
Abstract
Prion disease is a neurodegenerative malady, which is believed to be transmitted via a prion protein in its abnormal conformation (PrPSc). Previous studies have failed to demonstrate that prion disease could be induced in wild-type animals using recombinant prion protein (rPrP) produced in Escherichia coli. Here, we report that prion infectivity was generated in Syrian hamsters after inoculating full-length rPrP that had been converted into the cross-β-sheet amyloid form and subjected to annealing. Serial transmission gave rise to a disease phenotype with highly unique clinical and neuropathological features. Among them were the deposition of large PrPSc plaques in subpial and subependymal areas in brain and spinal cord, very minor lesioning of the hippocampus and cerebellum, and a very slow progression of disease after onset of clinical signs despite the accumulation of large amounts of PrPSc in the brain. The length of the clinical duration is more typical of human and large animal prion diseases, than those of rodents. Our studies establish that transmissible prion disease can be induced in wild-type animals by inoculation of rPrP and introduce a valuable new model of prion diseases.
Similar content being viewed by others

Introduction
Recent years have witnessed significant progress in providing strong support to the protein-only hypothesis that postulates that the prion diseases are transmitted via a prion protein that acquires an infectious self-replicating conformation (PrPSc) [26]. Previously, amyloid fibrils prepared in vitro from truncated recombinant PrP (rPrP) [4] were shown to induce transmissible disease in transgenic mice that express the same truncated cellular isoform of the prion protein (PrPC) at high levels [20]. However, the origin of transmissible prions in transgenic lines has been disputed due to concerns that the mice expressing PrPC at high levels are prone to neurological disorders with undetectably low levels of infectivity. While these concerns have been addressed [9], the objective of inducing prion disease in wild-type animals with rPrP still represents a great challenge.
Using an alternative approach that involves protein misfolding cyclic amplification (PMCA) conducted in crude brain homogenates, amplification of PrPSc was shown to be accompanied by an amplification of infectivity in vitro [8]. PMCA was also used to successfully generate prion infectivity in vitro de novo from minimal components including PrPC and polyanion molecules [11]. Attempts to replace PrPC with rPrP in PMCA, however, have failed, as rPrP was shown to inhibit PMCA reactions [24]. To date, attempts to generate transmissible prion disease in wild-type animals using rPrPs have not been successful despite numerous attempts in which rPrP was converted into a broad array of abnormal β-sheet rich conformations.
Here, we report that prion infectivity can be produced in Syrian hamsters by inoculating full-length rPrP converted into a cross-β-sheet amyloid conformation and subjected to an annealing procedure [6]. The clinical disease, while clearly recognizable as a transmissible spongiform encephalopathy (TSE) infection, nevertheless exhibited a highly unique clinical course and neuropathological features including large subependymal plaques, minor involvement of cerebellum and hippocampus, but consistent lesioning in thalamus and hypothalamus. Despite large amounts of PrPSc found in the brains, the new disease is characterized by a long clinical duration and very slow progression after the first clinical signs are observed, a feature that is more typical of human and large animal TSEs, than those of rodents.
Materials and methods
Expression and purification of PrP
Golden Syrian hamster full-length recombinant PrP encompassing residues 23–231 (rPrP) was expressed and purified according to the previously described procedure [5] with minor modifications. To eliminate adduct formation and increase protein yield, 5 mM DTT instead of 10 mM β-mercaptoethanol was used to solubilize inclusion bodies. Elution of PrP from the Ni-charged Sepharose resin was performed with 200 mM Imidasole. Oxidative refolding prior to HPLC was achieved by overnight dialysis of IMAC-purified PrP into 8 M Urea, 100 mM Tris, pH 7.5. During the last 3 h of dialysis, 10 mM EDTA was added to the dialysis buffer.
Formation of rPrP fibrils
Lyophilized rPrP was dissolved in 6 M GdnHCl to prepare a 3 mg/ml stock solution of rPrP. To form fibrils, the rPrP stock solution was diluted to a final protein concentration of 0.5 mg/ml and incubated at 37°C in 20 mM sodium acetate (pH 5.0), 1 M GdnHCl, 3 M urea, 150 mM NaCl, 10 mM EDTA for 4 days with continuous horizontal shaking at 600 rpm. Amyloid formation was confirmed by increased Thioflavin T fluorescence and electron microscopy [5]. Fibrils were dialyzed into 10 mM sodium acetate, pH 5.0.
Annealing of fibrils and inoculum preparation
The annealing was performed in two different ways: either in the presence of normal brain homogenate (NBH) or bovine serum albumin (BSA). The NBH used for annealing of rPrP fibrils was prepared from a 25-day-old Syrian hamster, in a facility and with equipment that has never been exposed to TSE agents. The brain was weighed, mixed with nine parts of ice-cold PBS, pH 7.2, and homogenized with a Branson Sonifier-450 tip sonicator until a homogeneous solution was formed (4 pulses of 15 s sonication with 15 s cooling on ice between the pulses). After incubation on ice for 30 min, the homogenate was transferred to a new tube leaving behind any crude material that had settled.
rPrP fibrils at 0.1 mg/ml were heat-treated in PBS, pH 7.4, in the presence of 5% NBH or 5 mg/ml BSA (Sigma, #A3294) and 0.1% Triton X-100 using the following procedure: 50 μl aliquots in thin-wall PCR tubes with domed caps were placed into the PCR machine and subjected to 5 cycles of 1 min incubation at 80°C followed by 1 min incubation at 37°C. The combined volume was immediately transferred to the animal facility for inoculation. To verify the result of annealing, an aliquot of the inoculum was supplemented with 100 mM Tris, pH 7.5, and subjected to digestion with 50 μg/ml PK for 1 h at 37°C. PK-resistant material was detected by Western immunoblotting with 3F4 antibody.
