
Abstract
Fungal pathogens need to contend with stresses including oxidants and antimicrobial chemicals resulting from host defenses. ChAP1 of Cochliobolus heterostrophus, agent of Southern corn leaf blight, encodes an ortholog of yeast YAP1. ChAP1 is retained in the nucleus in response to plant-derived phenolic acids, in addition to its well-studied activation by oxidants. Here, we used transcriptome profiling to ask which genes are regulated in response to ChAP1 activation by ferulic acid (FA), a phenolic abundant in the maize host. Nuclearization of ChAP1 in response to phenolics is not followed by strong expression of genes needed for oxidative stress tolerance. We, therefore, compared the transcriptomes of the wild-type pathogen and a ChAP1 deletion mutant, to study the function of ChAP1 in response to FA. We hypothesized that if ChAP1 is retained in the nucleus under plant-related stress conditions yet in the absence of obvious oxidant stress, it should have additional regulatory functions. The transcriptional signature in response to FA in the wild type compared to the mutant sheds light on the signaling mechanisms and response pathways by which ChAP1 can mediate tolerance to ferulic acid, distinct from its previously known role in the antioxidant response. The ChAP1-dependent FA regulon consists mainly of two large clusters. The enrichment of transport and metabolism-related genes in cluster 1 indicates that C. heterostrophus degrades FA and removes it from the cell. When this fails at increasing stress levels, FA provides a signal for cell death, indicated by the enrichment of cell death-related genes in cluster 2. By quantitation of survival and by TUNEL assays, we show that ChAP1 promotes survival and mitigates cell death. Growth rate data show a time window in which the mutant colony expands faster than the wild type. The results delineate a transcriptional regulatory pattern in which ChAP1 helps balance a survival response for tolerance to FA, against a pathway promoting cell death in the pathogen. A general model for the transition from a phase where the return to homeostasis dominates to a phase leading to the onset of cell death provides a context for understanding these findings.





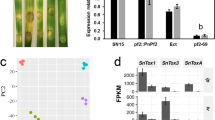
Adapted from a scheme of Galluzzi et al. (2016) comparing two alternative mechanisms; here, the diagram compares WT and mutant
Similar content being viewed by others


References
Asakura M, Okuno T, Takano Y (2006) Multiple contributions of peroxisomal metabolic function to fungal pathogenicity in Colletotrichum lagenarium. Appl Environ Microbiol 72:6345–6354. https://doi.org/10.1128/AEM.00988-06
Azevedo D, Tacnet F, Delaunay A et al (2003) Two redox centers within Yap1 for H2O2 and thiol-reactive chemicals signaling. Free Radic Biol Med. https://doi.org/10.1016/s0891-5849(03)00434-9
Bennet RN, Wallsgrove RM (1994) Secondary metabolites in plant defence mechanisms. New Phytol 127:617–633. https://doi.org/10.1111/j.1469-8137.1994.tb02968.x
Bhambra GK, Wang ZY, Soanes DM et al (2006) Peroxisomal carnitine acetyl transferase is required for elaboration of penetration hyphae during plant infection by Magnaporthe grisea. Mol Microbiol 61:46–60. https://doi.org/10.1111/j.1365-2958.2006.05209.x
Brive L, Takayama S, Briknarová K et al (2001) The carboxyl-terminal lobe of Hsc70 ATPase domain is sufficient for binding to BAG1. Biochem Biophys Res Commun 289:1099–1105. https://doi.org/10.1006/bbrc.2001.6087
Chen XL, Wang Z, Liu C (2016) Roles of peroxisomes in the rice blast fungus. Biomed Res Int. https://doi.org/10.1155/2016/9343417
Coleman JJ, Mylonakis E (2009) Efflux in fungi: La pièce de résistance. PLoS Pathog 5:e1000486. https://doi.org/10.1371/journal.ppat.1000486
Coleman JJ, White GJ, Rodriguez-Carres M, VanEtten HD (2011) An ABC transporter and a cytochrome P450 of Nectria haematococca MPVI are virulence factors on pea and are the major tolerance mechanisms to the phytoalexin pisatin. Mol Plant Microbe Interact 24:368–376. https://doi.org/10.1094/MPMI-09-10-0198
Condon BJ, Leng Y, Wu D et al (2013) Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. https://doi.org/10.1371/journal.pgen.1003233
Črešnar B, Petrič Š (2011) Cytochrome P450 enzymes in the fungal kingdom. Biochim Biophys Acta Proteins Proteom 1814:29–35
De Waard MA, Andrade AC, Hayashi K et al (2006) Impact of fungal drug transporters on fungicide sensitivity, multidrug resistance and virulence. Pest Manag Sci 62:195–207
Del Sorbo G, Schoonbeek HJ, De Waard MA (2000) Fungal transporters involved in efflux of natural toxic compounds and fungicides. Fungal Genet Biol 30:1–15. https://doi.org/10.1006/fgbi.2000.1206
