
Abstract
Background
Epilepsy, ataxia, sensorineural deafness, and tubulopathy (EAST syndrome) is a rare channelopathy due to KCNJ10 mutations. So far, only mild cerebellar hypoplasia and/or dentate nuclei abnormalities have been reported as major neuroimaging findings in these patients.
Methods
We analyzed the clinical and brain MRI features of two unrelated patients (aged 27 and 23 years) with EAST syndrome carrying novel homozygous frameshift mutations (p.Asn232Glnfs*14and p.Gly275Valfs*7) in KCNJ10, detected by whole exome sequencing.
Results
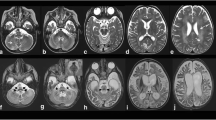
Brain MRI examinations at 8 years in Patient 1 and at 13 years in Patient 2 revealed a peculiar brain and spinal cord involvement characterized by restricted diffusion of globi pallidi, thalami, brainstem, dentate nuclei, and cervical spinal cord in keeping with intramyelinic edema. The follow-up studies, performed, respectively, after 19 and 10 years, showed mild cerebellar atrophy and slight progression of the brain and spinal cord T2 signal abnormalities with increase of the restricted diffusion in the affected regions.
Conclusion
The present cases harboring novel homozygous frameshift mutations in KCNJ10 expand the spectrum of brain abnormalities in EAST syndrome, including mild cerebellar atrophy and intramyelinic edema, resulting from abnormal function of the Kir4.1 inwardly rectifying potassium channel at the astrocyte endfeet, with disruption of water-ion homeostasis.


