
Abstract
The high-order structure of metaphase chromosomes remains still under investigation, especially the 30-nm structure that is still controversial. Advanced 3D imaging has provided useful information for our understanding of this detailed structure. It is evident that new technologies together with improved sample preparations and image analyses should be adequately combined. This mini review highlights 3D imaging used for chromosome analysis so far with future imaging directions also highlighted.
Similar content being viewed by others

Chromosome structure
The DNA in chromosomes holds the genomic information of all eukaryotes. Throughout the cell cycle progression, chromosomes condense into the higher-order compact structures until they reach mitosis. The biological implications of this chromosome higher-order structure is for packaging the long DNA molecules into chromatin fibers within chromosomes facilitating upon cell division, the separation of the chromatids for transportation of DNA fibers evenly to two new daughter cells without damage (Sumner 2003). The higher-order structure of mitotic chromosomes has been under investigation for more than three centuries with the full structural details still remaining unknown. Our current understanding is that the 2-nm-thick DNA with 146 base pairs (bp) wraps twice around an octamer of histone proteins forming a series of nucleosomes. Chromatin is defined as complexes of nucleosomes and their associated proteins (Alberts et al. 2002). The X-ray crystal structure of the nucleosome has been resolved down to Angstrom resolutions (Luger et al. 1997; Davey et al. 2002) at 3.1 Å and tetranucleosome structure at 9 Å (Arents et al. 1991; Schalch et al. 2005). Recently, Ekundayo et al. published tetranucleosome structures at 5.8 and 6.7 Å (Ekundo et al. 2017). These nucleosomes form a “bead on a string” like structure having 11 nm diameter (Olins and Olins 1974). The binding of the linker histone (H1 or H5) organizes the nucleosome arrays into a more condensed 30-nm chromatin fiber (Robinson et al. 2006). Linker histone (e.g., H1 or H5) bound to a single nucleosome is known as a chromatosome and have shown to be involved in the 30-nm structure that is compacted further into a mitotic chromosome.
The 30-nm chromatin fibers have frequently been observed by scanning electron microscopy (SEM) (Inaga et al. 2008); several models for the folding of the nucleosomes have been proposed for the 30-nm fiber. However, many studies now even question the existence of this structure especially in vivo (Maeshima et al. 2010; Nishino et al. 2012a; Fussner et al. 2011). Many models have been proposed since the discovery of the one-start helix/solenoid model. This model was proposed after purifying the 10-nm fiber with a low concentration of cations that showed linear arrangement of the nucleosomes stacked with their neighbors (Finch and Klug 1976). The other model proposed is the zigzag or two-start cross-linker model where the nucleosomes are not stacked with their neighbors but zigzag back and forth to form a two-start stack of nucleosomes (Woodcock et al. 1984; Dorigo 2004). EM studies led onto a two-start flat ribbon model that had about 5 nucleosomes per 11-nm length (Dorigo 2004). A zigzag conformation was proposed after crystallizing tetranucleosomes and performing X-ray crystallography that led onto a model having a twisted ribbon with 24–25-nm approximated diameter and a compaction density of five to six nucleosomes per 11-nm (Schalch et al. 2005). Further EM studies including cryo-EM supported the one-start solenoid model that showed 30-nm measurements. The study showed that the linker histone H5 and the length of different nucleosome repeats had an effect on chromatin fiber architecture (Robinson et al. 2006). Recently, a cryo-EM study using 12 × 187 and 12 × 177 bp nucleosomes with reconstituted fibers in the presence of histone H1 has also shown repeating tetra-nucleosomal structural units supported the zigzag two-start helix model (Song et al. 2014). Scanning transmission electron microscopy (STEM) has been used on intact chromosomes showing chromatin fibers with a fibrous structure of 20 to 30-nm (Fukui 2016). With a large number of studies performed, the exact structure of the 30-nm fiber still remains unclear (Maeshima et al. 2016a). Most of these studies have been done in vitro and the existence of the 30-nm structure in vivo is