
Abstract
Prostacyclin, or PGI2, is a product of PGI synthase (PGIS), down-stream of cyclooxygenase pathway. PGI2 has been demonstrated to play an important role in maintaining renal blood flow. Non-steroidal anti-inflammatory drugs (NSAIDs) that inhibit cyclooxygenase are reported to increase the susceptibility of patients to acute kidney injury (AKI). This study explores the role of endothelium-derived prostacyclin in ischemia-reperfusion injury (I/RI). The renal PGIS expression and PGI2 production markedly increased following I/RI. Loss of one allele of PGIS gene or selective endothelial PGIS deletion (TEK-CRE PGISfl/fl mice) caused more severe renal damage following I/RI than control mice. Iloprost, a PGI2 analog, administered 30 min before the I/R surgery, markedly attenuated the renal damage in both control mice and TEK-CRE PGISfl/fl mice. Renal p-PKA expression significantly increased after I/RI in wild-type mice but not in the PGIS deletion mice, consistent with IP receptor mediating the protective effect. Further studies showed that PGIS deficiency was associated with reduced fluorescence microsphere accumulation in the kidney following I/R. Folic acid also induced marked kidney injury; however, endothelial PGIS deletion did not worsen kidney injury compared with wild-type mice. These studies indicate that PGIS-derived PGI2 can protect the kidney from acute injury caused by ischemia and reperfusion and PGIS/PGI2 is a potential intervention target for AKI.
Similar content being viewed by others

Introduction
Acute kidney injury (AKI) is a global public concern that is associated with high morbidity, mortality, and healthcare costs, and is increasingly prevalent in both developing and developed countries [24, 27, 42]. Ischemia-reperfusion injury (I/RI) is the most common cause of AKI [2, 43]. Although extensive studies have been performed over the past decades, the incidence of AKI remains high, and therapies are limited. Furthermore, renal I/RI is almost unavoidable in renal transplantation [25], which may contribute significantly to delayed graft function (DGF). Additionally, up to 30% of the patients receiving cardiac surgery may experience acute kidney injury [36]. Other than dialysis, no pharmacological treatments reliably improve survival, limit injury, or accelerate recovery [47].
Prostacyclin, or PGI2, is a member of the prostaglandin family. PGI2 is one of the end metabolites of arachidonic acid that is produced via cyclooxygenase (COX) and PGI synthase (PGIS) [5, 32, 37]. PGI2 has a half-life of 30 s and is metabolically degraded to 6-Keto PGF1α. The short half-life limits its effect to the site of synthesis [19, 29]. As an endogenous vasodilator and inhibitor of leukocyte adhesion and platelet aggregation, PGI2 acts mainly on the membrane-bound IP receptor [9] and signals by increasing the intracellular cyclic AMP (cAMP) level [4, 9, 19, 31], which leads to the activation of protein kinase A (PKA) and further phosphorylation of related proteins that exert a variety of biologic functions [5, 12, 20, 21]. PGI2 can also bind to endogenous peroxisome proliferator-activated receptors (PPARs) and regulate the transcription of target genes [6, 40]. NSAIDs that inhibit COX and thus prostaglandin production have been reported to increase patient’s susceptibility to AKI [3, 10, 34, 35]. It has been reported that deletion of PGIS is associated with ischemic renal disorders, including nephrosclerosis and renal infarction [45]. Furthermore, in a study of rats, PGI2 protected the kidney from post-ischemic acute injury, although the mechanism is incompletely characterized [15]. These findings suggest that PGI2 plays an important role in maintaining renal blood flow and protecting the kidney from acute injury.
Therefore, the present study was designed to test the hypothesis that PGIS/PGI2 pathway is responsible for maintaining renal homeostasis during ischemic reperfusion and protecting the kidney from AKI. This study may shed light on the mechanism underlying AKI and the therapeutic potential of the PGI2 pathway during I/RI.
Materials and methods
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee of Fudan University. Wild-type (WT) C57BL/6 mice were purchased from Biomodel Organism (Shanghai, China). Mice were housed at a constant temperature and humidity room in the animal facility of Fudan University Medical Animal Center with a 12:12-h light-dark cycle and allowed free access to standard rodent chow and water.
The Cre/loxP and Flp/FRT recombination system was used to generate PGIS-floxed mouse line. The mouse genomic PGIS gene sequence was obtained from the UCSC Genome Browser. The targeting vector included a 4-kb 5′ homogenous arm, a reversed sequence including a splicing acceptor (SA) and an IRES (internal ribosome entrance site) tagged with EGFP (enhanced green fluorescence protein) and flanked by two reverse loxP sequence, a PGK-Neo selection cassette flanked by FRT, and a 2 kb 3′ homogenous arm. The IRES-EGFP cDNA in the targeting construct allows for detecting where the floxed PGIS is expressed in mice carrying the transgene. The target vector was transfected into embryonic stem cells following established protocols [39]. Neomycin-resistant embryonic cell clones were screened by Southern blot, and two correctly targeted clones were identified (Fig. 1b). Two target clones (1C11 and 3D8) were picked for blastocyst injection. Germline transmission of the transgene was obtained from the 1C11 line. Genotyping was achieved through PCR of tail DNA with primers: P1 5′- CTGTCCCTATCTAAACCTCACC-3′, P2 5′-TAGAGCGGCCATCATAACT-3′, P3 5′-GCCAAGCATTCGTAAAGCCC-3′. PCR was performed under the following conditions: denaturation at 95 °C for 3 min; 35 cycles of 94 °C for 30 s, 58 °C for 1 min, and 72 °C for 1 min; and 72 °C for 5 min. Primers P1 and P3 amplify an 857-bp wild-type allele fragment, and primers P1 and P2 amplify a 577-bp floxed allele fragment.