Similar content being viewed by others

References
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360:1960–1970
Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP (2009) Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci 106:5842–5847
Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366
Sicca F, Imbrici P, D’Adamo MC, Moro F, Bonatti F, Brovedani P, Grottesi A, Guerrini R, Masi G, Santorelli FM, Pessia M (2011) Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K + channel Kir4.1. Neurobiol Dis 43:239–247
Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y (2001) An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol 281:C922–C931
Sala-Rabanal M, Kucheryavykh LY, Skatchkov SN, Eaton MJ, Nichols CG (2010) Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10). J Biol Chem 285:36040–36048
Tanemoto M, Abe T, Uchida S, Kawahara K (2014) Mislocalization of K + channels causes the renal salt wasting in EAST/SeSAME syndrome. FEBS Lett 588:899–905
Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML (2016) The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol 132:1–21
van der Knaap MS, Bugiani M (2017) Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 134:351–382
Bugiani M, van der Knaap MS (2017) Childhood white matter disorders: much more than just diseases of myelin. Acta Neuropathol 134:329–330
Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P (2001) Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci 21:5429–5438
Djukic B, Casper KB, Philpot BD, Chin L-S, McCarthy KD (2007) Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27:11354–11365
Cross JH, Arora R, Heckemann RA, Gunny R, Chong K, Carr L, Baldeweg T, Differ A-M, Lench N, Varadkar S, Sirimanna T, Wassmer E, Hulton SA, Ognjanovic M, Ramesh V, Feather S, Kleta R, Hammers A, Bockenhauer D (2013) Neurological features of epilepsy, ataxia, sensorineural deafness, tubulopathy syndrome. Dev Med Child Neurol 55:846–856
Freudenthal B, Kulaveerasingam D, Lingappa L, Shah MA, Brueton L, Wassmer E, Ognjanovic M, Dorison N, Reichold M, Bockenhauer D, Kleta R, Zdebik AA (2011) KCNJ10 mutations disrupt function in patients with EAST syndrome. Nephron Physiol 119:p40–p48
Parrock S, Hussain S, Issler N, Differ A-M, Lench N, Guarino S, Oosterveld MJS, Keijzer-Veen M, Brilstra E, van Wieringen H, Konijnenberg AY, Amin-Rasip S, Dumitriu S, Klootwijk E, Knoers N, Bockenhauer D, Kleta R, Zdebik AA (2013) KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol 123:7–14
Abdelhadi O, Iancu D, Stanescu H, Kleta R, Bockenhauer D (2016) EAST syndrome: clinical, pathophysiological, and genetic aspects of mutations in KCNJ10. Rare Dis 4:e1195043
Benfenati V, Ferroni S (2010) Water transport between CNS compartments: functional and molecular interactions between aquaporins and ion channels. Neuroscience 168:926–940
Rash JE (2010) Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168:982–1008
Dubey M, Bugiani M, Ridder MC, Postma NL, Brouwers E, Polder E, Jacobs JG, Baayen JC, Klooster J, Kamermans M, Aardse R, de Kock CPJ, Dekker MP, van Weering JRT, Heine VM, Abbink TEM, Scheper GC, Boor I, Lodder JC, Mansvelder HD, van der Knaap MS (2015) Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Ann Neurol 77:114–131
Bugiani M, Dubey M, Breur M, Postma NL, Dekker MP, Ter Braak T, Boschert U, Abbink TEM, Mansvelder HD, Min R, van Weering JRT, van der Knaap MS (2017) Megalencephalic leukoencephalopathy with cysts: the Glialcam-null mouse model. Ann Clin Transl Neurol 4:450–465
Blanz J, Schweizer M, Auberson M, Maier H, Muenscher A, Hubner CA, Jentsch TJ (2007) Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci 27:6581–6589
van der Knaap MS, Boor I, Estévez R (2012) Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol 11:973–985
Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, van Berkel C, Polder E, Tollard E, Darios F, Brice A, de Die-Smulders CE, Vles JS, Vanderver A, Uziel G, Yalcinkaya C, Frints SG, Kalscheuer VM, Klooster J, Kamermans M, Abbink TE, Wolf NI, Sedel F, van der Knaap MS (2013) Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 12:659–668
Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL (2006) Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci 26:10984–10991
Acknowledgements
Patient samples were obtained from the “Cell Line and DNA Biobank from patients affected by Genetic Diseases” (Istituto Giannina Gaslini), member of Telethon Network of Genetic Biobanks (project no. GTB12001). This work was partially supported by unrestricted grants from “Cinque per mille e Ricerca Corrente, “Ministero della Salute” to MF and SL.
Author information
Authors and Affiliations
Contributions
MS: conception and design of the work, analysis and interpretation of brain MRI data. Drafting of manuscript, tables and figures. SL: acquisition, analysis and interpretation of mutation analysis data. Drafting of manuscript, tables and figures. CF: conception and design of the work. Collection and interpretation of patient 1 clinical data. Drafting the manuscript and revising critically for important intellectual content. PS: Collection and interpretation of patient 1 clinical data. Drafting the manuscript and revising critically for important intellectual content. TT: Collection and interpretation of patient clinical data. SP: Collection and interpretation of patient 2 clinical and neuroimaging data. GM: Drafting the manuscript and revising critically for important intellectual content. AR: acquisition of brain MRI data. Drafting the manuscript and revising critically. MF: acquisition of genetic results. Drafting the manuscript and revising critically for important intellectual content. CB: Drafting the manuscript and revising critically.
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical statement
No ethic committee approval is required for this retrospective study report.
Electronic supplementary material
Below is the link to the electronic supplementary material.
415_2018_8826_MOESM2_ESM.tiff
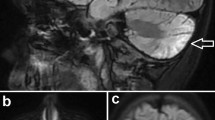
Online Figure 2: Brain MRI and MR spectroscopy of Patient 1 at the age of 27 years (A-C) and of Patient 2 at the age of 23 years (D-F). Coronal (A, D) and sagittal (B, E) T2-weighted images reveal high signal in the upper cervical spinal cord (arrows). The brainstem is slightly smaller, and the corpus callosum is thin (thick arrows). Note reduction of the vermian volume (arrowheads). (C, E) Proton MR spectroscopy of the brainstem shows slightly reduced NAA in Patient 1 (empty arrow) and absence of lactate peaks in both patients
415_2018_8826_MOESM3_ESM.tiff
Online Figure 3: Brain MRI examinations performed at 8 years of age (A) and 28 years of age (B, C) in Patient 1. The first MRI reveals normal cerebellar volume (thick arrows, A), while the follow-up study demonstrates mild and slowly progressive cerebellar atrophy, more pronounced at the level of superior cerebellar hemispheres (thick arrows, B). Note the evolution of the T2-hyperintensity changes at the level of the globi pallidi (arrows) and thalami (arrowheads).
415_2018_8826_MOESM4_ESM.tiff
Online Figure 4: Brain MRI examinations performed at 13 years of age (A) and 23 years of age (B, C) in Patient 2. The first MRI reveals normal cerebellar volume (thick arrows, A), while the follow-up study demonstrates mild and slowly progressive cerebellar atrophy, more pronounced at the level of superior cerebellar hemispheres (thick arrows, B). Note the evolution of the T2-hyperintensity changes at the level of the midbrain (arrows) and globi pallidi (arrowheads).
Rights and permissions
About this article

Cite this article
Severino, M., Lualdi, S., Fiorillo, C. et al. Unusual white matter involvement in EAST syndrome associated with novel KCNJ10 mutations. J Neurol 265, 1419–1425 (2018). https://doi.org/10.1007/s00415-018-8826-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-8826-7