debated. Several studies that do not support the 30-nm fiber existence (Maeshima et al. 2016a) include the small angle X-ray scattering (SAXS) studies done in solution that supports the 10-nm fiber (Joti et al. 2012; Nishino et al. 2012b). The disordered packing of 10-nm fibers has been suggested to be because of the face-to-face and edge-to-edge nucleosome–nucleosome interactions. This is known as the “polymer-melt” structure whereby the nucleosome fibers have been suggested to be moving and rearranging at the local level (Nishino et al. 2012b; Maeshima et al. 2014). Cryo-EM of frozen hydrated sections (close to native state) of chromosomes and nuclei has shown that the 30-nm exists in samples prepared after aldehyde fixation but not after cryo-preservation (Eltsov et al. 2014). Using electron spectroscopic imaging (ESI) with electron tomography showed 10-nm fibers in mouse euchromatic and heterochromatic regions (Fussner et al. 2012). Heterogeneous groups of nucleosomes called “nucleosome clutches” were visualized using super-resolution microscopy using mouse cells in situ and showed 10-nm fibers (Ricci et al. 2015). ChromEM that is a multitilt EM tomography and a labelling method that enhances the contrast of DNA showed that nucleosomes are organized into disordered chains. These have diameters ranging between 5 and 24-nm, highlighting that the 10-nm fiber is heterogeneous and varies in diameter (Ou et al. 2017). The 30-nm fiber structure formation is dependent on the condition the samples are prepared (Maeshima et al. 2016a). Divalent cations such as magnesium and calcium are involved in the chromatin organization and decondensation process (Dwiranti et al. 2016; Maeshima et al. 2016b; Phengchat et al. 2016). Compared to interphase nuclei, these cations are highly present in mitotic chromosomes. Lower concentrations of magnesium display the bead on a string like structure of 10-nm, but at higher concentrations, helical structures around ~ 30–40 nm in diameter have been observed (Dwiranti et al. 2016; Maeshima et al. 2018).
Proteins play an important role in the compaction of mitotic chromosome with over 158 proteins identified on chromosomes including the histones (Uchiyama et al. 2005; Ohta et al. 2010). The backbone structure of chromosomes after depletion of histone proteins is known as the chromosome scaffold (Paulson and Laemmli 1977). This consists of non-histone proteins, so-called scaffold proteins, which include condensin, topoisomerase IIα (Topo IIα), and kinesin family member 4 (Uchiyama et al. 2005; Earnshaw et al. 1985; Maeshima 2003; Samejima et al. 2012).
To fully understand the inner structure of the chromosome, both the chromatin fiber and the chromosome scaffold need to be understood (Fukui 2016). Different scientific approaches that have contributed hugely towards understanding how chromosomes are folded, compacted, and organized have been employed. Chromosome conformation capture techniques such as Hi C have been recently used to understand the contacts between chromatin fibers (Gibcus et al. 2018; Nagano et al. 2013; Naumova et al. 2013). Great efforts have also been made towards imaging chromosomes using different microscopy methods (Fukui 2016; Pollard and Earnshaw 2002; Flors and Earnshaw 2011). It is clear that three dimensional (3D) imaging is needed to study the intact mitotic chromosome. In this mini-review, we discuss the 3D imaging approaches used for investigating the higher-order chromosome structure. The limitations and future directions are also discussed.
Chromosome sample preparation procedures
Mammalian or animal chromosomes are generally prepared from live cells after growing in cell culture (Howe et al. 2014). At metaphase, they are at the most compact state making them easier for isolation and analysis. Cells are treated with a mitotic inhibitor, e.g., colcemid or nocodazole (Moralli et al. 2011) that disrupts the spindle fibers. After treatment with a hypotonic solution such as KCl that swells the cellular volume, the sample is then fixed using methanol-acetic acid (MAA) that preserves the chromosomes (Tobias et al. 2011) but causes partial denaturation and precipitation of nucleic acid and proteins (Shihab 2012). Chromosomes can also be prepared using polyamine buffer containing spermine and spermidine that are stable in-solution and have been used for a number of applications (Yusuf et al. 2014a; Kannan and Zilfalil 2009; Takata et al. 2007).