Generation and confirmation of PGIS-floxed mice. a Targeting strategy. The targeting construct has a 4-kb 5′ homogenous arm, a reversed sequence including an SA and an IRES-EGFP reporter flanked by two reverse loxP sequence, a PGK-Neo selection cassette flanked by FRT, and a 2-kb 3′ homogenous arm. b Southern blot showed two targeted ES cell clones that give expected hybridization patterns. The 3′ probe indicated in a was used to confirm that the correct target was obtained. c Identification of PGIS-floxed mice by PCR. d Determination of conventional knockout E16.5 embryos’ genotypes by PCR. PGIS production in the body of E16.5 embryos was analyzed by real-time PCR (e) and Western blot (f). A total of 11 mice were included (n = 4 in the WT group; n = 4 in the PGIS+/− group, and n = 3 in the PGIS−/− group). The data are expressed as the mean ± SEM and were analyzed using one-way ANOVA among multiple groups followed by Scheffe post hoc testing. ***P < 0.001. WT, wild-type; KO, knockout
Mice with PGIS gene deletion were obtained by crossing the floxed PGIS mouse with a universal Cre mouse (the EIIA-Cre mouse on the B6 background). Most of the homozygous PGIS knockout mice (PGIS−/−) experienced embryonic lethality. The heterozygous (PGIS+/−) mice were further bred with WT C57BL/6 mice to expand the heterozygous PGIS knockout mice (PGIS+/−) and WT littermates (PGIS+/+). Genotyping was achieved with primers P4 5′-ACAGGGTTTCATCCTCAATT-3′, P5 5′- GTCCTTATCATCGTCGTCTTT-3′, which amplified a 900-bp knockout allele fragment.
The endothelial-specific PGIS-deficient mouse line was generated by mating the PGISfl/fl mice with the endothelial Cre TEK-CRE mouse line. Mice with both floxed PGIS alleles and the TEK-CRE transgene were generated (TEK-CRE PGISfl/fl). TEK-CRE PGISfl/fl mice were then backcrossed to the PGISfl/fl mice to generate both deletable and nondeletable littermates.
A smooth muscle cell-specific PGIS-deficient mouse line was generated by mating the PGISfl/fl mice with the SMMHC-iCRE mouse line. Mice with both floxed PGIS alleles and the SMMHC-iCRE transgene were generated (SMMHC-iCRE PGISfl/fl). SMMHC-iCRE PGISfl/fl mice were then backcrossed to the PGISfl/fl mice to generate both deletable and nondeletable littermates.
Experimental model of AKI and drug treatment
Warm ischemia-reperfusion surgery was performed on 8- to 10-week-old male C57BL/6 mice. Mice were subjected to flank incisions. Ischemia was induced after right-sided uninephrectomy by clipping the pedicles of the remaining left kidney for 25 min. Reperfusion was confirmed visually. The color of the kidney turned from dark purple to pink. Sham surgery was performed in a similar manner, except for clamping of the renal vessels. Iloprost (50 mg/kg body wt) or an equivalent amount of PBS was administered 30 min before the surgery by intraperitoneal injection. In the nephrotoxic model, folic acid was dissolved in 0.2 ml of 0.3 mM sodium bicarbonate (NaHCO3) and administered intraperitoneally at a concentration of 250 mg/kg/wt. To collect blood and tissue samples, mice were anesthetized with chloral hydrate (400 mg/kg, intraperitoneal injection).
Renal blood flow evaluation
Red fluorescent 15 μm FluoSpheres (Molecular Probes) were used. Fifty microliters of the microsphere mixture was injected into the left atrium of live mice after IR surgery.
In vivo imaging system
Mice were anesthetized with 5% chloral hydrate. The abdomen was opened to fully expose the kidney. In vivo images were acquired with using an In Vivo Imaging System (NightOWL LB 983, Berthold Technologies, Germany).
Quantitation of fluorescence
The animals were sacrificed, and the kidneys were removed for fluorescence quantification after digestion and filtration, according to the manufacturer’s instructions. The intensity of the fluorescence signal was determined with an EnSpire luminescence spectrophotometer (Perkin-Elmer, Beaconsfield, UK) equipped with a 96-well microplate reader. The fluorescence intensity was measured at the optimal excitation-emission wavelengths of red dye (570 and 598 nm).
Prostaglandin production assessment
The production of PGI2 was assessed by measuring 6-Keto PGF1α using an ELISA Kit (No. 515211; Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer’s instructions. Thromboxane A2 was assessed by measuring thromboxane B2 using an ELISA Kit (No. 501020; Cayman Chemical). PGE2 was also measured with an ELISA Kit (514,010; Cayman Chemical).