Bioassay
For the first passage, weanling golden Syrian hamsters were inoculated intracerebrally with the preparation of rPrP fibrils described above. Each animal was anesthetized with interperitoneal pentobarbital before receiving 50 μl of inoculum. Hamsters were observed daily for disease starting from the third month postinoculation. Two out of eight inoculated hamsters died intercurrently at 409 and 652 days postinoculation without signs of any disease. The six remaining hamsters were still healthy at 661 days postinoculation when they were euthanized and their brains removed aseptically and saved for subsequent passage and analysis.
For the second passage, 10% BHs prepared by sonication in PBS, pH 7.4, were dispersed by an additional 30 s of sonication immediately before inoculation. Each hamster received 50 μl of inoculum intracerebrally under general anesthesia (2% O2/4 MAC isoflurane). After inoculation, hamsters were observed daily for disease.
Proteinase K resistance assay
Brains were collected aseptically and cut in half with disposable scalpels. One half was used to prepare 10% BHs, while the second half was stored at −80°C for future analysis or fixed in formalin for histopathology. Homogenization was performed on ice, in PBS, pH 7.4, with Sonics vibra cell VCX750 (Newtown, CT) tip sonicator fitted with a stepped microtip by 2 pulses of 30 s sonication with 30 s cooling on ice between the pulses. An aliquot of 10% brain homogenate was mixed with an equal volume of 4% sarcosyl in PBS, supplemented with 50 mM Tris, pH 7.5, and digested with 20 μg/ml PK for 30 min at 37°C with 1,000 rpm shaking (Eppendorf thermomixer). The reaction was stopped by adding 2 mM PMSF and SDS sample buffer. Samples were boiled for 10 min and loaded onto NuPAGE 12% BisTris gels. After transfer to PVDF membrane, PrP was detected with R1/R2 or 3F4 antibody, as indicated. To achieve a comparable intensity of bands, samples without PK were usually loaded at a 1:4 to 1:20 dilution as indicated in the figure legends.
To analyze PK resistance as a function of PK concentration, 10% BHs prepared by sonication in PBS (see above) were diluted to 1% using 1% BH from healthy hamsters in PBS, pH 7.5, 0.15 M NaCl, 1% Triton, and 0.25% SDS. Lyophilized Proteinase K (Sigma P6556) was dissolved in the PK buffer containing 20 mM Tris, pH 7.5, and 1 mM CaCl2, and the same buffer was used to prepare serial dilutions of PK. Each aliquot of brain homogenate was supplemented with the same volume, but increasing concentration of PK, with the final concentration ranging from 0 to 2.5 mg/ml. After incubation at 37°C for 1 h, the reaction was stopped by addition of SDS-sample buffer and boiling the samples for 10 min. Samples were loaded onto NuPAGE 12% BisTris gels, transferred to PVDF membrane, and detected with 3F4 antibody. To generate average curves and standard deviation errors, all samples were assayed at least twice.
Deglycosylation of PrPSc
Removal of N-linked glycans was performed as previously described [10], using glycerol-free PNGase F and supplied buffers (New England BioLabs). After termination of PK digestion with 2 mM PMSF, the resulting 36 μl of digest was supplemented with 4 μl of 10× glycoprotein buffer and heated at 95°C for 10 min. After heat-treatment, 5 μl of 10× G7 reaction buffer, 5 μl of 10% NP-40 and 3 μl of PNGase F enzyme were added, and the samples were incubated overnight at 37°C. The next day, the samples were boiled for 10 min in SDS sample buffer (Invitrogen) and loaded onto NuPAGE 12% BisTris gels. After transfer to PVDF, PrP was detected with 3F4 antibody.
Conformational stability assay
10% brain homogenates were incubated with various concentrations of GdnHCl for 2 h at room temperature. After incubation, all samples were diluted with 2% Sarcosyl and PBS to the final concentration of GdnHCl of 0.4 M, digested with 20 μg/ml PK for 1 h at 37°C with shaking at 1,000 rpm (Eppendorf thermomixer). The reaction was stopped by 2 mM PMSF. After addition of a proteinase inhibitors cocktail (Complete, EDTA-free, Roche), samples were precipitated by 0.3% sodium phosphotungstate, pH 7.4 as previously described [27]. After overnight incubation at 37°C with shaking 1,000 rpm (Eppendorf thermomixer), samples were centrifuged for 30 min at 13,000 rpm (accuSpin microR centrifuge, Fisher Scientific). The pellet was resuspended in 0.2% Sarcosyl/PBS, followed by addition of SDS sample buffer and shaking for 15 min at 99°C, 1,000 rpm. Samples were loaded into NuPAGE 12% BisTris gels, transferred to PVDF membrane, and detected with 3F4 antibody. Signal intensities were calculated using WCIF ImageJ. The experimental data were fitted using a two-state model and C50 values calculated from fitting parameters. To generate average values and standard deviation errors, all samples were assayed twice.