3D imaging of chromosomes
Chromosome size, number, and morphology have been well characterized using standard 2D optical microscopy (Tobias et al. 2011). Banding methods allow investigation of structural alterations and numerical abnormalities (Kannan and Zilfalil 2009). G-banding that distinguishes the states of chromatin being heterochromatin and euchromatin can be identified showing as dark and light bands on the chromosome respectively (Tobias et al. 2011). The use of fluorescent dyes further allowed genes to be localized using fluorescence in situ hybridization (FISH), (Volpi and Bridger 2008). Determination of the higher-order mitotic chromosome structure using the optical microscope is not possible as the resolution is limited to the diffraction limit of light (~ 200-nm), making it difficult to resolve the smaller structures (Abbe 1873). This has made it difficult to study chromatin live cell structures at the nanoscale (Mora-Bermúdez and Ellenberg 2007). It has been reported that enhanced resolution can be beyond the Abbe diffraction limit of light (200-nm) with wavelength-scale solid immersion lenses (Mason et al. 2010) and through the assistance of InSb thin layers (Ding et al. 2016). Live cell studies use GFP to label the proteins of interest such as specific histones (Kanda et al. 1998) have been hindered over the years due to absence of methods for labelling of specific DNA sequences in morphologically intact chromosomes (Struckov and Belmont 2009). Recent studies have shown live cell imaging of a single labelled chromosome using CRISPR/Cas 9 technology (Zhou et al. 2017). This technology has also been used to multicolor label specific chromosomal loci in live cells (Ma et al. 2015).
Ultra-high-resolution microscopy has been used to investigate mitotic chromosomes at high resolution. Even though the transmission electron microscope (TEM) has Angstrom resolution, it is not a suitable microscopy for studying intact mitotic chromosomes because chromosomes are too thick for the electrons to penetrate through biological samples over 1 micron thick (Ris 1981). Whole human chromosomes have been imaged and reconstructed in 3D by TEM tomography after the sample was chemically prepared (Engelhardt 2000; Harauz et al. 1987). Forty-one projection images were obtained after tilting the sample from − 60° to + 60° to reconstruct. This led onto the reconstruction of a 3D chromosome in which internal structure showed 26 to 58-nm features and clearly showed distribution of 30-nm fiber that is consistent with the published looping model (Engelhardt 2000).
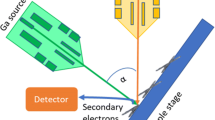
Scanning electron microscopy (SEM) images the sample surface by scanning it with a focused electron beam and collection of backscattered electrons. The major limitation has been due to sample preparation as MAA chromosomes show an artificial surface “skin” layer over the chromosomes following air-drying (Shemilt et al. 2014). A “drop-cryo” method was developed for barley chromosomes that allowed routine investigation of the surface structure information. On the arms of the chromosomes, chromomeres of around 200–300-nm were observed that had a knot-like structure representing highly condensed regions while the centromeres showed parallel fibers (Wanner and Formanek 2000; Iwano et al. 1997). Removal of nucleoplasm from the surface of MAA human chromosomes has been investigated after treating the chromosomes using a commercial enzyme product, cytoclear (Shemilt et al. 2014).
Barley chromosome structure has been investigated after focused ion beam SEM (FIB-SEM) (Schroeder-Reiter et al. 2009). After performing the drop-cryo method, the samples were critical-point dried. Platinum blue staining was performed, and the phosphorylated histone H3 (serine 10) was labelled with immunogold. Slices were taken using a Ga+ beam from the sample surface after it was tilted 54°. The dissected chromosome showed many cavities within the cross section. A full 3D-reconstructed chromosome was achieved from a series of 198 cross sections at resolutions between 1.5 and 3-nm but blurred by a labelling diameter between 15 and 30-nm (see Fig. 1) (Schroeder-Reiter et al. 2009). This method has a great advantage to obtain images with high resolution brought from direct observation of exposed inner structure; however, sample damage caused by accelerated Ga+ beam cannot be ignored.

Reconstruction of a Barley chromosome after 3D FIB-SEM. 198 sections were aligned after imaging. The chromosome was immunogold labeled with phosphorylated histone H3 (ser 10) (yellow) (Schroeder-Reiter et al. 2009).
Serial block face SEM (SBF-SEM) allows 3D information and is similar to FIB-SEM but uses a diamond knife instead of focused G+ ions to cut the sample (Denk and Horstmann 2004). The sample is first embedded into a resin and dissected by ultra-microtome within the microscopy chamber, allowing serial inner structure images by SEM. Human chromosomes have been investigated using this method that allowed full 3D images of polyamine preparations (see Fig. 2) (Yusuf et al. 2014b). SBF-SEM has been used on a human prophase nucleus where 36 out of 46 were analyzed. Chromosomes showed porous network structure after MAA treatment and platinum blue staining. With a resolution of around 50-nm in 3D, the internal structure could not be determined; however, chromosomes showed parallel arrangement of chromatids. Sister chromatids showed curved cylindrical shapes with a well-conserved diameter of around 765 nm that were 2 to 3 μm long (Chen et al. 2017). A correlative approach known as 3D combined light microscopy and serial block face-scanning electron microscopy (CLEM) was used to study mitotic chromosomes. After analysis of wild-type and Ki-67-depleted chromosomes, it was shown that the periphery was 30–47% of the entire chromosome volume with more than 33% of the protein mass of isolated mitotic chromosomes determined by quantitative proteomics. This study concluded that the chromatin made a small percentage of the total mass of metaphase chromosomes (Booth et al. 2016). SBF-SEM and FIB-SEM both involve serial cutting of the sample that destroys the original sample. Both the diamond knife and Ga+ beam can leave an amorphous layer after sectioning and also leave damage.