Measurement of blood pressure
Mice were restrained in a mouse pocket and maintained at 37 °C. Each mouse had their blood pressure continuously measured 10 times per day for a week by the tail-cuff method with a noninvasive automatic blood pressure analyzer (BP-2000 Blood Pressure Analysis System, Visitech Systems, USA), and the mean results were used.
Renal function evaluation
Renal function was evaluated by blood urea nitrogen (BUN). BUN was measured using a UREA KIT (liquid; UV-GLDH method; Shanghai Kehua Bioengineering Co., Ltd.) according to the manufacturer’s instructions.
Kidney histology
Kidney samples were fixed in 4% paraformaldehyde, and paraffin-embedded, 2-μm sections were stained with periodic acid-Schiff. Over six low-power fields of each section were reviewed and scored for tubular injury in a single-blind fashion using a modified one to four scoring system, as described previously [1].
Immunofluorescence
Paraformaldehyde-fixed, paraffin-embedded, 2-μm-thick sections were deparaffinized in xylene and rehydrated in a graded alcohol series. To retrieve the antigen, sections were heated in citrate buffer (pH 6.0) for 15 min at 120 °C by means of an autoclave. Sections were then blocked with 5% BSA and incubated overnight at 4 °C with the following primary antibodies: mouse anti-PGIS (1:100; Cayman Chemical) and rat anti-CD34 (1:200; Abcam, Cambridge, UK). For fluorescent visualization of the bound primary antibodies, sections were further incubated with the appropriate Cy3-conjugated or FITC-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories) for 1 h at room temperature.
Electron microscopy
Kidney tissues were fixed in 2.5% glutaraldehyde and 2.5% paraformaldehyde in phosphate buffer. After a standard embedding procedure, the peritubular capillaries were examined and photographed under a transmission electron microscope.
TUNEL assay
Apoptotic cells were detected by TUNEL assay (G3250; Promega, Fitchburg, WI) according to the manufacturer’s instruction, and the number of TUNEL-positive cells in five sections per kidney was quantified. DAPI was used as a counterstain.
Real-time PCR
Total RNA was extracted from the kidney tissue using TRIzol Reagent (Invitrogen, Carlsbad, CA), and 1 μg RNA was reverse transcribed using oligo dT as the primer according to the manufacturer’s instructions (RR037; Takara Bio, Dalian, China). Real-time PCR was performed using the SYBR Green Premix Kit (RR820; Takara Bio, Dalian, China). The expression level of genes of interest was calculated with a comparative method (2−DDCT) using β-actin as an internal control. The following primers were obtained from the primer bank: PGIS, forward 5′-CTGGTTGGGGTATGCCTTGG and reverse 5′- TCATCACTGGGGCTGTAATGT; β-actin, forward 5′- GGCTGTATTCCCCTCCATCG and reverse 5′- CCAGTTGGTAACAATGCCATGT.
Western blotting
Protein was extracted from frozen kidney tissues using RIPA lysis buffer (P0013B; Beyotime, Nantong, China) with a phosphatase inhibitor tablet (Roche Diagnostics GmbH, Mannheim, Germany), a proteinase inhibitor cocktail tablet (Roche), and 100 mM phenylmethanesulfonyl fluoride (ST506; Beyotime), and the ratio of the listed chemicals to the RIPA lysis buffer was 1:100. Protein concentration was determined using the Bradford protein assay (P0011; Beyotime). After the proteins were separated via SDS-PAGE, they were transferred electrophoretically to PVDF membranes (Millipore, Billerica, MA) and blocked with 5% nonfat milk or BSA. Then, immunoblotting was performed using specific antibodies overnight at 4 °C: rabbit anti-PGIS (1:500; Abcam), anti-PKA (1:1000; CST, Danvers, MA), anti-P-PKA (1:1000; CST), anti-β-actin (1:5000; CST), and anti-α-tubulin (1:8000; Abcam). Then, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse Ig for 1 h at room temperature. Antibody labeling on the Western blots was visualized using a chemiluminescence reagent (WBKLS0100; Millipore) with GE ImageQuant LAS 4000. Densitometry analysis was performed using ImageJ software.
Statistical analyses
All values are expressed as the mean ± SEM. Statistical comparisons were made by Student’s unpaired t test or one-way ANOVA with post hoc analysis. GraphPad Prism 7 and SPSS 24 software were used for all calculations. A two-sided P value of less than 0.05 was considered significant.
Results
Generation of PGIS-floxed mice
To examine the role of PGIS and PGI2 during kidney I/RI, we developed a PGIS-floxed mouse line. The SA-IRES-EGFP fragment and a PGK-Neo selection cassette were inserted between PGIS exon 1 and exon 2 to inactivate the PGIS gene (Fig. 1a). Neo-resistant embryonic cell clones were screened by Southern blot (Fig. 1b). The mouse genotype was identified by PCR (Fig. 1c). To confirm whether the PGIS-floxed mice were successfully generated, we mated the PGISfl/+ mouse with the EIIA-cre mouse to produce conventional PGIS knockouts. The homozygous PGIS knockout mice were embryonic lethal after E16.5 days. We analyzed the expression of PGIS mRNA and PGIS protein in the E16.5 embryos by real-time PCR and Western blot, respectively (Fig. 1d–f). The expression of PGIS mRNA was significantly reduced in heterozygous embryos and was absent in homozygous embryos. Likewise, the PGIS protein was undetectable in homozygous embryos, indicating that the PGIS-floxed mice were successfully established.