Doehlemann G, Wahl R, Horst RJ et al (2008) Reprogramming a maize plant: transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis. Plant J 56:181–195. https://doi.org/10.1111/j.1365-313X.2008.03590.x
Fernandez J, Wright JD, Hartline D et al (2012) Principles of carbon catabolite repression in the rice blast fungus: Tps1, Nmr1-3, and a MATE-family pump regulate glucose metabolism during infection. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002673
Ferrer-Sevillano F, Fernández-Cañón JM (2007) Novel phacB-encoded cytochrome P450 monooxygenase from Aspergillus nidulans with 3-hydroxyphenylacetate 6-hydroxylase and 3,4-dihydroxyphenylacetate 6-hydroxylase activities. Eukaryot Cell 6:514–520. https://doi.org/10.1128/EC.00226-06
Fujihara N, Sakaguchi A, Tanaka S et al (2010) Peroxisome biogenesis factor PEX13 is required for appressorium-mediated plant infection by the anthracnose fungus Colletotrichum orbiculare. Mol Plant Microbe Interact 23:436–445. https://doi.org/10.1094/MPMI-23-4-0436
Galluzzi L, Bravo-San Pedro JM, Kepp O, Kroemer G (2016) Regulated cell death and adaptive stress responses. Cell Mol Life Sci 73:2405–2410
Gao S, Li Y, Gao J et al (2014) Genome sequence and virulence variation-related transcriptome profiles of Curvularia lunata, an important maize pathogenic fungus. BMC Genom. https://doi.org/10.1186/1471-2164-15-627
Goncalves AP, Cordeiro JM, Monteiro J et al (2014) Activation of a TRP-like channel and intracellular Ca2+ dynamics during phospholipase-C-mediated cell death. J Cell Sci 127:3817–3829. https://doi.org/10.1242/jcs.152058
Gonçalves AP, Heller J, Daskalov A et al (2017) Regulated forms of cell death in fungi. Front Microbiol. https://doi.org/10.3389/fmicb.2017.01837
Heller J, Clavé C, Gladieux P et al (2018) NLR surveillance of essential SEC-9 SNARE proteins induces programmed cell death upon allorecognition in filamentous fungi. Proc Natl Acad Sci USA 115:E2292–E2301. https://doi.org/10.1073/pnas.1719705115
Kameshwar AKS, Qin W (2017) Gene expression metadata analysis reveals molecular mechanisms employed by Phanerochaete chrysosporium during lignin degradation and detoxification of plant extractives. Curr Genet 63:877–894. https://doi.org/10.1007/s00294-017-0686-7
Kim JH, Campbell BC, Mahoney N et al (2007) Enhanced activity of strobilurin and fludioxonil by using berberine and phenolic compounds to target fungal antioxidative stress response. Lett Appl Microbiol 45:134–141. https://doi.org/10.1111/j.1472-765X.2007.02159.x
Kimura A, Takano Y, Furusawa I, Okuno T (2001) Peroxisomal metabolic function is required for appressorium-mediated plant infection by Colletotrichum lagenarium. Plant Cell 13:1945–1957. https://doi.org/10.1105/TPC.010084
Klose J, Kronstad JW (2006) The multifunctional β-oxidation enzyme is required for full symptom development by the biotrophic maize pathogen Ustilago maydis. Eukaryot Cell 5:2047–2061. https://doi.org/10.1128/EC.00231-06
Kretschmer M, Klose J, Kronstad JW (2012) Defects in mitochondrial and peroxisomal β-oxidation influence virulence in the maize pathogen Ustilago maydis. Eukaryot Cell 11:1055–1066. https://doi.org/10.1128/EC.00129-12
Kuroda T, Tsuchiya T (2009) Multidrug efflux transporters in the MATE family. Biochim Biophys Acta Proteins Proteom 1794:763–768
Lamping E, Baret PV, Holmes AR et al (2010) Fungal PDR transporters: phylogeny, topology, motifs and function. Fungal Genet Biol 47:127–142. https://doi.org/10.1016/j.fgb.2009.10.007
Lanoue A, Burlat V, Henkes GJ et al (2010) De novo biosynthesis of defense root exudates in response to Fusarium attack in barley. New Phytol 185:577–588. https://doi.org/10.1111/j.1469-8137.2009.03066.x
Leach J, Lang BR, Yoder OC (1982) Methods for selection of mutants and in vitro culture of Cochliobolus heterostrophus. Microbiology 128:1719–1729. https://doi.org/10.1099/00221287-128-8-1719
Lev S, Hadar R, Amedeo P et al (2005) Activation of an AP1-like transcription factor of the maize pathogen Cochliobolus heterostrophus in response to oxidative stress and plant signals. Eukaryot Cell 4:443–454. https://doi.org/10.1128/EC.4.2.443-454.2005
Liu W, Saint DA (2002) Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Commun 294:347–353. https://doi.org/10.1016/S0006-291X(02)00478-3
Liu S, Hou Y, Liu W et al (2015) Components of the calcium-calcineurin signaling pathway in fungal cells and their potential as antifungal targets. Eukaryot Cell 14:324–334. https://doi.org/10.1128/EC.00271-14
