
Abstract
The diagnosis of rare inherited diseases is becoming more and more complex as an increasing number of clinical conditions appear to be genetically heterogeneous. Multigenic inheritance also applies to the autosomal recessive progressive cerebellar ataxias (ARCAs), for which 14 genes have been identified and more are expected to be discovered. We used homozygosity mapping as a guide for identification of the defective locus in patients with ARCA born from consanguineous parents. Patients from 97 families were analyzed with GeneChip Mapping 10K or 50K SNP Affymetrix microarrays. We identified six families homozygous for regions containing the autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) gene, two families homozygous for the ataxia-telangiectasia gene (ATM), two families homozygous for the ataxia with oculomotor apraxia type 1 (AOA1) gene, and one family homozygous for the AOA type 2 (AOA2) gene. Upon direct gene testing, we were able to identify a disease-related mutation in all families but one of the two kindred homozygous at the ATM locus. Although linkage analyses pointed to a single locus on chromosome 11q22.1-q23.1 for this family, clinical features, normal levels of serum alpha-foetoprotein as well as absence of mutations in the ATM gene rather suggest the existence of an additional ARCA-related gene in that interval. While the use of homozygosity mapping was very effective at pointing to the correct gene, it also suggests that the majority of patients harbor mutations either in the genes of the rare forms of ARCA or in genes yet to be identified.



Similar content being viewed by others
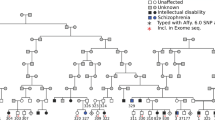

References
Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G, Ginglinger E, Boulay C, Courtois S, Drouot N, Fritsch M, Delaunoy JP, Stoppa-Lyonnet D, Tranchant C, Koenig M (2010) Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics 11:1–12
Assoum M (2010) Rundataxin, a novel protein with RUN and diacylglycerol binding domains, is mutant in a new recessive ataxia. Brain 133:2439–2447
Barbot C, Coutinho P, Chorao R, Ferreira C, Barros J, Fineza I, Dias K, Monteiro J, Guimaraes A, Mendonca P, do Ceu Moreira M, Sequeiros J (2001) Recessive ataxia with ocular apraxia: review of 22 Portuguese patients. Arch Neurol 58:201–205
Ben Hamida M, Belal S, Sirugo G, Ben Hamida C, Panayides K, Ionannou P, Beckmann J, Mandel JL, Hentati F, Koenig M et al (1993) Friedreich’s ataxia phenotype not linked to chromosome 9 and associated with selective autosomal recessive vitamin E deficiency in two inbred Tunisian families. Neurology 43:2179–2183
Bouhlal Y, El-Euch-Fayeche G, Amouri R, Hentati F (2005) Distinct phenotypes within autosomal recessive ataxias not linked to already known loci. Acta Myol 24:155–161
Bouhlal Y, El Euch-Fayeche G, Hentati F, Amouri R (2009) A novel SACS gene mutation in a Tunisian family. J Mol Neurosci 39:333–336
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427
Criscuolo C, Mancini P, Menchise V, Sacca F, De Michele G, Banfi S, Filla A (2005) Very late onset in ataxia oculomotor apraxia type I. Ann Neurol 57:777
De Michele G, Coppola G, Cocozza S, Filla A (2004) A pathogenetic classification of hereditary ataxias: is the time ripe? J Neurol 251:913–922
Dupre N, Bouchard JP, Gros-Louis F, Rouleau GA (2007) Mutations in SYNE-1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Med Sci (Paris) 23:261–262
Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, Bouchard JP, Mathieu J, Melancon SB, Schalling M, Lander ES, Morgan K, Hudson TJ, Richter A (2000) ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 24:120–125
Fogel BL, Perlman S (2007) Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 6:245–257
Garcia A, Criscuolo C, de Michele G, Berciano J (2008) Neurophysiological study in a Spanish family with recessive spastic ataxia of Charlevoix-Saguenay. Muscle Nerve 37:107–110
Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, Sanes JR, Bouchard JP, Rouleau GA (2007) Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet 39:80–85