PGIS deficiency aggravates I/R-induced AKI
The PGIS expression significantly increased at 48 h after I/R (Fig. 2a). To determine the role of PGIS in kidney injury, studies were performed in the PGIS heterozygous mice. Deletion of one allele of the PGIS gene significantly aggravated renal damage compared with wild-type littermates at 48 h after ischemia, resulting in a higher BUN levels and more severe tubular injury (Fig. 2b, c).

The PGIS-deficient kidney is susceptible to AKI. WT and PGIS+/− mice were subjected to right uninephrectomy and sham surgery or 25 min of left renal ischemia. a The relative mRNA expression of PGIS was measured at the indicated times. A total of 20 mice were included (n = 4 per time point). b BUN concentrations were measured at 48 h after ischemia. A total of 10 mice were included (n = 4 in the WT group and n = 6 in the PGIS+/− group). c Renal tubular injury is indicated by representative periodic acid-Schiff staining of kidney sections at both low and high magnification. The data are expressed as the mean ± SEM and were analyzed by Student’s unpaired t test between two groups and one-way ANOVA among multiple groups followed by Dunnett’s post hoc testing. ***P < 0.001; WT, wild type; CON, control
Role of endothelial PGIS in AKI
By immunofluorescence, PGIS was found colocalized with CD34, an endothelial cell marker. PGIS was expressed in the cytoplasm of endothelial cells both in the peritubular capillaries and the glomeruli (Fig. 3a). To determine the specific role of endothelial PGIS and PGI2 during renal I/RI, we generated a mouse that was deficient in PGIS specifically within the endothelium by mating the floxed PGIS mouse with an endothelial Cre mouse, the TEK-CRE mouse. PGIS expression was significantly decreased both at the mRNA and protein levels in endothelial-specific PGIS-deficient mice (TEK-CRE PGISfl/fl) compared with control mice (Fig. 3b, c). The production of PGI2 was also significantly reduced both in the blood and kidney compared with the control group, as assessed by 6-Keto PGF1α, a stable metabolite of PGI2 (Fig. 3d, e). We also measured TXB2, a stable metabolite of TXA2, and PGE2 in kidneys (Fig. 3f, g). Deleting the PGIS gene in endothelial cells did not influence the expression of other genes in the prostaglandin family.

Prostanoid production in endothelial-specific PGIS-deficient mice. a Costaining of PGIS with endothelial cell marker CD34 in wild-type kidney sections. b Relative mRNA expression level of PGIS in the kidneys of control (CON) and endothelial-specific PGIS-deficient (TEK-CRE PGISfl/fl) mice (P = 0.001). c Representative Western blot image of PGIS protein expression in the kidney. GAPDH was used as a loading control. Band density was measured using ImageJ software (P = 0.003). d 6-Keto PGF1α level in the plasma (P = 0.020). 6-Keto PGF1α (e) (P = 0.001), TXB2 (f), and PGE2 (g) levels in the kidney. The experiments were repeated twice. A total of 12 mice were included (n = 6 per group). The data are expressed as the mean ± SEM and were analyzed by Student’s unpaired t test. *P < 0.05; **P < 0.01; NS, not significant
No developmental abnormalities were observed in the kidneys of TEK-CRE PGISfl/fl mice (Fig. 4). The blood pressure and heart rates were comparable between wild-type mice and TEK-CRE PGISfl/fl mice (Table 1). Following ischemia and reperfusion, the TEK-CRE PGISfl/fl mice had higher BUN levels than control mice (Fig. 5a). The endothelial-specific PGIS knockout also abolished the increase of PGIS expression and PGI2 production after ischemia and reperfusion (Fig. 5b, c). Deletion of the PGIS gene specifically in smooth muscle cells did not worsen the kidney injury compared with wild-type littermates after ischemia and reperfusion (Fig. 5d).