Protein misfolding cyclic amplification
Substrate was prepared following the protocol kindly provided by J. Castilla. Healthy hamsters were euthanized and immediately perfused with PBS, pH 7.4, supplemented with 5 mM EDTA. Brains were dissected, 10% brain homogenate (w/v) was prepared using ice-cold conversion buffer and glass/Teflon tissue grinders cooled on ice and attached to a constant torque homogenizer (Heidolph RZR2020). The brains were ground at low speed until homogeneous, then 5 additional strokes completed the homogenization. The composition of conversion buffer was as previously described [8]: Ca2+- and Mg2+-free PBS, pH 7.5, supplemented with 0.15 M NaCl, 1.0% Triton, and 1 tablet of complete protease inhibitors cocktail (Roche, Cat# 1836145) per 50 ml of conversion buffer. The resulting 10% normal BH in conversion buffer was used as the substrate in PMCA reactions. To prepare seeds, 10% scrapie BH in PBS was diluted 100-fold in the conversion buffer and 10 μl of the dilution were used to seed 90 μl of NBH in PMCA. Samples in 0.2 ml thin-wall PCR tubes were placed in a floating rack inside Misonix-3000 cup-horn sonicator, filled with 300 ml water. Two coils of rubber tubing attached to a circulating water bath were installed for maintaining 37°C inside the sonicator chamber. The standard sonication program consisted of 40 s sonication pulses delivered at 80% efficiency applied every hour during a 48-h period.
Brain homogenates from the first passage animals were subjected to a single round of PMCA in an attempt to increase the sensitivity of detection. In this case, the BH served both as seed and as substrate for the amplification. A half brain from each animal (non-perfused) was homogenized in the conversion buffer and aliquots of 100 μl were subjected to 40 s sonication pulses applied every hour during 48-h period as described above.
To analyze production of PK-resistant PrP material in PMCA, 15 μl of the sample was supplemented with 2.5 μl SDS and 2.5 μl PK, to the a final concentration of SDS and PK of 0.25% and 50 μg/ml, respectively, followed by incubation at 37°C for 1 h. The digestion was terminated by addition of SDS-sample buffer and boiling for 10 min. Samples were loaded onto NuPAGE 12% BisTris gels, transferred to PVDF membrane, and detected with 3F4 antibody.
Histopathological studies
Tissues from three diseased hamsters from the second passage of the NBH-annealed fibril preparation, four same age controls that had been inoculated with BHs from the uninoculated same age controls of the first passage, and three uninoculated age-matched controls from the cohort of animals used for the second passage were examined. Formalin fixed brain halves divided at the midline (right hemisphere), spinal cord, and internal organs, including tongue, testis, fat tissue, intestine at different levels, stomach, spleen, liver, pancreas, kidney, bladder, adrenals, heart, lung, trachea, skin, hind leg muscle, salivary and mesenteric lymph nodes, thymus, and sciatic nerve were processed for conventional stains including hematoxylin–eosin and luxol fast blue-nuclear fast red, Alcian-blue and Periodic acid Schiff (PAS), as well as for immunohistochemistry for PrP, using the monoclonal anti-PrP antibody 3F4 (1:1000, Covance, Berkeley, CA, USA), and monoclonal anti-glial fibrillar acidic protein (GFAP; 1:3000, Dako, Glostrup, Denmark). Blocks were treated in formic acid (96%) prior to embedding in paraffin. For detection of disease-associated PrP, we applied a pretreatment of 30-min hydrated autoclaving at 121°C followed by 5 min in 96% formic acid. We evaluated all tissues for the presence of inflammation and PrP immunoreactivity, and the brain for the presence of spongiform change and degree of gliosis. Degree of spongiform change, neuronal loss and gliosis, and intensity of PrP immunostaining was semiquantitatively evaluated (0 none, 1 mild, 2 moderate, and 3 severe). Lesion profiles were obtained by averaging scores of spongiform change, neuronal loss, and gliosis for each anatomical region and animal group [25]. For ultrastructural investigation, small samples of spinal cord periaqueductal regions obtained by needle biopsy from the paraffin block, were deparaffinized, fixed in glutaraldehyde and embedded in epoxy resin. Semi-thin sections were cut at a thickness of 0.3–0.5 μm and were stained with toluidine blue. Ultrathin sections of 80–90 nm were analyzed with a Zeiss electron microscope. In addition, we compared brain pathology with three Syrian hamsters (Harlan, WI) that had been infected by intracranial injection with 10% (w/v) scrapie brain homogenate (263K strain) and that showed clinical evidence of disease.
Results
De novo generation of PrPSc in wild-type animals
Full-length golden Syrian hamster rPrP was converted into amyloid fibrils and subjected to annealing prior to inoculation [5, 6]. Previously we showed that annealing (heating to 80°C in the presence of normal brain homogenate (NBH)) extends the short proteinase K (PK)-resistant region in rPrP fibrils encompassing residues 152/162–231 to residues ~97–231 [5, 6]. The annealing was performed in the presence of NBH prepared from 25-day-old golden Syrian hamsters or with bovine serum albumin (BSA). The NBH-assisted annealing produced a 16 kDa PK-resistant band, whereas in BSA-assisted annealing a 16 kDa band was not detectable (Fig. 1a). All experiments on expression and purification of rPrP, its conversion into fibrils, annealing and preparation of inocula were performed in a facility and with equipment that have never been exposed to TSEs (concerns regarding possible cross-contamination are addressed in SI).