3D SBF-SEM of human mitotic chromosome. X-shaped human reconstructed after 13-nm × 100-nm sections. Box size 4.253 × 3.741 × 1.6 μm (extracted from (Yusuf et al. 2014b))
In contrast to TEM, X-rays have the ability to penetrate through whole intact chromosomes. Imaging intact chromosomes is advantageous and simplifies the procedure saving time for sample preparation. Coherent diffraction imaging (CDI) using X-ray diffraction was achieved on intact mitotic chromosomes in both 2D and 3D (Nishino et al. 2009). The sample preparation involved fixing intact chromosomes onto thin silicon membranes. Another huge advantage is that this study did not stain the sample with any heavy metals. The 2D resolution achieved was 38-nm. 3D was achieved after tilting the sample to different angles from − 70° to + 70° at intervals of 2.5° or 5°. For 3D data analysis, coherent diffraction data sets at 38 incident angles were used resulting in 120-nm spatial resolution in 3D. The study showed not only the surface of the chromosome but also high electron density around the centromere and chromosome axis. No significant internal structure was observed (see Fig. 3) (Nishino et al. 2009). This method has a disadvantage because the spatial resolution is often limited by the X-ray radiation damage and/or by statistical precision at high angles (Nishino et al. 2009).

X-ray diffraction of unstained human chromosome with reconstructed projection image. a Coherent diffraction pattern of an human chromosome. Chromosome image b in gray scale with arrow indicating the centromere region. and Chromosome image c in color scale showing a high-intensity region on the chromatids (extracted from (Nishino et al. 2009))
Atomic force microscopy (AFM) may be another strong tool to visualize 3D chromosome structure. AFM gives us images which are similar to those obtained using SEM by scanning the solid sample surface with a sharp probe tip and detecting the interaction force between the tip and the sample to get height information. From this principle, AFM does not require metal coating and the observation can be performed either in vacuum, air, or liquid condition (Ushiki et al. 1996, 2008). Human chromosomes isolated by hexylene glycol method were dropped onto a glass slide and observed in hexylene glycol buffer. No fixation was performed. The obtained images showed 400–800-nm thick chromosomes whose surface was covered with globular or fibrous structures with about 50-nm in thickness (Hoshi et al. 2006).
3D structured illumination microscopy (SIM), a super-resolution technique, has been performed on mitotic chromosomes (Carlton 2008). SIM allows us to get higher-resolution images by illuminating a sample with a striped pattern of visible light, which is rotated and scanned to give a number of images, obtained in different phases and direction conditions. After processing, the image resolution is said to reach approximately 120-nm (lateral), being twice the optical diffraction limit (Schermelleh et al. 2008). It has been shown that the axial distributions of scaffold proteins in metaphase chromatids are composed of two twisted double strands. It was suggested that this allows both chromosomal bending/flexibility and rigidity to occur (Fig. 4) (Poonperm et al. 2015).

Imaging of metaphase chromosomes investigating double strands after immunostaining for hCAP-E and Topo IIα. a Wide-field microscopy after applying deconvolution (DC) of a immunostained chromosome shows difficulty in imaging the double strands. b 3D-SIM images of the same chromosome as a. Arrowheads show double stands. a, b Scale bars 1 μm. Insets A and B that are magnified views of a and b (white box) and clearly show double strands on the length of the chromosome that represents the chromosome scaffold (red dotted line). Scale bar for A and B insets is 250 nm (extracted from (Poonperm et al. 2015))
Future direction
Despite the impressive nature of imaging methods mentioned above, the detailed higher-order structure is still under debate. Optical super-resolution microscopy techniques are under development and present an opportunity to study the structure of the chromosomes below the diffraction limit. Such methods should allow the study of live cells that has not been possible with any previous high-resolution method and with development in 3D. Optical super-resolution techniques are already being applied to study chromatin contacts (Wang et al. 2017) and will prove useful for studying chromosomes. This technique is limited to the number of dyes used and the thickness of the sample. STEM allows visualization of 3D structures without sectioning the sample and does not require staining. The limitation is in the z-direction thickness (Fukui 2016).
