
Abstract
Electrooxidation of phenol was studied on platinum electrode in five aprotic non-aqueous solvents (acetonitrile, dimethyl sulfoxide, dimethyl formamide, acetone, and tetrahydrofuran) with cyclic and normal pulse voltammetry. The cyclic voltammetric results showed that fouling of the electrode surface by the polyphenol took place continuously in dimethyl formamide and a weak passivation of the electrode could be observed in dimethyl sulfoxide. From this solvent, a coherent and mechanically removable, weakly adsorbed layer could be obtained. In the other three solvents, the electrode passivated completely after five scans. Diffusion barrier properties of the polymer formed in acetone were the most pronounced of all solvents towards a redox probe. Normal pulse voltammetric investigations showed that the extent of electrode passivation is insignificant in three of the solvents used. It was due to the application of the short anodic potential pulses where the oxidation of phenol occurred and the formed oligomers diffused into the bulk of solution.
Similar content being viewed by others


Introduction
Phenol is used in industrial processes, and it is also an environmentally hazardous material. Several works aimed to explore the electrochemical behaviour of different phenols for several reasons but mostly in aqueous solutions. Generally, the problem is during their electrooxidation the film formation at the surface of the commonly used bare electrodes (glassy carbon, Pt, Au) and they foul quickly [1,2,3,4,5,6,7]; thus, many limitations arise from this fact. For example, electrochemical determination of the most phenolic compounds in different samples encounters difficulties as reactivation of electrode surface makes the procedure more lengthy. In aqueous solutions, phenol can be oxidized in parallel to the polyphenol film also to 1,4-dihydroxibenzene which oxidizes further to the corresponding benzoquinone [8].
However, in many cases electrode fouling is a problem; in respect of some applications the formed film might have advantageous properties. In the literature, a large amount of reports can be found in concern of aqueous solutions, but less article is about the electrochemistry of phenols in non-aqueous solvents, particularly where the study of unsubstituted phenol was the subject of the work. Especially quinones and non-fouling phenols were studied with voltammetric techniques in acetonitrile [9, 10]. There are works where tetramethylammonium phenolate was electrochemically oxidized in acetonitrile resulting a poly(phenylene oxide) layer on Pt and Au electrode [11]. During electrooxidation of phenol PPO film was also deposited onto ITO surface and the film exhibited electrically non-conductive properties; thus, it was tested in design of nanoscale structures [12]. Investigation of electrode fouling processes in different media explores the properties of the formed films which might be interesting due to many applications, mainly corrosion protection [13, 14], size exclusion properties (molecular sieving) [15, 16]. The latter is widely investigated, and the structure of the deposited polymer film can vary depending on the solvent. Film porosity determines the diffusion transport barrier properties, and redox species can be selected to estimate the permeability. There are some works about the electropolymerization of halogenated phenols from their transition metal complexes and electroinitiated polymerization of phenols having unsaturated side chain mainly in acetonitrile and dimethyl formamide [17,18,19,20,21,22,23,24,25,26]. Application of a strong pyridine-type base determines the product of electrooxidation of phenols; for example, in case of 4-tercbutylphenol the corresponding dimer forms [27]. Bisphenol A was also studied with anodic oxidation in aprotic organic solvents [28]. Ionic liquids provide also an appropriate medium for electrooxidation processes due to their wide potential window. Some phenols showed reversible electrochemical behaviour using ionic liquids as solvent [29, 30].
Further electrochemical importance, our previous investigations have shown that deposition of other aromatics onto nanostructured surface of semiconductors, such as adsorption of carbon nanotubes onto CeO2 or ZnO surfaces, shows itself strong dependence on the solvents used [31, 32].
Another significant aspect of the investigation of surface polymer films in non-aqueous solvents is the electrochemical renewing of the working electrode after their deposition. In contrast to water many other solvents can provide wider potential window; thus, reactivation of electrode is also possible [30].
In this work, we present the results of the electrooxidation of phenol in five non-aqueous solvents using two techniques, cyclic voltammetry and normal pulse voltammetry. As permittivity of the solvent is a critical factor by the electrochemistry of compounds in aprotic non-aqueous solvents, they were selected according to their different dielectric constants. Emphasis is on the film formation, film compactness and composition.