De novo generation of PrPSc in Syrian hamsters. a Western blotting of rPrP fibrils annealed in the presence of NBH or BSA. NBH-annealed fibrils showed a PK-resistant product of 16 kDa. Undigested samples were loaded at 1/20th the amount of the digested samples. b Western blotting of BHs from animals inoculated with NBH-annealed fibrils. Animal #36798 showed PK-resistant PrPSc with a characteristic band shift. c Western blotting of BHs from animals inoculated with BH from animal #36798. Without PK treatment, 1/10th of a sample was loaded. d Western blotting of BHs from animals inoculated with 10% BH from animal #36793. Undigested sample was loaded at 1/10th the amount of the digested samples. e Western blotting of BHs from animals inoculated with NBH-annealed fibrils. BHs were subjected to a single round of PMCA that consisted of 40-s sonication pulses applied every hour during a 48-h period, where each sample was its own substrate. Samples in a, b, e were treated with 50 μg/ml PK, and samples in c and d with 20 μg/ml PK. 3F4 antibody was used for a, c–e and R1 antibody was used for b
The NBH- and BSA-annealed rPrP fibrils were inoculated intracerebrally into golden Syrian hamsters along with the following controls: α-rPrP-monomer mock-annealed with NBH, mock-annealed NBH alone, and non-annealed rPrP fibrils. The flow chart outlining primary inoculations and serial passages is presented in Fig. 2. None of the animals developed any signs of disease for up to 661 days after inoculation when they were euthanized (Table 1). One hamster inoculated with NBH-annealed fibrils (animal #36798) showed PK-resistant PrP with a band-shift typical for PrPSc (Fig. 1b). One animal inoculated with BSA-annealed rPrP amyloid (animal #36787) also showed PK-resistance, however, with atypical, low molecular weight (LMW) PK-resistant bands (Fig. S1). No PK-resistant PrPs were found in animals inoculated with any of the controls or in uninoculated age-matched controls (Table 1, Fig. S2).

A flow chart outlining the serial passages of NBH- or BSA-annealed rPrP fibrils. Animal numbers are shown in the boxes. The animals that produced PrPSc detectible by Western blotting are in dark gray boxes, and the animals where PrPSc was detected after a single round of PMCA are in light gray boxes. Animal #36787 showed atypical, low molecular weight PK-resistant bands
Brain homogenates (BHs) from six animals that represent three groups were selected for a second passage: (1) BH with PrPSc (animal #36798) and BH that lacked detectible PrPSc (animal #36793), both from the group inoculated with NBH-annealed fibrils; (2) BH with LMW PK-resistant (animal #36787) and BH that lacks detectible PK-resistant PrPs (animal #36788), both from the group inoculated with BSA-annealed fibrils; and (3) two uninoculated age-matched controls (Table 1, Fig. 2). All hamsters inoculated with BH of animal #36798 from the NBH-annealed group showed clinical signs of disease at 481 ± 4 days after inoculation (Table 1). The clinical signs and progression were highly unique (detail description is in SI, movie clips 1–4). The clinical disease progressed unusually slowly, therefore one animal (#43902) was euthanized 42 days after clinical onset to establish whether this was indeed a TSE infection. The conditions of the remaining animals deteriorated very slowly and all were euthanized from 80 to 120 days after onset of clinical symptoms before any had reached a terminal moribund stage. All BHs from this group including animal #43902 showed large amounts of PK-resistant PrPSc (Fig. 1c). As judged from Western blotting, the amount of PK-resistant PrPSc exceeded by several fold the amount of PrPSc in 263K-inoculated hamsters at the terminal stage (Fig. S3).
Surprisingly, all animals inoculated with BH of animal #36793 that received NBH-annealed fibrils also developed clinical disease, however, at an older age (565 ± 14 days after inoculation, Table 1). This group had the same symptoms as those inoculated with the BH of animal #36798 and showed similarly large amounts of PK-resistant PrPSc in their brains (Fig. 1d). Puzzled by these results, we went back and analyzed PK-resistance in BHs of animals that received NBH-annealed fibrils again. Prior to PK-digestion, however, 10% BHs were subjected to a sonication procedure similar to a single round of protein misfolding cyclic amplification (PMCA) that consisted of 48 cycles. The BH of animal #36793 that was selected for the second passage showed a small amount of PK-resistant PrPSc after sonication (Fig. 1e, Fig. S4a). This small, initially undetectable quantity of PK-resistant PrPSc cannot be due to horizontal transmission of prions from the animal #36798, because these two animals were housed in different cages. When the conditions for PMCA reaction were optimized for the newly generated TSE strain, two additional BHs (#36799 and #36791), one from the group inoculated with NBH-annealed fibrils and another from the group inoculated with BSA-annealed fibrils showed PK-resistant PrPSc (Fig. S4b). In summary, 3 out of 6 animals inoculated with NBH-annealed fibrils showed infectivity/PrPSc as detected by either second passage or by a single-round PMCA. PMCA has been used to generate prion infectivity de novo from normal brain homogenate but only after 10 rounds of 240 sonication cycles per round. After 50 days of sonication only 1 BH out of 10 showed PrPSc [2]. In contrast, we used only a single round of 48 cycles, where seed and substrate were in the same homogenate, and where our objective was to increase sensitivity of detection. There was no possibility of de novo synthesis as born out by controls. None of the BHs from any of the negative control groups that included uninoculated age-matched controls, second passages of two uninoculated age-matched controls, animals inoculated with mock-annealed NBH, or with α-rPrP-monomer mock-annealed with NBH produced any PK-resistant PrP after PMCA (Table 1, Figs. S2, S4b).