3D studies done to date have tended to require harsh chemical fixation, dehydration, and drying steps that may not represent the true nature of the sample under investigation. Cryo-electron microscopy enables us to observe biological samples close to the native state (Eltsov et al. 2008). Chromosome samples are rapidly cooled down to cryogenic temperature that allows samples to be embedded in vitreous ice, which appears to be featureless. In principle, there is no morphology destruction caused by water volume expansion. Development of new technologies that will maintain the chromosome condition as close to its native state would be useful to obtain information of chromosome surface and interior details. Such methods would include 3D Cryo TEM, SEM, or X-ray imaging. To date, there is no report showing 3D metaphase chromosomes imaged with such techniques but these are under development. 3D Cryo FIB-SEM and Cryo 3D CDI have already been performed on cells (Schertel et al. 2013; Rodriguez et al. 2015), and Cryo X-ray Ptychographic imaging has been attempted on nuclei (Yusuf et al. 2017). Super resolution microscopy is proven promising for investigation chromatin structure (Sydor et al. 2015). Photoactivated localization microscopy (PALM) together with single molecule tracking has been applied to study chromatin dynamics in live cells that showed nucleosome domains during cell cycle (Nozaki et al. 2017). PALM has also been used to image Drosophila mitotic chromosomes at high resolution (∼ 30 nm) after labelling with H2AvD-EGFP, a histone H2A variant in the presence of background (Matsuda et al. 2010). Using new and advanced 3D imaging tools, the higher-order chromosome structure enigma should be resolved after a century of research.
References
Abbe E (1873) Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch Mikrosk Anat 9:413–418
Alberts B, Johnson A, Lewis J et al (2002) Molecular biology of the cell, 4th edn. Garland Science. Chromosomal DNA and Its Packaging in the Chromatin Fiber, New York Available from: https://www.ncbi.nlm.nih.gov/books/NBK26834/
Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN (1991) The nucleosomal core histone octamer at 3.1 a resolution: a tripartite protein assembly and a left-handed. Proc Natl Acad Sci U S A 22:10148–10152
Booth DG, Beckett AJ, Molina O, Samejima I, Masumoto H, Kouprina N, Larionov V, Prior IA, Earnshaw WC (2016) 3D CLEM reveals that a major portion of mitotic chromosomes is not chromatin. Mol Cell 64(4):790–802
Carlton PM (2008) Three-dimensional structured illumination microscopy and its application to chromosome structure. Chromosom Res 16:351–353
Chen B, Yusuf M, Hashimato T, Estandarte AK, Thompson GE, Robinson IK (2017) Three-dimensional positioning and structure of chromosomes in a human prophase nucleus. Sci Adv 3(7):e1602231
Davey CA, Sargent DF, Luger K, Mäder AW, Richmond TJ (2002) Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol 319(5):1097–1113
Denk W, Horstmann H (2004) Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol 2:e329
Ding C, Wei J, Li Q, Liang X, Wei T (2016) Extending resolution of scanning optical microscopy beyond the Abbe limit through the assistance of InSb thin layers. Opt Lett 41(7):1550–1553
Dorigo B (2004) Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306:1571–1573
Dwiranti A, Takata H, Uchiyama S, Fukui K (2016) The effect of magnesium ions on chromosome structure as observed by scanning electron microscopy (SEM) and scanning transmission electron microscope (STEM) tomography. Chromosome Sci 19(1–4):19–23
Earnshaw WC, Halligan B, Cooke CA, Heck MM, Liu LF (1985) Topoisomerase II is a structural component of mitotic chromosome scaffolds. J Cell Biol 100:1706–1715
Ekundo B, Richmond J, Schalch T (2017) Capturing structural heterogeneity in chromatin fibers. J Mol Biol 429(20):3031–3042