Morphology of the kidneys of wild-type and TEK-CRE PGISfl/fl mice (periodic acid-Schiff stain)

Endothelial PGIS protects the kidney from AKI. The control mice, TEK-CRE PGISfl/fl mice, and SMMHC-iCRE PGISfl/fl mice were subjected to either renal ischemia or a sham operation. a BUN concentrations at the indicated times. A total of eight mice were included (n = 4 per group). b Representative Western blot images of PGIS protein expression in the kidney (in the control group, P = 0.033 Sham vs. I/RI). β-Actin was used as a loading control. Band density was measured using ImageJ software. c The production of 6-Keto PGF1α in the kidneys (in the control group, P = 0.001 Sham vs. I/RI). For b, c, 24 mice were included (CON mice: n = 7 in the Sham group, n = 6 in the I/RI group; TEK-CRE PGISfl/fl mice: n = 5 in the Sham group, n = 6 in the I/RI group). d BUN concentrations were measured at 48 h after ischemia. A total of eight mice were included (n = 4 per group). The data are expressed as the mean ± SEM and were analyzed by Student’s unpaired t test between two groups and one-way ANOVA among multiple groups followed by Scheffe post hoc testing. For a, the data were analyzed by Student’s unpaired t test followed by Bonferroni correction (a two-sided P value less than 0.01 was considered significant). *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant
The PGIS/PGI2/IP receptor signaling pathway is activated during I/R-induced AKI
The IP receptor signals mainly through the cAMP/PKA signaling pathway. Following I/R, the level of phosphorylated PKA was significantly increased in the control kidneys but not in the endothelial-specific PGIS knockout kidneys (Fig. 6), consistent with IP/PKA signaling in PGI2-associated effect on kidney injury. We then examined the role of PGIS/PGI2 in a nephrotoxic AKI induced by folic acid. PGIS deficiency did not worsen renal damage in the folic acid model (Fig. 7).

Representative Western blot images of phosphorylated protein kinase A (p-PKA) (in the control group, P = 0.006 Sham vs. I/RI). α-Tubulin was used as a loading control. Band density was measured using ImageJ software. A total of 24 mice were included (CON mice: n = 7 in the Sham group, n = 6 in the I/RI group; TEK-CRE PGISfl/fl mice: n = 5 in the Sham group, n = 6 in the I/RI group). The data are expressed as the mean ± SEM and were analyzed by one-way ANOVA among multiple groups followed by Scheffe post hoc testing. *P < 0.05; **P < 0.01

PGI2 has no protective effects in a folic acid-induced AKI model. a BUN concentrations at the indicated times. b Renal tubular injury as indicated by representative periodic acid-Schiff staining of kidney sections at 48 h after folic acid treatment. A total of 10 mice were included (n = 5 per group). The data are expressed as the mean ± SEM and were analyzed by Student’s unpaired t test between two groups
Treatment with a PGI2 analog protects the kidney from I/R injury
We further examined whether a PGI2 analog, iloprost, can rescue the effect of knocking out PGIS. Iloprost or PBS was administered 30 min before the surgery. BUN measurement, histology analysis, and TUNEL staining were performed at 48 h after ischemia. As shown in Fig. 8, mice treated with iloprost had a significantly lower BUN and improved tubular damage compared with mice treated with PBS both in the control group and the TEK-CRE PGISfl/fl group, indicating a potential target for AKI treatment.

PGI2 protects kidneys from I/R-induced AKI. a Renal function as evaluated by serum BUN in control and TEK-CRE PGISfl/fl mice at 48 h after ischemia with or without treatment with the PGI2 analog, iloprost (control group: P = 0.001 I/RI + PBS vs. I/RI + Iloprost; TEK-CRE PGISfl/fl group: P = 0.002 I/RI + PBS vs. I/RI + Iloprost). Renal tubular injury as indicated by representative periodic acid-Schiff staining of kidney sections and evaluated based on a pathological score (control group: P = 0.026 I/RI + PBS vs. I/RI + Iloprost; TEK-CRE PGISfl/fl group: P = 0.019 I/RI + PBS vs. I/RI + Iloprost) (b) and TUNEL staining (c). A total of 24 mice were included (CON mice: n = 6 in the I/RI + PBS group, n = 7 in the I/RI + Iloprost group; TEK-CRE PGISfl/fl mice: n = 6 in the I/RI + PBS group, n = 5 in the I/RI + Iloprost group). The data are expressed as the mean ± SEM and were analyzed by one-way ANOVA among multiple groups followed by Scheffe post hoc testing. *P < 0.05; **P < 0.01; ***P < 0.001
Deletion of PGIS gene in endothelial cells reduced renal blood flow after reperfusion
In the kidneys of TEK-CRE PGISfl/fl mice, red blood cells were readily observed in the peritubular capillaries at 24 h after ischemia (Fig. 9a). We then examined renal blood flow using a fluorescent microspheres technique. The fluorescent intensity of the kidney was markedly less in the TEK-CRE PGISfl/fl mice compared with the control mice (Fig. 9b, c).

Renal blood flow after reperfusion. a Electron microscopy images of the renal peritubular capillaries of the indicated groups are shown. Scale bars, 5 μm. b In vivo whole-body imaging of fluorescent microspheres following I/R surgery. c Quantification of the fluorescence intensity. A total of 10 mice were included (n = 6 in the CON group and n = 4 in the TEK-CRE PGISfl/fl group). The data are expressed as the mean ± SEM and were analyzed by Student’s unpaired t test between two groups (P = 0.044). *P < 0.05
Discussion
In the present study, we demonstrated that (1) I/R stimulated the expression of PGIS and PGI2; (2) global PGIS deficiency or endothelial-specific PGIS deletion exacerbated I/R-induced injuries; (3) an exogenous PGI2 analog protected the kidney from I/R-induced injury; and (4) PGI2 deficiency was associated with reduced renal blood flow following reperfusion. These results strongly suggest that endothelium-derived PGI2 plays a critical role in protecting the kidney from I/R injury.