None of the hamsters from the second passage of BSA-annealed fibrils developed clinical disease before they were euthanized at 661 days after inoculation. Despite the lack of clinical signs, 6 out of 7 hamsters inoculated with LMW PK-resistant BH showed PK-resistant PrP in amounts similar to those found at the terminal stage of 263K-inoculated hamsters and with a band-shift typical for PrPSc (Fig. S5a).
Unique biochemical features of the de novo generated prions
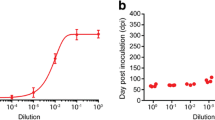
Biochemical assays were employed to characterize the newly generated TSE strain that was designated SSLOW (Synthetic Strain Leading to OverWeight). While the PrPSc glycosylation pattern of SSLOW appeared to be similar to that of 263K, the PrPSc PK-resistant core was slightly shorter in SSLOW (by ~0.5–1 kDa) than in 263K, as judged from the mobility after deglycosylation by PGNase (Fig. 3a). The slightly shorter size was a characteristic feature of PrPSc produced in the first and second passage of NBH-annealed fibrils (Fig. 3a). The conformational stability assay revealed that the resistance of SSLOW to GdnHCl-induced denaturation decreased gradually over the course of serial transmission. The [GdnHCl]1/2 values were 4.1 ± 0.05 M, 3.0 ± 0.1 M, and 2.0 ± 0.2 M for the NBH-annealed fibrils used for inoculations, PrPSc from the first passage (animal # 36798), and PrPSc from the second passage, respectively (Fig. 3b). After the second passage, the [GdnHCl]1/2 values for SSLOW were similar to that of 263K (Fig. 3b). Despite similar conformational stabilities, SSLOW had a much longer incubation time than 263K. Furthermore, the yield of in vitro replication of SSLOW was persistently lower than that of 263K, as judged from the kinetics of PrPSc amplification measured within a single round of PMCA reactions (Fig. 3c). The resistance of PrPSc to proteolytic digestion was similar for SSLOW and 263K as evaluated by the assays conducted at increasing concentrations of PK (Fig. S6).

Biochemical characterization of the de novo generated prions. a BHs from the first (animal #36798) or second passage (animals #43902 and #43908) of NBH-annealed fibrils were treated with PK and PNGase and analyzed by Western blotting. Arrows indicate the center of the deglycosylated PrPSc from 263K-inoculated hamsters. BH #43902 was loaded at 1/4th of the amount of 263K BH. b Western blotting and the conformational stability profiles (iv) of the GdnHCl-induced transitions of the NBH-annealed fibrils (i), or PrPSc from first (ii) or second (iii) passage of NBH-annealed fibrils. NBH-annealed fibrils (blue circles), PrPSc from first passage (green square, animal #36798), second passage (yellow, orange, brown, or pink triangles—animals #43902, 43904, 43908, or 43909), or 263K-inoculated animals (red diamonds). Each BH was loaded onto the gel twice and the data were averaged. c The amplification kinetics for 263K or PrPSc from second passage of NBH-annealed fibrils (BH #43902) as monitored by Western blotting during a single round of PMCA that consisted of 100 cycles. Samples in a, b (ii and iii) were treated with 20 μg/ml PK; samples in c with 50 μg/ml PK; and samples in b (i) with 2 μg/ml PK
The deglycosylation with PGNase of the PK-resistant PrP from the second passage of BSA-annealed fibrils revealed that the PK-resistant core was similar to that of SSLOW strain being slightly shorter than the deglycosylated PrPSc from 263K (Fig. S5b). While the similarity in PK-resistant core suggests that the same strain is emerging in animals inoculated with BSA-annealed fibrils, it remains to be determined whether a clinical disease will appear in a third passage of BSA-annealed fibrils.
SSLOW strain displays unique neuropathological profile
Histopathological studies of hamsters from the second passage of NBH-annealed fibrils revealed characteristic signs of TSE infection including spongiform degeneration, neuronal loss, reactive astrogliosis, and deposition of disease-associated PrP in the brains and spinal cords (Fig. 4, Figs. S7, S8). No inflammatory changes or immunoreactivity for disease-associated PrP were found in any peripheral tissues examined (complete list of tissues is in Methods). Spongiform change, neuronal loss, and reactive astrogliosis were uniformly prominent in the thalamus, caudate-putamen, brain stem, spinal cord, and deeper layers of frontal cortex. However, they were very mild in the hippocampus and cerebellum (Fig. 4a, c–e, Fig. S7). In contrast to animals infected with SSLOW strain, the brains of 263K-infected hamsters showed uniform involvement of the cerebellum, hippocampus and all layers of the frontal cortex (Fig. 4a, c–e, Fig. S7).