Materials and methods
All chemicals and solvents were analytical grade. As the electrochemistry of phenols is influenced by water content in non-aqueous solvents, freshly purchased anhydrous liquids were used without purification and the solutions were freshly prepared. The water was present in the liquids in concentrations around 10−3 M. As working electrode Pt disc (1 mm in diameter) was used, sealed in polyetheretherketone (product of eDAQ). The counter electrode was a Pt wire with diameter of 1 mm and a silver wire served as reference electrode in non-aqueous solvents. By measurements taken in aqueous solutions, saturated calomel (SCE) electrode was the reference. Before use in most of investigations, the Pt electrode was wet polished on a polishing cloth with 1 and 0.05 μm alumina powder. Then, it was thoroughly washed with deionized water. Then, the electrode was placed in doubly distilled water and its surface was cleaned in an ultrasonic bath to remove the physically adsorbed species and thoroughly washed with deionized water. All electrodes were rinsed with dry acetone directly before measurements to remove the traces of water and again left to dry. Tetrabutylammonium perchlorate (TBuClO4) was used as supporting electrolyte in non-aqueous systems and KCl in aqueous solutions. The solid materials were stored in a desiccator placed into the dark to minimize the introduction of water into the prepared solutions. The prepared solutions of phenol were stored in dark as it is a light-sensitive material. Before the experiments, oxygen purging was not applied.
The electrochemical experiments were carried out at ambient temperature (25 ± 2 °C) with a potentiostat (eDAQ Pty. Ltd., Australia) which was connected to a computer for data acquisition.
Micro-Raman investigations were carried out with a LABRAM HR spectrometer (Horiba Jobin–Yvon, Lille, France). Before all spectroscopic studies, the working electrode was cleaned according to the procedure described previously. After the cleaning, electrodeposition of phenol was accomplished from its solution made with the corresponding solvent in the potential range where phenol electrooxidation took place by application of ten subsequent cyclic voltammetric scans with scan rate of 0.1 V/s. Finally, the electrode was washed with the pure solvent to remove the unreacted phenol and supporting electrolyte from the polymer film. An exception was the polymer formed in dimethyl sulfoxide where it was soaked in pure dimethyl sulfoxide for 5 min and finally thoroughly washed with doubly deionized water to remove completely the solvent. For the complete solvent evaporation the electrode was kept in air for 10 min. The electrode was fixed under the objective of the spectrometer, and the ocular was focused to a point at the platinum surface where a significant amount of deposited polymer could be found. It was illuminated to a red laser light (λ = 632.81 nm) to investigate the electrodeposited polymer.
For the fluorescence measurements, a Fluorolog3 Horiba Jobin–Yvon spectrofluorimeter (Lille, France) was used and a Specord Analytik Jena photometer was applied for taking the absorbance spectra.
Results and discussion
Cyclic voltammetric studies of phenol in the different solvents
An important property of solvents is the potential window where they are stable and can be used to study different soluted compounds. Insets of Fig. 1 show the corresponding cyclic voltammetric curves for the pure solvents containing 0.1 M TBuClO4 as supporting electrolyte. Significant current raising up could be observed close to 2 V in case of acetonitrile; thus, this solvent provides a wide potential window for studying different compounds. Due to this property, acetonitrile is widely used as an inert non-aqueous solvent [33,34,35]. Compared with the other selected solvents, it has far the smallest background current except for tetrahydrofuran. The current increased slowly in tetrahydrofuran due to the large ohmic drop caused by its low permittivity, and therefore the extent of ion association of supporting electrolyte was high. This observation was certified by adding a non-fouling and soluble redox active compound, hydroquinone (1,4-dihydroxibenzene) to the solution in 50 mM concentration and no voltammetric peak appeared in the selected potential window (not shown). The other solvents oxidize above 2 V, so they can be used between 0 and 2 V to study oxidization processes. In the investigated solvents, the platinum electrode is stable during anodic polarization in the applied potential regions.