It is well documented that prostanoids play an important role in maintaining local homeostasis [18]. PGIS expression markedly increased following I/R injury, and PGIS deficiency significantly exacerbated kidney I/R injury. These results suggest that PGIS/PGI2 is involved in the cellular response to I/R stress and protects the kidney from injury. COX inhibitors, which are widely used anti-inflammatory drugs and pain relievers, are associated with AKI. COX-derived products include PGE2, PGI2, and TXA2. The present study suggests that PGI2 is an important protective factor, particularly under I/R conditions, and its inhibition may be, at least in part, responsible for COX inhibitor-induced AKI. Importantly, several studies have shown that the ability of the kidney to produce PGI2 decreases with aging [7, 33]. Whether reduced PGI2 production in aged people contributes to their increased susceptibility to AKI remains to be investigated.
For the first time, the present study shows in vivo that endothelial PGI2 plays a critical role in the cellular response to ischemic reperfusion and protects the kidney from injury using our floxed PGIS mice. By costaining PGIS with CD34, an endothelial marker, we show that PGIS is extensively expressed in the endothelial cells of the kidney. Our floxed PGIS mice allowed us to specifically explore the role of endothelial PGIS in renal I/R injury. We selectively deleted the PGIS gene in endothelial cells by crossing the floxed PGIS mice with an endothelial cell Cre mouse (TEK-CRE PGISfl/fl). In an I/R-induced AKI model, the PGIS and PGI2 expression levels significantly increased after ischemia in the control kidneys but not in endothelial PGIS knockout kidneys. Selective deletion of endothelial PGIS markedly worsened the kidney damage following I/R, suggesting a protective role of endothelial PGI2 in the kidney. Deletion of PGIS did not alter the production of TXA2 and PGE2, suggesting that the worsened renal damage was not caused by TXA2 or other prostanoids. Furthermore, administration of iloprost significantly attenuated the renal damage both in the control mice and endothelial PGIS knockout mice, supporting the protective role of PGI2. However, iloprost binds with equal affinity to IP and prostaglandin EP1 receptors and with lower affinity to EP3 receptors. Several studies show that high concentration of PGI2 can cause vasoconstriction under such conditions as NO or IP deficiency, and the vasoconstrictor effect of PGI2 is blocked by TP receptor antagonist [13, 26, 28]. Although these studies are important for us to understand the mechanism underlying hypertension or COX1-mediated vasoconstriction, whether the interaction of PGI2 and TP is involved in the protective effect of PGIS/PGI2 in AKI remains to be explored.
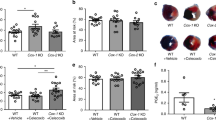
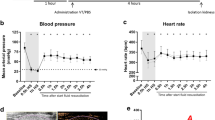
PGI2 has been reported to play an important role in modulating blood pressure by coupling with the IP receptor to relax vascular smooth muscle and control renin release [16, 22]. NSAIDs that inhibit COX and thus endogenous prostanoid synthesis may compromise the control of blood pressure in subjects with pre-existing salt-sensitive hypertension [14, 22]. In the present study, we examined the effect of PGIS deletion on systemic blood pressure. The result showed that the blood pressure of the TEK-CRE PGISfl/fl mice was not significantly different compared with wild-type mice, suggesting that the observed effect in the endothelial PGIS knockout was not mediated via its effect on blood pressure.
In our study, global deletion of the PGIS gene led to embryonic lethality, suggesting that the PGIS product is required for development. Mice with heterozygous PGIS deletion or with selective endothelial PGIS deletion were fertile and developed normally. No ischemic renal disorders were observed in heterozygous PGI2-deficient mice or endothelial PGI2-deficient TEK-CRE PGISfl/fl mice. Therefore, the observed effect of PGI2 deficiency on the kidney following I/R injury does not seem to be caused by abnormal renal development, although subtle developmental abnormality that may cause abnormal response to injury can not be excluded.
The protective effect of PGI2 against cellular stress has been reported [17, 23, 44]. However, the mechanism underlying its protective effect is incompletely understood and may vary according to the injury stresses. Several mechanisms through which PGIS and PGI2 exert their protective effects have been proposed, including anti-inflammatory mechanisms [9, 30, 38, 46], reducing oxidative stress [11, 41], and regulating renal blood flow [45]. It has been reported that prostacyclin and PPAR protect against I/RI by inducing an anti-inflammatory pathway [6]. Although we can show that PGIS deletion is associated with worsened kidney injury in an I/R model, PGIS deletion did not worsen the kidney injury in folic acid-induced AKI, which is a nephrotoxic model. Then, we used fluorescent microspheres to assess the effect of PGI2 on renal blood flow. Deletion of the PGIS gene in endothelial cells significantly reduced fluorescent microsphere accumulation in kidneys after reperfusion. A recent study showed that the restoration of microvascular perfusion is associated with reduced kidney injury [8]. However, this result is inconsistent with published results suggesting that the beneficial effect of PGI2 in renal I/R injury is not related to changes in renal blood flow [15]. The difference between these results may be due to how PGI2 was administered. Continuous infusion may cause hypotension and stimulate renin release, which would then block the reperfusion.