Lagier-Tourenne C, Tazir M, Lopez LC, Quinzii CM, Assoum M, Drouot N, Busso C, Makri S, Ali-Pacha L, Benhassine T, Anheim M, Lynch DR, Thibault C, Plewniak F, Bianchetti L, Tranchant C, Poch O, DiMauro S, Mandel JL, Barros MH, Hirano M, Koenig M (2008) ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet 82:661–672
Lagier-Tourenne C, Tranebaerg L, Chaigne D, Gribaa M, Dollfus H, Silvestri G, Betard C, Warter JM, Koenig M (2003) Homozygosity mapping of Marinesco-Sjogren syndrome to 5q31. Eur J Hum Genet 11:770–778
Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, Pandolfo M, Schulz JB, Pouget J, Calvas P, Shizuka-Ikeda M, Shoji M, Tanaka M, Izatt L, Shaw CE, M’Zahem A, Dunne E, Bomont P, Benhassine T, Bouslam N, Stevanin G, Brice A, Guimaraes J, Mendonca P, Barbot C, Coutinho P, Sequeiros J, Durr A, Warter JM, Koenig M (2004) Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 36:225–227
Nikali K, Suomalainen A, Saharinen J, Kuokkanen M, Spelbrink JN, Lonnqvist T, Peltonen L (2005) Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet 14:2981–2990
Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, Sokol R, Arai H, Inoue K, Mandel JL, Koenig M (1995) Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet 9:141–145
Ouyang Y, Takiyama Y, Sakoe K, Shimazaki H, Ogawa T, Nagano S, Yamamoto Y, Nakano I (2006) Sacsin-related ataxia (ARSACS): expanding the genotype upstream from the gigantic exon. Neurology 66:1103–1104
Savitsky K, Sfez S, Tagle DA, Ziv Y, Sartiel A, Collins FS, Shiloh Y, Rotman G (1995) The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum Mol Genet 4:2025–2032
Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG, Haites N, Byrd PJ, Taylor AM (1998) ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet 62:334–345
Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99:577–587
Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, Badano JL, Leitch CC, Salem N, Chouery E, Corbani S, Jalk N, Vicaire S, Sarda P, Hamel C, Lacombe D, Holder M, Odent S, Holder S, Brooks AS, Elcioglu NH, Silva ED, Rossillion B, Sigaudy S, de Ravel TJ, Lewis RA, Leheup B, Verloes A, Amati-Bonneau P, Megarbane A, Poch O, Bonneau D, Beales PL, Mandel JL, Katsanis N, Dollfus H (2006) BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet 38:521–524
Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, Vicaire S, Jacquelin C, Plewniak F, Leitch CC, Sarda P, Hamel C, de Ravel TJ, Lewis RA, Friederich E, Thibault C, Danse JM, Verloes A, Bonneau D, Katsanis N, Poch O, Mandel JL, Dollfus H (2007) Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet 80:1–11
Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR (2002) Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32:267–272
Taylor AM, Byrd PJ (2005) Molecular pathology of ataxia telangiectasia. J Clin Pathol 58:1009–1015
Van Goethem G, Martin JJ, Dermaut B, Lofgren A, Wibail A, Ververken D, Tack P, Dehaene I, Van Zandijcke M, Moonen M, Ceuterick C, De Jonghe P, Van Broeckhoven C (2003) Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord 13:133–142
Acknowledgments
We wish to thank Catherine Dubois d’Enghien for ATM gene sequencing and Mustafa A. Salih for sharing material and information on the families from Saudi Arabia. This study was financially supported by funds from the Institut National de la Santé et de la Recherche Scientifique (INSERM), the Centre National de la Recherche Scientifique (CNRS), and the Agence Nationale pour la Recherche-Maladies Rares (ANR-05-MRAR-013-01) to M.K., and the E-Rare EUROSPA network (to F.M.S.). D.H–B.B. was supported by the French association “Connaître les Syndromes Cérébelleux”; M.A. is supported by a BDI fellowship from CNRS; F.F. is a fellow of the Roma Tre University-IRCCS Bambino Gesù Hospital joint PhD programme.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
H’mida-Ben Brahim, D., M’zahem, A., Assoum, M. et al. Molecular diagnosis of known recessive ataxias by homozygosity mapping with SNP arrays. J Neurol 258, 56–67 (2011). https://doi.org/10.1007/s00415-010-5682-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-010-5682-5