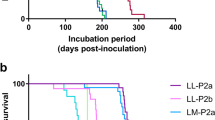
SSLOW strain displays unique neuropathological profile. a, b Lesion profile (a) and PrP immunopositivity score (b) in hamsters inoculated with SSLOW (2nd passage of NBH-annealed fibrils) or 263K. The lesion profile was obtained by averaging the scores for spongiform change, neuronal loss, and gliosis for three animals within each group. c–e Comparison of spongiform changes in the hippocampus (c), thalamus (d), or cerebellum (e) stained with hematoxylin and eosin (upper panels) or anti-PrP 3F4 antibody (lower panels) in a representative hamsters inoculated with SSLOW (animal # 43902, left panels) or 263K (right panels). f Low-magnification (×20) overview of frontal cortex (enlarged at ×100 in right upper inset) and caudate-putamen (enlarged at ×100 in right lower inset) using immunostaining for disease-associated PrP. g, h High-magnification images (×400) of PrP immunoreactive plaques in the periventricular subependymal region (g), and synaptic and perineuronal PrP immunoreactivity in the frontal cortex (h). i Overview of PrP immunostaining (×50) of spinal cord demonstrates prominent plaque deposition (enlarged right lower inset) in the midline and in periaqueductal region. j, k Electron microscopy of a plaque from the periaqueductal region from the spinal cord (scale bar 10 μm in j or 0.4 μm in k)
The distribution and pattern of PrP immunostaining were remarkably different for SSLOW- and 263K-infected brains (Fig. 4b–e, Fig. S7). While both strains showed synaptic immunoreactivity for PrP deposits, SSLOW-infected animals were characterized by more intensive perineuronal deposits and large plaques with a diameter of 20–60 μm in the subpial, periventricular, and periaqueductal subependymal regions (Fig. 4f–h, Fig. S7, S8). In contrast, 263K-infected brains showed focal patchy or smaller plaque-like deposits with a diameter of 5–15 μm in gray and white matter structures. Both strains displayed immunoreactivity in subependymal regions, whereas perivascular PrP deposits were found only in hamsters infected by the 263K strain. While synaptic and perineuronal immunoreactivity showed anatomical variability between SSLOW-infected animals, the most striking histopathological feature of SSLOW strain was the uniform deposition of large plaques in the subpial, periventricular, and periaqueductal subependymal regions spanning all locations including brain and spinal cord (Fig. 4, Figs. S7, S8). These plaques stained with Alcian blue and with PAS at their borders (Fig. S8). Electron microscopy revealed that these plaques consist of a loose meshwork of randomly orientated filaments of 10–25 nm in diameter (Fig. 4j, k). Animals from control groups lacked any spongiform degeneration or deposition of disease-associated PrP (Fig. S9).
Discussion
The current studies demonstrate that recombinant PrP in its fibrillar β-sheet rich conformation subjected to annealing induces a transmissible form of prion disease in wild-type animals. In the first passage, NBH-annealed fibrils were found to seed formation of PrPSc in hamster brains, but failed to cause clinical disease, which was only observed at the second passage. The lack of symptomatic disease by end of life is consistent with a very slow replication of PrPSc and that the newly emerged strain is intrinsically slow. Previous work has shown that the incubation period of a prion disease can exceed the life span of an animal even when induced by natural prion strains [13–16, 19]. In classical studies by Dickinson et al. [13], mice inoculated with high titers of certain strains of mouse-adapted scrapie prions remained free of clinical symptoms for their entire life span despite histopathological changes and accumulation of PrPSc in their brains at the end of their life span. Because incubation time, long or short, is an intrinsic function of TSE strain, it cannot be used to deduce the concentration of infectious material in the inoculum without more knowledge of the dose response. A number of lines of evidence in the current study suggest that the newly generated strain that we call SSLOW is intrinsically very slow. When the incubation time exceeds the life span of the animal it may be difficult to determine the actual transmission rate because one can detect only those infections that have progressed to a detectable endpoint by the end of life. To increase the sensitivity at the endpoint, we employed a single-round PMCA. Using this procedure, PrPSc was detected in two additional animals inoculated with NBH-annealed fibrils. We do not know whether or not all of the animals from the first passage would have proved to be infected, had we carried all of them forward in a second passage.
The apparent 50% efficiency of infection in animals inoculated with NBH-annealed fibrils could be attributed to several factors. First, because rPrP fibrils lack a GPI-anchor, they do not attach to the cell surface as efficiently as authentic PrPSc and, therefore, might display reduced ability to invade cells and seed prion replication. Second, rPrP fibrils and PrPSc appear to have fundamentally different global structures [29]. Because PrPC might not be fully compatible with the structure of rPrP fibrils, seeding of PrPC conversion with rPrP fibrils might proceed through slow conformational adaptation [3]. The gradual change in conformational stability during serial transmission observed here supports the hypothesis of conformational adaptation. Previous studies on synthetic mouse prions revealed a similar trend, where conformational stability of the de novo generated synthetic strains was found to decrease gradually during their serial transmission [9, 21]. The conformational adaptation could involve a conformational switch within individual fibrils [23], where rPrP fibrils with one global structure might recruit PrPC, elongate and give a rise to PrPSc with different global structure. Third, as postulated by the “unified theory” of prion propagation [28], efficient replication of prions appears to require cellular cofactors, such as RNAs [11, 12]. The 50% efficiency of the infection in the first passage indicates that the annealing protocol might not yet faithfully mimic the interaction of rPrP with a co-factor for generating authentic PrPSc in vitro. The fact that PrPSc was found in both animal groups, inoculated either with NBH- or BSA-annealed fibrils, suggests that the role of NBH-annealing is non-specific and that, perhaps, it plays a role of a stabilizer. By analogy, numerous viruses must be stabilized with milk, serum, albumin, or gelatin to survive inoculation.