In summary, PGI2 expression was increased after I/R-induced AKI. The PGIS and PGI2 deficits in mouse kidneys increased vulnerability to I/R-induced AKI. Iloprost, an analog of PGI2, rescued the kidney from I/R-induced injury. This study provides evidence suggesting that PGIS-derived PGI2 can protect the kidney from acute injury caused by ischemic reperfusion and could be a potential preventive intervention for AKI.
References
Basile DP, Dwinell MR, Wang SJ, Shames BD, Donohoe DL, Chen S, Sreedharan R, Van Why SK (2013) Chromosome substitution modulates resistance to ischemia reperfusion injury in Brown Norway rats. Kidney Int 83:242–250
Bonventre JV, Weinberg JM (2003) Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14:2199–2210
Bonventre JV, Yang L (2011) Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121:4210–4221
Breyer MD, Breyer RM (2000) Prostaglandin receptors: their role in regulating renal function. Curr Opin Nephrol Hypertens 9:23–29
Breyer MD, Breyer RM (2001) G protein-coupled prostaniod receptors and the kidney. Annu Rev Physiol 63:579–605
Chen HH, Chen TW, Lin H (2009) Prostacyclin-induced peroxisome proliferator-activated receptor-α translocation attenuates NF-κB and TNF-α activation after renal ischemia-reperfusion injury. Am J Physiol Ren Physiol 297:F1109–F1118
Choi JH, Yu BP (1998) Dietary restriction as a modulator of age-related changes in rat kidney prostaglandin production. J Nutr Health Aging 2:167–171
Collett JA, Corridon PR, Mehrotra P, Kolb AL, Rhodes GJ, Miller CA, Molitoris BA, Pennington JG, Sandoval RM, Atkinson SJ, Campos-Bilderback SB, Basile DP, Bacallao RL (2017) Hydrodynamic isotonic fluid delivery ameliorates moderate-to-severe ischemia-reperfusion injury in rat kidneys. J Am Soc Nephrol 28:2081–2092
Dorris SL, Peebles RS Jr (2012) PGI2 as a regulator of inflammatory diseases. Mediat Inflamm 2012:926968
Dreischulte T, Morales DR, Bell S, Guthrie B (2015) Combined use of nonsteroidal anti-inflammatory drugs with diuretics and/or renin–angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney Int 88:396–403
Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, Fitzgerald GA (2004) COX-2-derived prostacyclin confers atheroprotection on female mice. Science 306:1954–1957
El-Haroun H, Clarke DL, Deacon K, Bradbury D, Clayton A, Sutcliffe A, Knox AJ (2008) IL-1 beta, BK, and TGF-beta1 attenuate PGI2-mediated cAMP formation in human pulmonary artery smooth muscle cells by multiple mechanisms involving p38 MAP kinase and PKA. Am J Phys Lung Cell Mol Phys 294:L553–L562
Feletou M, Verbeuren TJ, Vanhoutte PM (2009) Endothelium-dependent contractions in SHR: a tale of prostanoid TP and IP receptors. Br J Pharmacol 156:563–574
Fierro-Carrion GA, Ram CV (1997) Nonsteroidal anti-inflammatory drugs (NSAIDs) and blood pressure. Am J Cardiol 80:775–776
Finn WF, Hak LJ, Grossman SH (1987) Protective effect of prostacyclin on postischemic acute renal failure in the rat. Kidney Int 32:479–487
Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim HS, Rockman HA, Fitzgerald GA, Koller BH, Coffman TM (2005) Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab 2:201–207
Goto Y, Yamaguchi S, Tamura M, Mochizuki H, Kurumatani H, Okano K, Miyamoto M (2014) A prostacyclin analog prevents the regression of renal microvascular network by inhibiting mitochondria-dependent apoptosis in the kidney of rat progressive glomerulonephritis. Prostaglandins Other Lipid Mediat 112:16–26
Hao CM, Breyer MD (2007) Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int 71:1105–1115
Hao CM, Breyer MD (2008) Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol 70:357–377
Harris PC, Torres VE (2014) Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124:2315–2324
Imig JD (2006) Eicosanoids and renal vascular function in diseases. Clin Sci 111:21–34
Jackson EK (1989) Relation between renin release and blood pressure response to nonsteroidal anti-inflammatory drugs in hypertension. Hypertension 14:469–471
Johannes T, Ince C, Klingel K, Unertl KE, Mik EG (2009) Iloprost preserves renal oxygenation and restores kidney function in endotoxemia-related acute renal failure in the rat. Crit Care Med 37:1423–1432
Kam Tao Li P, Burdmann EA, Mehta RL, World Kidney Day Steering Committee (2013) Acute kidney injury: global health alert. J Nephropathol 2(90–97):2013
Lameire N, Van Biesen W, Vanholder R (2005) Acute renal failure. Lancet 365:417–430