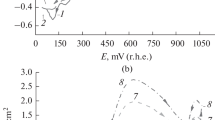
Subsequent cyclic voltammetric curves for 50 mM phenol dissolved in the non-aqueous solvents taken with scan rate of 0.1 V/s (a acetonitrile, b acetone, c dimethyl formamide, d dimethyl sulfoxide, e tetrahydrofuran). Inset graphs show the cyclic voltammograms of the pure solvents containing 0.1 M TBuClO4 supporting electrolyte
By the electrochemical oxidation of phenol, a phenoxy radical forms which couples with another radical or unreacted phenol. The electrode fouling process is based on the continuous entrapment of phenolic units to the formed polymer. In non-aqueous solvents, the pathway leading to the formation of poly(phenylene oxide) is valid to describe the mechanism for the film formation [3, 28, 36,37,38]. The layers which can be electrodeposited are electrically non-conductive and depending on their permeability they can isolate the electrode from the solution containing the electroactive compound under study.
Because of the appearance of the voltammetric peak of phenol in the first scan, a wide window was selected for the solvents, between 0 and 2 or 2.5 V. Five subsequent cycles are shown in Fig. 1 for 50 mM phenol solutions prepared with the selected non-aqueous solvents, respectively. In acetonitrile, the current raised sharply during the first scan resulting an anodic peak around 1.5 V. The fouling of the electrode surface occurred, and during the next scans very small current intensities could be obtained due to the poly(phenylene oxide) formation in accordance with the finding in the literature [11].
The blocking effect caused by polymer formation could be observed also in acetone indicating also that the product of the electrode process is sparingly soluble in this solvent. However, after the large anodic peak at 1.73 V in the first scan there was a small peak in the second scan. Smaller peaks appeared also in the further scans suggesting that the formed film is permeable for the phenol molecules. To verify this hypothesis, the recording of subsequent voltammograms was repeated immediately after each other between 1 and 2 V, but from the second scan peaks did not appear showing that shorter times were available for the filling up of the pores of polymer film by diffusion with phenol molecules.
In dimethyl formamide, the first voltammetric curve does not contain any peak due to the overlapping of the oxidation of phenol and the solvent. Formation of polyphenol took place at the electrode surface which is indicated by the appearance of peaks in the next curves around 1.7 V which is lower than the potential where dimethyl formamide begins to oxidize. Their height decreased slowly from the second to the fifth curve showing that the formed product does not passivate strongly the working electrode. Compared with the cyclic voltammogram of the pure solvent with the ones recorded in the presence of phenol, the current measured at 2.5 V was sufficiently smaller in the solution of phenol showing that only a part of the electrode surface was accessible for the solvent molecules due to the formed polyphenol.
Dimethyl sulfoxide proved to be an appropriate solvent to minimize the fouling effect, but the voltammograms do not contain any qualitative information regarding the phenol electrooxidation as solvent oxidization significantly overlapped with the studied charge transfer process. Around 1.7 V oxidation waves showed up that are attributed to the oxidation of phenol. The voltammograms are almost the same indicating that the electrode passivated weakly. The explanation for the results might be the structure of the formed polymer in dimethyl sulfoxide as it is very permeable. After thorough washing, a light brown layer was visible on the platinum surface and a coherent film consisting of polymers formed during the repetitive scans. The oxidation reactions could take place underneath the layer.
The curves recorded in tetrahydrofuran show ohmic behaviour in accordance with the previously obtained results with 1,4-dihydroxybenzene. The electrode reaction resulted also in an electrode fouling polymer, and electrode reaction was shut off by the fifth scan. Contrarily to the low currents, a thin and coherent film deposits from tetrahydrofuran.
Spectroscopic studies of the electrodeposited films and other products
According to the infrared absorption of the aromatic systems around 1600 cm−1 (stretching vibration), micro-Raman spectra were recorded between 1430 and 1730 cm−1 for each polymer electrodeposited from the corresponding solvent. An image from each polymer coated surface and the spectra are collected in Fig. 2. The images reveal that the surface is covered by polyphenol in various thicknesses. The film deposited from tetrahydrofuran has an arbitrary structure showing that electrodeposition of polymer was favoured in the surface inhomogeneities. Around 1600 cm−1, a broad peak appears that might be attributed to the aromatic stretch whose intensity is in correlation with the density of the layer. Around 1670 cm−1, a smaller peak shows up attributable to the C=O and conjugated C=C double bond stretch in different intensities by all films. There are also compounds having peaks of these functional groups in the region of 1670 and 1715 cm−1 [39,40,41,42,43]. Their intensities indicate that the cyclohexadienone units are also present in the films which form from the phenoxy radical and then, the other phenol molecules join to it. According to other works carried out in acetonitrile, it can be concluded that the electrodeposited product is poly(phenylene oxide) in each solvent as the spectra are similar, but the film formed in tetrahydrofuran has more cyclohexadienone unit than the other ones.