The 50% transmission at first passage is also consistent with a very low titer of the active species in the original inoculum. However, if one assumes that only a small subpopulation of fibrils is infectious, it would be difficult to explain a correlation between conformational stability of rPrP fibrils, which is a bulk property of fibril preparations, and the incubation times to disease observed in the previous study on synthetic prions [9].
One of the most remarkable features of the prion disease caused by SSLOW strain is its long clinical duration after the first clinical signs are observed. In fact, the actual duration to a death endpoint is not known as the animals, while seriously compromised, were still mobile, eating and drinking at the time of euthanasia. It is conceivable that animals may have endured longer than 120 days after first clinical signs, if they had not been euthanized. Such a long duration is more typical of human and large animal TSEs, than those of rodents. The slow progression is surprising considering that large amounts of PrPSc were already present at the initial stages of clinical disease. The slow progression may be attributable to the very peculiar pattern of deposition of PrPSc which was found predominantly in the form of large plaques. The PrPSc deposition patterns and brain histopathology differed prominently between SSLOW and 263K or other hamster strains. One of the most intriguing features of the SSLOW strain was the very minor involvement of the cerebellum highlighting a unique neurotropism of this strain. Very mild PrPSc deposition in the cerebellum correlated well with apparent lack of the severe motor dysfunctions typical for 263K. The sparing of the hippocampus in SSLOW is intriguing and reminiscent of sparing of the hippocampus in the most common subtype (MM/MV-1) of human sporadic Creutzfeldt–Jakob disease (CJD) [7, 25]. In contrast, the large subpial and subependymal plaques were distinct not only from plaque-like deposits in 263K scrapie of hamsters but also from human TSEs with plaques including the Kuru plaque variant (MV-2) of sporadic CJD or Gerstmann–Straussler–Scheinker disease.
In second passage of NBH-annealed fibrils, the two groups of animals inoculated with BHs from #36793 and #36798 showed different incubation times to onset of disease. This difference can be attributed to a lower infectivity titer in BH from #36793, consistent with the substantially lower amount of PK-resistant PrPSc in this brain. As judged from clinical signs and biochemical assays, however, the same strain appears to have emerged in both groups. Remarkably, in a second passage of BSA-annealed fibrils, PK-resistant PrP showed a band shift identical to that of SSLOW PrPSc raising the possibility that the same stain might emerge in serial passages of two independently prepared inoculums: NBH- or BSA-annealed fibrils. The third passage of NBH- and BSA-annealed fibrils should clarify this question.
The conformational stability of SSLOW PrPSc that formed in a second passage was very similar to that of 263K, yet these two strains showed markedly different incubation times arguing that factors other than conformational stability also contribute to the rate of prion replication. Nevertheless, the substantially longer incubation time for SSLOW correlated well with the lower yield of SSLOW replication in PMCA as compared to 263K (Fig. 3c).
Much of the public health risk from TSE diseases derives from their long asymptomatic incubation times. During this period infected humans (or animals) appear normal and their underlying disease is currently undetectable. Such individuals pose a real risk of spreading the infection through invasive medical procedures or tissue or blood donation. Variant CJD infections of humans heterozygous at the codon 129 of the prion gene may present such a risk [18]. The SSLOW strain offers an experimental model to investigate this problem.
In pursuing the objective of generating synthetic prions in vitro, it is useful to consider how prions originate in the natural environment. Spontaneous occurrence of transmissible prion disease has never been documented in wild-type rodents including golden Syrian hamsters or wild-type mice, nor has prion disease occurred upon serial inoculations of brain homogenates from old to young animals [17]. All experimental hamster or mouse strains have been derived by serial transmission of TSE agents from species with substantially longer life span than rodents. While other neurodegenerative diseases can be triggered as a result of proteostasis deficiency at old age [1], the genesis of transmissible prions appears to require a very specific trigger and/or conditions that are much more peculiar than imbalance of proteostasis. Surprisingly, the current study suggests that transmissible prion disease can be induced by a preparation of rPrP fibrils, which appear to be fundamentally different with respect to their global structure from PrPSc. One can speculate that only partial or distant similarities between fibrillar rPrP and PrPSc structures may be sufficient for triggering conversion of PrPC into PrPSc by fibrillar rPrP. We do not know whether the emergence of highly efficient prion strains in nature occurs within a single life span or require evolution, adaptation, and selection of PrPSc structures. Prion genesis in nature might require a series of transmissions from animal to animal as may have happened with Bovine Spongiform Encephalopathy. In this regard, it would be interesting to learn whether clinical disease appears in a third passage of BSA-annealed fibrils.