Levy JV (1980) Prostacyclin-induced contraction of isolated aortic strips from normal and spontaneously hypertensive rats (SHR). Prostaglandins 19:517–525
Lewington AJ, Cerda J, Mehta RL (2013) Raising awareness of acute kidney injury: a global perspective of a silent killer. Kidney Int 84:457–467
Liu B, Zhang Y, Zhu N, Li H, Luo W, Zhou Y (2013) A vasoconstrictor role for cyclooxygenase-1-mediated prostacyclin synthesis in mouse renal arteries. Am J Physiol Ren Physiol 305:F1315–F1322
Majed BH, Khalil RA (2012) Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev 64:540–582
Müller T, Dürk T, Blumenthal B, Herouy Y, Sorichter S, Grimm M, Panther E, Cicko S, Norgauer J, Idzko M (2010) Iloprost has potent anti-inflammatory properties on human monocyte-derived dendritic cells. Clin Exp Allergy 40:1214–1221
Narumiya S, FitzGerald GA (2001) Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest 108:25–30
Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B (1976) Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature 261:558–560
Okumura M, Imanishi M, Yamashita T, Yamamura Y, Kim S, Iwao H, Tanaka S, Fujii S (2000) Renal production of thromboxane and prostaglandins in a rat model of type 2 diabetes. Life Sci 66:371–377
Oni L, Hawcutt DB, Turner MA, Beresford MW, McWilliam S, Barton C, Park BK, Murray P, Wilm B, Copple I, Floyd R, Peak M, Sharma A, Antoine DJ (2017) Optimising the use of medicines to reduce acute kidney injury in children and babies. Pharmacol Ther 174:55–62
Prieto-García L, Pericacho M, Sancho-Martínez SM, Sánchez Á, Martinez-Salgado C, López-Novoa JM, López-Hernández FJ (2016) Mechanisms of triple whammy acute kidney injury. Pharmacol Ther 167:132–145
Rosner MH, Okusa MD (2006) Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 1:19–32
Smith WL (1992) Prostanoid biosynthesis and mechanisms of action. Am J Phys 263:F181–F191
Strassheim D, Riddle SR, Burke DL, Geraci MW, Stenmark KR (2009) Prostacyclin inhibits IFN-gamma-stimulated cytokine expression by reduced recruitment of p300/CBP to STAT1 in a SOCS-1-independent manner. J Immunol 183:6981–6988
Tompers DM, Labosky PA (2004) Electroporation of murine embryonic stem cells: a step-by-step guide. Stem Cells 22:243–249
Vamecq J, Colet JM, Vanden Eynde JJ, Briand G, Porchet N, Rocchi S (2012) PPARs: interference with Warburg’ effect and clinical anticancer trials. PPAR Res 2012:304760
Yang C, DeMars KM, Alexander JC, Febo M, Candelario-Jalil E (2017) Sustained neurological recovery after stroke in aged rats treated with a novel prostacyclin analog. Stroke 48:1948–1956
Yang L, Xing G, Wang L, Wu Y, Li S, Xu G, He Q, Chen J, Chen M, Liu X, Zhu Z, Yang L, Lian X, Ding F, Li Y, Wang H, Wang J, Wang R, Mei C, Xu J, Li R, Cao J, Zhang L, Wang Y, Xu J, Bao B, Liu B, Chen H, Li S, Zha Y, Luo Q, Chen D, Shen Y, Liao Y, Zhang Z, Wang X, Zhang K, Liu L, Mao P, Guo C, Li J, Wang Z, Bai S, Shi S, Wang Y, Wang J, Liu Z, Wang F, Huang D, Wang S, Ge S, Shen Q, Zhang P, Wu L, Pan M, Zou X, Zhu P, Zhao J, Zhou M, Yang L, Hu W, Wang J, Liu B, Zhang T, Han J, Wen T, Zhao M, Wang H (2015) Acute kidney injury in China: a cross-sectional survey. Lancet 386:1465–1471
Yang Y, Song M, Liu Y, Liu H, Sun L, Peng Y, Liu F, Venkatachalam MA, Dong Z (2016) Renoprotective approaches and strategies in acute kidney injury. Pharmacol Ther 163:58–73
Yano T, Itoh Y, Kubota T, Sendo T, Oishi R (2004) A prostacyclin analog beraprost sodium attenuates radiocontrast media-induced LLC-PK1 cells injury. Kidney Int 65:1654–1663
Yokoyama C, Yabuki T, Shimonishi M, Wada M, Hatae T, Ohkawara S, Takeda J, Kinoshita T, Okabe M, Tanabe T (2002) Prostacyclin-deficient mice develop ischemic renal disorders, including nephrosclerosis and renal infarction. Circulation 106:2397–2403
Zhou W, Hashimoto K, Goleniewska K, O'Neal JF, Ji S, Blackwell TS, Fitzgerald GA, Eqan KM, Geraci MW, Peebles RS Jr (2007) Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol 178:702–710
Zuk A, Bonventre JV (2016) Acute kidney injury. Annu Rev Med 67:293–307
Funding
This study was supported by the National Natural Science Foundation of China (grants 81130075 and 31471101) and 985 project 985 III-YFX0302.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cao, Y., Guan, Y., Xu, YY. et al. Endothelial prostacyclin protects the kidney from ischemia-reperfusion injury. Pflugers Arch - Eur J Physiol 471, 543–555 (2019). https://doi.org/10.1007/s00424-018-2229-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2229-6