Eltsov M, Maclellan KM, Maeshima K, Frangakis AS, Dubochet J (2008) Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc Natl Acad Sci U S A 105(50):19732–19737
Eltsov M, Sosnovski S, Olins AL, Olins AL (2014) ELCS in ice: cryo-electron microscopy of nuclear envelope-limited chromatin sheets. Chromosoma 123:303–312
Engelhardt P (2000) In: Meyers RA (ed) Electron tomography of chromosome structure in encyclopedia of analytical chemistry, vol 6. Wiley, Chichester, pp 4948–4984
Finch JT, Klug A (1976) Solenoidal model for superstructure in chromatin. Proc Natl Acad Sci U S A 73:1897–1901
Flors C, Earnshaw WC (2011) Super-resolution fluorescence microscopy as a tool to study the nanoscale organization of chromosomes. Curr Opin Chem Biol 15(6):838–844
Fukui K (2016) Contribution of nanotechnology to chromosome science. Chromosome Sci 19:51–56
Fussner E, Ching RW, Bazett-Jones DP (2011) Living without 30 nm chromatin fibers. Trends Biochem Sci 36:1–6
Fussner E, Strauss M, Djuric U, Li R, Ahmed K, Hart M, Ellis J, Bazett-Jones DP (2012) Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep 13:992–996
Gibcus JH, Samejima K, Goloborodko A, Samejima I, Naumova N, Nuebler J, Kanemaki MT, Xie L, Paulson JR, Earnshaw WC, Mirny LA, Dekker J (2018) A pathway for mitotic chromosome formation. Science 359(6376):eaao6135
Harauz G, Borland L, Bhar GF, Zeitler E, Hell MV (1987) Three-dimensional reconstruction of a human metaphase chromosome from electron micrographs. Chromosoma 95:366–374
Hoshi O, Shigeno M, Ushiki T (2006) Atomic force microscopy of native human metaphase chromosomes in a liquid. Arch Histol Cytol 69(1):73–78
Howe B, Umrigar A, Tsien F (2014) Chromosome preparation from cultured cells. J Vis Exp 83:50203
Inaga S, Tanaka K, Ushiki T (2008) Transmission and scanning electron microscopy of mammalian metaphase chromosomes. In: Fukui K, Ushiki T (eds) Chromosome nanoscience and technology. CRC Press, Boca Raton, pp 93–104
Iwano M, Fukui K, Takaichi S, Isogai A (1997) Globular and fibrous structure in barley chromosomes revealed by high-resolution scanning electron microscopy. Chromosom Res 5:341–349
Joti Y, Hikima T, Nishino Y, Kamada F, Hihara S, Takata H, Ishikawa T, Maeshima K (2012) Chromosomes without a 30-nm chromatin fiber. Nucleus 3:404–410
Kanda T, Sullivan KF, Wahl GF (1998) Histone–GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8(7):377–385
Kannan TP, Zilfalil BA (2009) Cytogenetics: past, present and future. Malays J Med Sci 16(2):4–9
Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389(6648):251–260
Ma H, Naseri A, Reyes-Gutierrez P, Wolfe SA, Zhang S, Pederson T (2015) Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci U S A 112:3002–3007
Maeshima K, Laemmli UK (2003) A two-step scaffolding model for mitotic chromosome assembly. Dev Cell 4:467–480
Maeshima K, Hihara S, Eltsov M (2010) Chromatin structure: does the 30-nm fibre exist in vivo? Curr Opin Cell Biol 22:291–297
Maeshima K, Imai R, Tamura S, Nozaki T (2014) Chromatin as dynamic 10-nm fibers. Chromosoma 123:225–237
Maeshima K, Ide S, Hibino K, Sasai M (2016a) Liquid-like behavior of chromatin. Curr Opin Genet Dev 37:36–45
Maeshima K, Rogge R, Tamura S, Joti Y, Hikima T, Szerlong H, Krause C, Herman J, Seidel E, DeLuca J, Ishikawa T, Hansen JC (2016b) Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J 35(10):1115–1132
Maeshima K, Matsuda T, Shindo Y, Imamura H, Tamura S, Imai R, Kawakami S, Nagashima R, Soga T, Oka K, Nagai T (2018) A transient rise in free Mg2+ ions released from ATP-Mg hydrolysis contributes to mitotic chromosome condensation. Curr Biol 28(3):444–451
Mason DR, Jouravlev MV, Kim KS (2010) Enhanced resolution beyond the Abbe diffraction limit with wavelength-scale solid immersion lenses. Opt Lett 35(12):2007–2009
Matsuda A, Shao L, Boulanger J, Kervrann C, Carlton PM, Kner P, Agard D, Sedat JW (2010) Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP—histones. PLos One 5(9):e12768