In summary, this work represents an important advance in prion science, in that (i) it is the first time that a wild-type recombinant PrP has been shown to be able to induce a TSE disease in a wild-type animal; (ii) the disease generated by the in vitro refolded rPrP while clearly recognizable as a TSE disease is unique in its clinical presentation, pathology, and biochemistry and may prove of great interest. Because of its unique features, the SSLOW strain may prove to be a valuable model for addressing a number of puzzling questions of prion biology: what factors determine prion neurotropism; how strain-specific PrPSc structures control the rate of prion replication; why clinical disease could progresses so slowly despite large amounts of PrPSc deposition; and the physical nature of toxic versus infectious PrP isoforms. Further studies on synthetic prions will likely provide unique insights into the genesis and evolution of natural prion strains.
Abbreviations
- PrPC :
-
Normal cellular isoform of the prion protein
- PrPSc :
-
Abnormal, disease-associated isoform of the prion protein
- rPrP:
-
Recombinant PrP
- PMCA:
-
Protein misfolding cyclic amplification
- TSE:
-
Transmissible spongiform encephalopathy
- BH:
-
Brain homogenate
- NBH:
-
Normal brain homogenate
- PK:
-
Proteinase K
- BSA:
-
Bovine serum albumin
- LMW:
-
Low molecular weight
References
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319:916–919
Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C (2009) De novo generation of infectious prions in vitro produces a new disease phenotype. PLOS Pathog 5:e1000421
Baskakov IV, Breydo L (2007) Converting the prion protein: what makes the protein infectious. Biochim Biophys Acta (Molecular Basis of Disease) 1772:692–703
Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE (2002) Pathway complexity of prion protein assembly into amyloid. J Biol Chem 277:21140–21148
Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV (2005) In vitro conversion of full length mammalian prion protein produces amyloid form with physical property of PrPSc. J Mol Biol 346:645–659
Bocharova OV, Makarava N, Breydo L et al (2006) Annealing PrP amyloid firbils at high temperature results in extension of a proteinase K resistant core. J Biol Chem 281:2373–2379
Budka H, Head MW, Ironside JW et al (2003) Prion disorders. In: Dickson DW (ed) Neurodegeneration: the molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 287–297
Castilla J, Saa P, Hetz C, Soto C (2005) In vitro generation of infectious scrapie prions. Cell 121:195–206
Colby DW, Giles K, Legname G et al (2009) Design and construction of diverse mammalian prion strains. Proc Acad Natl Sci USA 106:20417–20422
Deleault AM, Deleault NR, Harris BT, Rees JR, Supattapone S (2008) The effects of prion protein proteolysis and disaggregation on the strain properties of hamster scrapie. J Gen Virol 89:2642–2650
Deleault NR, Harris BT, Rees JR, Supattapone S (2007) Formation of native prions from minimal components in vitro. Proc Acad Natl Sci USA 104:9741–9746
Deleault NR, Lucassen RW, Supattapone S (2003) RNA molecules stimulate prion protein conversion. Nature 425:717–720
Dickinson AG, Fraser H, Outram GW (1975) Scrapie incubation time can exceed natural lifespan. Nature 256:732–733
Frigg R, Klein MA, Hegyi I, Zinkernagel RM, Aguzzi A (1999) Scrapie pathogenesis in subclinically infected B-cell-deficient mice. J Virol 73:9584–9588
Hill A, Collinge J (2003) Subclinical prion infection in human and animals. Br Med Bull 66:161–170
Hill AF, Collinge J (2003) Subclinical prion infection. Trends Microbiol 11:578–584
Hill AF, Joiner S, Linehan J et al (2000) Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci USA 97:10248–10253
Kaski D, Mead S, Hyare H et al (2009) Variant CJD in an individual heterozygous for PRNP codon 129. Lancet 374:2128
Klein MA, Frigg R, Raeber AJ et al (1998) PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat Med 4:1429–1433
Legname G, Baskakov IV, Nguyen H-OB et al (2004) Synthetic mammalian prions. Science 305:673–676
Legname G, Nguyen H-OB, Baskakov IV et al (2005) Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci USA 102:2168–2173
Makarava N, Baskakov IV (2008) The same primary structure of the prion protein yields two distinct self-propagating states. J Biol Chem 283:15988–15996
Makarava N, Ostapchenko VG, Savtchenko R, Baskakov IV (2009) Conformational switching within individual amyloid fibrils. J Biol Chem 284:14386–14395
Nishina K, Deleault NR, Mahal S et al (2006) The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry 45:14129–14139
Parchi P, Giese A, Capellari S et al (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Safar J, Wille H, Itri V et al (1998) Eight prion strains have PrPSc molecules with different conformations. Nat Med 4:1157–1165
Weissmann C (1991) A “unified theory” of prion propagation. Nature 352:679–683
Wille H, Bian W, McDonald M et al (2009) Natural and synthetic prion structure from X-ray fiber diffraction. Proc Acad Natl Sci USA 106:16990–16995
Acknowledgments
We thank Pamela Wright for editing the manuscript. This work was supported by University of Maryland Biotechnology Institute and Baltimore Research and Education Foundation. Parts of this work were done in the framework of the EU Network of Excellence NeuroPrion, Subproject PrioGen.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 3 (mpg 13.3 Mb)
Supplementary material 4 (mpg 44.5 Mb)
Supplementary material 5 (mpg 45.6 Mb)
Supplementary material 6 (mpg 6.43 Mb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Makarava, N., Kovacs, G.G., Bocharova, O. et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 119, 177–187 (2010). https://doi.org/10.1007/s00401-009-0633-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0633-x