Micro-Raman spectra and optical microscopic images for polyphenol films electrodeposited from the different solvents (a acetonitrile, b acetone, c dimethyl formamide, d dimethyl sulfoxide, e tetrahydrofuran)
The micro-Raman investigations for the polymer formed in dimethyl sulfoxide were extended to other spectral ranges for additional information about polymer composition (not shown). Previously, a large overlapping of solvent and phenol electrooxidation could be observed and coupling of the different radicals might be also possible. This is important to mention as electrooxidation of the solvent can also lead to the formation of radicals. No additional peaks appeared in the range of C–H stretching vibration (between 2800 and 3000 cm−1) characteristic for the CH2 and CH3 groups reinforcing that moieties of solvent molecules are not present in the polyphenol film. Furthermore, spectra were recorded also between 500 and 700 cm−1 where characteristic peak for the S=O double bond stretching appears. In this spectral range, there was no any peak. However, the formation of dimers and oligomers by coupling of a phenoxy radical and a radical formed from dimethyl sulfoxide molecules cannot be excluded. This reaction can result in a cyclohexadienone derivative which can dissolve in the solution phase, and after its production it can diffuse away from the polymer film. Six subsequent constant potential electrolysis was carried out in a dimethyl sulfoxide solution containing 0.1 M phenol. Their time was set to 90 s keeping the potential of the working electrode at 2 V in accordance with the appearance of oxidation waves of phenol. After electrolysis, a light brown layer formed which could be taken off mechanically from the electrode surface which shows that the in situ growth of polyphenol-based membranes might be possible in concentrated monomer solutions. Between the electrolysis times, the electrode surface was polished and thoroughly washed with doubly deionized water and dried according to the procedure described in “Materials and methods” section. The colour of the solution changes during the electrolysis from colourless to light brown suggesting the formation of solution of the polymer.
For complementary information concerning the products, the solutions were ten times diluted with pure dimethyl sulfoxide and then, absorbance measurements were taken between 280 and 500 nm in pure dimethyl sulfoxide, in the initial and electrolyzed solution prepared with dimethyl sulfoxide. The part of the spectrum containing the range where the difference between the two solutions is the most remarkable can be seen in Fig. 3. In that wavelength range, dimethyl sulfoxide has weak absorption and the absorbance of the electrolyzed solution was larger than the absorbance of the solution without electrolysis in the wavelength range between 310 and 400 nm. This is due to the soluble polymer molecules.

Absorbance curves in pure dimethyl sulfoxide (curve a), in the solution containing 0.01 M phenol without electrolysis (curve b) and after electrolysis (curve c)
To get deeper insight into the identity of products fluorescence spectroscopic investigations were carried out between 300 and 500 nm with the electrolyzed and original solution by using 310 nm excitation wavelength (Fig. 4). The reason was for that the difference between the two curves was the highest at 310 nm. The curves clearly show that the oxidized forms of phenol presenting in solution have a large fluorescence peak which is shifted to larger wavelength compared with solution containing phenol monomer indicating the presence of soluble polymeric products.