Mora-Bermúdez F, Ellenberg J (2007) Measuring structural dynamics of chromosomes in living cells by fluorescence microscopy. Methods 41:158–167
Moralli D, Yusuf M, Mandegar MA, Khoja S, Monaco ZL, Volpi EV (2011) An improved technique for chromosomal analysis of human ES and iPS cells. Stem Cell Rev 7(2):471–477
Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Ernest DL, Tanay A, Fraser P (2013) Single cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502(7469):59–64
Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, Dekker J (2013) Organization of the mitotic chromosome. Science 342(6161):948–953
Nishino Y, Takahashi Y, Imamoto N, Ishikawa T, Maeshima K (2009) Three-dimensional visualization of a human chromosome using coherent X-ray diffraction. Phys Rev Lett 102:018101
Nishino Y, Eltsov M, Joti Y, Ito K, Takata H, Takahashi Y, Hihara S, Frangakis AS, Imamoto N, Ishikawa T, Maeshima K (2012a) Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J 31:1644–1653
Nishino Y, Eltsov M, Joti Y, Ito K, Takata H, Takahashi Y, Hihara S, Frangakis AS, Imamoto N, Ishikawa T, Maeshima K (2012b) Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J 31:1644–1653
Nozaki T, Imai R, Tabo M, Nagashima R, Tamura S, Tani T, Joti Y, Tomita M, Hibino K, Kanemaki MT, Wendt KS, Okada Y, Nagai T, Maeshima K (2017) Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol Cell 67(2):282–293.e7
Ohta S, Bukowski-Wills JC, Sanchez-Pulido L, Alves Fde L, Wood L, Chen ZA, PLatani M, Fischer L, Hudson DF, Ponting CP, Fukagawa T, Earnshaw WC, Rappsilber J (2010) The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 142(5):810–821
Olins AL, Olins DE (1974) Spheroid chromatin units (v bodies). Science 183:330–332
Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, O’Shea CC (2017) ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357(6349):eaag0025
Paulson JR, Laemmli UK (1977) The structure of histone-depleted metaphase chromosomes. Cell 12:817–828
Phengchat R, Takata H, Morii K, Inada N, Inada N, Murakoshi H, Uchiyama S, Fukui K (2016) Calcium ions function as a booster of chromosome condensation. Sci Rep 6:38281
Pollard TD, Earnshaw WC (2002) Cell biology. Elsevier, New York
Poonperm R, Takata H, Hamano T, Matsuda A, Uchiyama S, Hiraoka Y, Fukui K (2015) Chromosome scaffold is a double-stranded assembly of scaffold proteins. Sci Rep 5:11916
Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, Cosma MP (2015) Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160:1145–1158
Ris H (1981) Stereoscopic electron microscopy of chromosomes. Methods Cell Biol 1981(22):77–96
Robinson PJ, Fairall L, Huynh VA, Rhodes D (2006) EM measurements define the dimensions of the “30-nm” chromatin fiber: evidence for a compact, interdigitated structure. Proc Natl Acad Sci U S A 103:6506–6511
Rodriguez JA, Xu R, Chen CC, Huang Z, Jiang H, Chen AL, Raines KS, Pryor A Jr, Nam D, Wiegart L, Song C, Madsen A, Chushkin Y, Zontone F, Bradley PJ, Miao J (2015) Three-dimensional coherent X-ray diffractive imaging of whole frozen-hydrated cells. IUCrJ 2(Pt 5):575–583
Samejima K, Samejima I, Vagnarelli P, Ogawa H, Vargiu G, Kelly DA, de Lima AF, Kerr A, Green LC, Hudson DF, Ohta S, Cooke CA, Farr CJ, Rappsilber J, Earnshaw WC (2012) Mitotic chromosomes are compacted laterally by KIF4 and condensin and axially by topoisomerase IIα. J Cell Biol 199:755–770
Schalch T, Duda S, Sargent DF, Richmond T (2005) X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 436:138–141
Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, Kner P, Burke B, Cardoso MC, Agard DA, Gustafsson MGL, Leonhardt H, Sedat JW (2008) Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 320(5881):1332–1336
Schertel A, Snaidero N, Han HM, Ruhwedel T, Laue M, Grabenbauer M, Möbius W (2013) Cryo FIB-SEM: volume imaging of cellular ultrastructure in native frozen specimens. J Struct Biol 184(2):355–360