Longo VD, Mitteldorf J, Skulachev VP (2005) Opinion: programmed and altruistic ageing. Nat Rev Genet 6:866–872
Mandal SM, Chakraborty D, Dey S (2010) Phenolic acids act as signaling molecules in plant-microbe symbioses. Plant Signal Behav 5:359–368
Nicholson RL, Hammerschmidt R (1992) Phenolic compounds and their role in disease resistance. Annu Rev Phytopathol 30:369–389. https://doi.org/10.1146/annurev.py.30.090192.002101
Omote H, Hiasa M, Matsumoto T et al (2006) The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol Sci 27:587–593. https://doi.org/10.1016/j.tips.2006.09.001
Palková Z, Váchová L (2006) Life within a community: benefit to yeast long-term survival. FEMS Microbiol Rev 30:806–824
Podobnik B, Stojan J, Lah L et al (2008) CYP53A15 of Cochliobolus lunatus, a target for natural antifungal compounds. J Med Chem 51:3480–3486. https://doi.org/10.1021/jm800030e
Roohparvar R, De Waard MA, Kema GHJ, Zwiers LH (2007) MgMfs1, a major facilitator superfamily transporter from the fungal wheat pathogen Mycosphaerella graminicola, is a strong protectant against natural toxic compounds and fungicides. Fungal Genet Biol 44:378–388. https://doi.org/10.1016/j.fgb.2006.09.007
Shalaby S, Horwitz BA (2015) Plant phenolic compounds and oxidative stress: integrated signals in fungal–plant interactions. Curr Genet 61:347–357. https://doi.org/10.1007/s00294-014-0458-6
Shalaby S, Horwitz BA, Larkov O (2012) Structure–activity relationships delineate how the maize pathogen Cochliobolus heterostrophus uses aromatic compounds as signals and metabolites. Mol Plant Microbe Interact 25:931–940. https://doi.org/10.1094/MPMI-01-12-0015-R
Shalaby S, Larkov O, Lamdan NL, Horwitz BA (2014) Genetic interaction of the stress response factors ChAP1 and Skn7 in the maize pathogen Cochliobolus heterostrophus. FEMS Microbiol Lett 350:83–89
Shalaby S, Larkov O, Lamdan NL et al (2016) Plant phenolic acids induce programmed cell death of a fungal pathogen: MAPK signaling and survival of Cochliobolus heterostrophus. Environ Microbiol 18:4188–4199. https://doi.org/10.1111/1462-2920.13528
Shamir R, Maron-Katz A, Tanay A et al (2005) EXPANDER—an integrative program suite for microarray data analysis. BMC Bioinform. https://doi.org/10.1186/1471-2105-6-232
Shanmugam V, Ronen M, Shalaby S et al (2010) The fungal pathogen Cochliobolus heterostrophus responds to maize phenolics: novel small molecule signals in a plant–fungal interaction. Cell Microbiol 12:1421–1434. https://doi.org/10.1111/j.1462-5822.2010.01479.x
Sharon A, Shlezinger N (2013) Fungi infecting plants and animals: killers, non-killers, and cell death. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1003517
Sharon A, Finkelstein A, Shlezinger N, Hatam I (2009) Fungal apoptosis: function, genes and gene function. FEMS Microbiol Rev 33:833–854
Shlezinger N, Minz A, Gur Y et al (2011) Anti-apoptotic machinery protects the necrotrophic fungus Botrytis cinerea from host-induced apoptotic-like cell death during plant infection. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1002185
Shlezinger N, Goldfinger N, Sharon A (2012) Apoptotic-like programed cell death in fungi: the benefits in filamentous species. Front Oncol. https://doi.org/10.3389/fonc.2012.00097
Shlezinger N, Irmer H, Dhingra S et al (2017) Sterilizing immunity in the lung relies on targeting fungal apoptosis-like programmed cell death. Science 357:1037–1041. https://doi.org/10.1126/science.aan0365
Sukhanova EI, Rogov G, Severin FF, Zvyagilskaya R (2012) Phenoptosis in yeasts. Biochemistry (Moscow) 77:761–775. https://doi.org/10.1134/S0006297912070097
Tobin MB, Peery RB, Skatrud PL (1997) Genes encoding multiple drug resistance-like proteins in Aspergillus fumigatus and Aspergillus flavus. Gene 200:11–23. https://doi.org/10.1016/S0378-1119(97)00281-3
Zheng P, Xia Y, Xiao G et al (2011) Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. https://doi.org/10.1186/gb-2011-12-11-r116
Acknowledgements
We thank Amir Sharon for discussions of fungal RCD, and Laura D. Cohen for critical reading of the manuscript. We are grateful to Nitsan Dahan for expert assistance with confocal microscopy at the Technion Life Sciences and Engineering Infrastructure Center Life Sciences (LS&E) Center, and to Tal Katz-Ezov (Director, Technion Genome Center, TGC), Roni Koren (Horwitz lab) and Guy Horev (Bioinformatics Knowledge Unit) for their advice and suggestions. This work was supported in part by Grant 332/13 from the Israel Science Foundation and by the Russell Berrie Nanotechnology Institute, Technion. H. S. graduate student fellowship was funded in part by the Irwin and Joan Jacobs Graduate School, Technion.
