong WH, Guo M, Zhang M, Schimmel P (2014) Human tRNA synthetase catalytic nulls with diverse functions. Science 345:328–332. https://doi.org/10.1126/science.1252943
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Geer LY, Bryant SH (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucl Acid Res 45:D200–D203. https://doi.org/10.1093/nar/gkw1129
Merritt EA, Bacon DJ (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol 277:505–524
Musante L, Puttmann L, Kahrizi K, Garshasbi M, Hu H, Stehr H, Lipkowitz B, Otto S, Jensen LR, Tzschach A, Jamali P, Wienker T, Najmabadi H, Ropers HH, Kuss AW (2017) Mutations of the aminoacyl-tRNA-synthetases SARS and WARS2 are implicated in the aetiology of autosomal recessive intellectual disability. Hum Mutat. https://doi.org/10.1002/humu.23205
Oprescu SN, Griffin LB, Beg AA, Antonellis A (2017) Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods 113:139–151. https://doi.org/10.1016/j.ymeth.2016.11.013
Pang YLJ, Martinis SA (2009) A paradigm shift for the amino acid editing mechanism of human cytoplasmic leucyl-tRNA synthetase. Biochemistry 48:8958–8964. https://doi.org/10.1021/bi901111y
Singh H, Zhao M, Chen Q, Wang Y, Li Y, Kaitu’u-Lino TJ, Tong S, Nie G (2015) Human HtrA4 expression is restricted to the placenta, is significantly up-regulated in early-onset preeclampsia, and high levels of HtrA4 cause endothelial dysfunction. J Clin Endocrinol Metab 100:E936–E945. https://doi.org/10.1210/jc.2014-3969
Stephen J, Vilboux T, Haberman Y, Pri-Chen H, Pode-Shakked B, Mazaheri S, Marek-Yagel D, Barel O, Di Segni A, Eyal E, Hout-Siloni G, Lahad A, Shalem T, Rechavi G, Malicdan MC, Weiss B, Gahl WA, Anikster Y (2016) Congenital protein losing enteropathy: an inborn error of lipid metabolism due to DGAT1 mutations. Eur J Hum Genet 24:1268–1273. https://doi.org/10.1038/ejhg.2016.5
Stephen J, Vilboux T, Mian L, Kuptanon C, Sinclair CM, Yildirimli D, Maynard DM, Bryant J, Fischer R, Vemulapalli M, Mullikin JC, Huizing M, Gahl WA, Malicdan MCV, Gunay-Aygun M (2017) Mutations in KIAA0753 cause Joubert syndrome associated with growth hormone deficiency. Hum Genet 136:399–408. https://doi.org/10.1007/s00439-017-1765-z
Taft RJ, Vanderver A, Leventer RJ, Damiani SA, Simons C, Grimmond SM, Miller D, Schmidt J, Lockhart PJ, Pope K, Ru K, Crawford J, Rosser T, de Coo IF, Juneja M, Verma IC, Prabhakar P, Blaser S, Raiman J, Pouwels PJ, Bevova MR, Abbink TE, van der Knaap MS, Wolf NI (2013) Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet 92:774–780. https://doi.org/10.1016/j.ajhg.2013.04.006
Tamura K, Himeno H, Asahara H, Hasegawa T, Shimizu M (1991) Identity determinants of E. Coli tRNA-Val. Biochem Biophys Res Commun 177:619–623
Tsai PC, Soong BW, Mademan I, Huang YH, Liu CR, Hsiao CT, Wu HT, Liu TT, Liu YT, Tseng YT, Lin KP, Yang UC, Chung KW, Choi BO, Nicholson GA, Kennerson ML, Chan CC, De Jonghe P, Cheng TH, Liao YC, Zuchner S, Baets J, Lee YC (2017) A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain. https://doi.org/10.1093/brain/awx058
Wolf NI, Salomons GS, Rodenburg RJ, Pouwels PJ, Schieving JH, Derks TG, Fock JM, Rump P, van Beek DM, van der Knaap MS, Waisfisz Q (2014) Mutations in RARS cause hypomyelination. Ann Neurol 76:134–139. https://doi.org/10.1002/ana.24167
Zhang X, Ling J, Barcia G, Jing L, Wu J, Barry BJ, Mochida GH, Hill RS, Weimer JM, Stein Q, Poduri A, Partlow JN, Ville D, Dulac O, Yu TW, Lam AT, Servattalab S, Rodriguez J, Boddaert N, Munnich A, Colleaux L, Zon LI, Soll D, Walsh CA, Nabbout R (2014) Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am J Hum Genet 94:547–558. https://doi.org/10.1016/j.ajhg.2014.03.003
Acknowledgements
We thank patient and family for participating in this study.
Funding
This work was supported by the Intramural Research Programs of the National Human Genome Research Institute and the Center for Information Technology, National Institutes of Health, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Stephen, J., Nampoothiri, S., Banerjee, A. et al. Loss of function mutations in VARS encoding cytoplasmic valyl-tRNA synthetase cause microcephaly, seizures, and progressive cerebral atrophy. Hum Genet 137, 293–303 (2018). https://doi.org/10.1007/s00439-018-1882-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-018-1882-3