Schroeder-Reiter E, Pérez-Willard F, Zeile U, Wanner G (2009) Focused ion beam (FIB) combined with high resolution scanning electron microscopy: a promising tool for 3D analysis of chromosome architecture. J Struct Biol 165(2):97–106
Shemilt L, Estandarte AKC, Yusuf M, Robinson IK (2014) Scanning electron microscope studies of human metaphase chromosomes. Phil Trans R Soc A 372:20130144
Shihab SNA (2012) Your easy way to chromosomes. Author House, Bloomington
Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G (2014) Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344:376–380
Struckov AG, Belmont AS (2009) Mitotic chromosome structure: reproducibility of folding and symmetry between sister chromatids. Biophys J 96(4):1617–1628
Sumner AT (2003) Chromosomes: organization and function. Blackwell Publishing Ltd., Oxford, pp 5–142
Sydor AM, Czymmek KJ, Puchner EM, Mennella V (2015) Super-resolution microscopy: from single molecules to supramolecular assemblies. Trends Cell Biol 25(12):730–748
Takata H, Uchiyama S, Nakamura N, Nakashima S, Kobayashi S, Sone T, Kimura S, Lahmers S, Granzier H, Labeit S, So M, Fukui K (2007) A comparative proteome analysis of human metaphase chromosomes isolated from two different cell lines reveals a set of conserved chromosome-associated proteins. Genes Cells 12(3):269–284
Tobias ES, Connor M, Ferguson-Smith M (2011) Essential medical genetics, includes desktop edition. Wiley, Hoboken
Uchiyama S, Kobayashi S, Takata H, Ishihara T, Hori N, Higashi T, Hayashihara K, Sone T, Higo D, Nirasawa T, Takao T, Matsunaga S, Fukui K (2005) Proteome analysis of human metaphase chromosomes. J Biol Chem 280(17):16994–17004
Ushiki T, Hitomi J, Ogura S, Umemoto T, Shigeno M (1996) Atomic force microscopy in histology and cytology. Arch Histol Cytol 59(5):421–431
Ushiki T, Shigeno M, Hoshi O (2008) Techniques for imaging human metaphase chromosomes in liquid conditions by atomic force microscopy. Nanotechnology 19:384022
Volpi EV, Bridger JM (2008) FISH glossary: an overview of the fluorescence in situ hybridization technique. Biotechniques 45(4):385–386
Wang Y, Ratna P, Shivashankar GV (2017) Superresolution imaging of nanoscale chromosome contacts. Sci Rep 7:42422
Wanner G, Formanek H (2000) A new chromosome model. J Struct Biol 132:147–161
Woodcock CL, Frado LL, Rattner JB (1984) The higher-order structure of chromatin: evidence for a helical ribbon arrangement. J Cell Biol 99:42–52
Yusuf M, Parmar N, Bhella GK, Robinson IK (2014a) A simple filtration technique for obtaining purified human chromosomes in suspension. Biotechniques 56:257–261
Yusuf M, Chen B, Hashimato T, Estandarte AK, Thompson GE, Robinson IK (2014b) Staining and embedding of human chromosomes for 3D serial block face scanning electron microscopy. Biotechniques 57(6):302–307
Yusuf M, Zhang F, Chen B, Bhartiya A, Cunnea K, Wagner U, Cacho-Nerin F, Schwenke J, Robinson IK (2017) Procedures for cryogenic X-ray ptychographic imaging of biological samples. IUCrJ 4(2):147–151
Zhou Y, Wang P, Tian F, Gao G, Huang L, Wei W, Xie XS (2017) Painting a specific chromosome with CRISPR/Cas9 for live-cell imaging. Cell Res 27:298–301
Acknowledgements
The authors would like to acknowledge Brookhaven National Laboratory supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-SC00112704. The authors would also like to acknowledge the support of Professor El-Nasir MA Lalani of the Centre for Regenerative Medicine and Stem Cell Research at the Aga Khan University, Karachi.
Funding
This work was supported under grant number BB/H022597/1 by the BBSRC for a “Diamond Professorial Fellowship for imaging chromosomes by coherent X-ray diffraction.”
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yusuf, M., Kaneyoshi, K., Fukui, K. et al. Use of 3D imaging for providing insights into high-order structure of mitotic chromosomes. Chromosoma 128, 7–13 (2019). https://doi.org/10.1007/s00412-018-0678-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-018-0678-5