Author information
Authors and Affiliations
Contributions
HS, SS, and BAH designed the study and wrote the manuscript. HS, SS, and OG prepared the samples and carried out the experiments. OK and MH performed the RNA-Seq data analysis.
Corresponding author
Additional information
Communicated by M. Kupiec.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
294_2019_1012_MOESM2_ESM.tif
Supplementary material 2 (TIFF 258 kb) Fig. S1. PCA plots for WT (A) and ∆chap1 (B); the biological replicates (five for WT, three for ∆chap1) for WT are labeled (r1-r5). Replicates for the same treatment are represented by symbols of the same color (for color coding see legend at top right) (C0: control (DMSO), C0.5: 0.5 mM FA, C1: 1 mM FA, C2: 2 mM FA
294_2019_1012_MOESM3_ESM.tif
Supplementary material 3 (TIFF 180 kb) Fig. S2. Transcript levels for selected genes, measured by qRT-PCR. A. Transcript levels for control (DMSO), 0.5, 1, and 2 mM FA. B. Transcript levels for control (DMSO), and 2 mM FA. The asterisks *, **, ***, **** indicate significance at P < 0.05, 0.01, 0.001, and 0.0001, respectively, in WT and ∆chap1 and between them. The y-axis indicates transcript levels on a logarithmic scale normalized by actin transcript levels. Error bars indicate SD for three biological replicates. C. Gene expression correlation between qRT-PCR (x-axis) and RNA-Seq data (y-axis). Coordinates of each point are the fold changes (FA treatment/control) for the same treatment measured by the two methods, plotted on log2-log2 scale. The correlation coefficient (R2) and linear regression line are shown
294_2019_1012_MOESM4_ESM.tif
Supplementary material 4 (TIFF 390 kb) Fig. S3. MA plots of differentially expressed genes identified in each treatment (C0.5, C1, C2) versus control (C0) for WT (A) and ∆chap1 (B). Data represent individual gene responses plotted as log2 fold change (y-axis) versus the mean of normalized counts (x-axis). The red dots indicate transcripts whose levels differ significantly from the control at adjusted p value < 0.05, with a negative change representing the down-regulated genes and a positive change representing the up-regulated genes. C0: control, C0.5: 0.5 mM FA, C1: 1 mM FA, C2: 2 mM FA
294_2019_1012_MOESM5_ESM.tif
Supplementary material 5 (TIFF 219 kb) Fig. S4. GO (A) and EuKaryotic Orthologous Groups (KOG) (B) functional enrichment analysis of DEGs in cluster 1 and cluster 2. The x-axis indicates functional groups. The y-axis indicates –log(p value). The threshold p value ≤ 0.01 was corrected for multiple testing using the FDR correction. The enrichment factor (frequency of genes of a functional class within the annotated genes in the examined set, divided by frequency within the background set of all the genes in the genome), indicated at the top of each bar. Blue bars, ‘Molecular Function’ (MF) categories in GO; green bars, ‘Cellular Component’ (CO) categories in GO; red bars, ‘Biological Process’ (BP) categories in GO
294_2019_1012_MOESM6_ESM.tif
Supplementary material 6 (TIFF 267 kb) Fig. S5. The expression of the first ten genes that are regulated significantly only in 2 mM FA and have annotation by KOG/Interpro description, with the highest (A) and lowest (B) fold change in WT, according to RNA-Seq data. The x-axis indicates the ID number for each gene. The y-axis indicates fold change in gene expression at 2 mM FA (C2) compared to the control (C0) in WT and ∆chap1
Rights and permissions
About this article

Cite this article
Simaan, H., Shalaby, S., Hatoel, M. et al. The AP-1-like transcription factor ChAP1 balances tolerance and cell death in the response of the maize pathogen Cochliobolus heterostrophus to a plant phenolic. Curr Genet 66, 187–203 (2020). https://doi.org/10.1007/s00294-019-01012-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-019-01012-7