Fluorescence spectra taken with 310 nm excitation wavelength for the original solution of phenol (initial concentration: 0.01 M) prepared with dimethyl sulfoxide (curve a) and for the electrolyzed solution (curve b)
The formed polymers are scarcely soluble in the selected solvents except for dimethyl sulfoxide as the light brown colour appeared during the electrolysis indicated it. In this solvent, the polymer solubility was evaluated by using the absorbance curves taken in the ultraviolet region for phenol. Due to the electrolysis, the concentration decreased by 37% of the initial value (diluted to 10 mM) which composed of the polymer quantity precipitated forming membrane at the electrode surface and the polymer particles dissolved in the solution. The membrane was peeled off from the electrode surface and washed thoroughly with distilled water to remove the unreacted phenol and supporting electrolyte from it. After drying, its mass was weighted. By taking the molar mass of phenoxy units, the molar quantity of phenol could be calculated and then the equivalent concentration change of the solution knowing its volume. The difference between the change calculated directly from the calibration curve and the equivalent change due to the membrane formation results in the concentration of phenol presenting in the solution in form of polymer. Due to the presence of different length chains, the polymer concentration can be added in the form of concentrated solute mass. Multiplying by the molar mass of phenoxy unit, the solubility is 34.44 mg/L.
Complementary voltammetric studies in acetonitrile
As acetonitrile has a wide potential window for oxidation processes, the cyclic voltammetric experiments with phenol were extended to 3 V (Fig. 5). At potentials higher than 2 V, the current raised sharply at each scan which is due to the electrooxidation of the formed film at 1.4 V and unreacted phenol molecules. The previously (around 1.4 V) formed polyphenol film was removed from the electrode surface caused by the dissolving by acetonitrile. In another work, effect of addition of acetonitrile to ionic liquids was investigated on boron-doped diamond electrode [30]. It was found that acetonitrile breaks attractive forces between the formed polymer and electrode surface at higher potentials. At platinum electrode similar phenomena play a key role in the surface activation. So, instead of the continuous decreasing the currents between 2 and 3 V increased continuously in the subsequent scans until a saturation could be reached. In contrast to the voltammetric peaks obtained in the subsequent scans between 0 and 2 V, the peak heights of peaks appeared at 1.4 V from the second scans were almost identical. It evidences that the electrode surface was anodically cleaned partially and the peak heights in cycles 2–5 were present and are remarkably higher than by cycling between 0 and 2 V. It shows that a significant portion of the electrode surface area was accessible for the further electrooxidation process.

Subsequent cyclic voltammograms for 50 mM phenol in acetonitrile in the extended potential window to 3 V (v = 0.1 V/s, supporting electrolyte 0.1 M TBuClO4)
Performance of the electrode coated with the polyphenol films in electrode reactions of another redox active substance
In order to get more insights into the properties of the electropolymerized polyphenol films, their diffusion barrier properties were tested by selecting K3[Fe(CN)6] as a redox probe and its 5 mM aqueous solution was used containing 50 mM KCl as supporting electrolyte. The film electrodeposition from the different non-aqueous solvents was carried out between 0 and 2 V or between 0 and 2.5 V similarly to the electrodeposition illustrated in “Cyclic voltammetric studies of phenol in the different solvents” Section (v = 0.1 V/s). Ten cyclic voltammetric scans were applied consecutively to cover completely the Pt surface with polyphenol layer. After washing with deionized water linear sweep voltammograms were taken with the electrode covered by polymer film started from 0.8 to 0 V versus SCE in the aqueous solution of the redox probe with scan rate of 0.1 V/s.
The recorded linear voltammetric curves of [Fe(CN)6]3− ion in its aqueous solution are collected in Fig. 6. Curve a is related to the bare platinum electrode where a cathodic voltammetric peak shows up at 0.17 V. The curves taken with the electrode coated with the different polyphenol layers show smaller peak indicating that the layers formed act as diffusion barrier for the [Fe(CN)6]3− ions. In this potential range, the surface of the previously cleaned electrode and polyphenol-coated platinum electrode is stable, so the observed diminished currents are due to the polyphenol films. The polyphenol formed in acetone exhibited the highest resistance to the redox reaction. Previously in the electrodeposition process, very small peaks appeared also in the last cycles indicating that phenol molecules can enter the pores, but the permeation of larger [Fe(CN)6]3− ions is prevented. The curves taken with the electrode coated with layer formed in dimethyl sulfoxide have a small reduction wave in the corresponding potential range of charge transfer process of [Fe(CN)6]3− ion. It can be attributed to the higher pores presenting in the film permeable for the redox probe molecules.

Linear sweep voltammetric curves for aqueous solution of 5 mM [Fe(CN)6]3− in presence of 50 mM KCl supporting electrolyte (v = 0.1 V/s) taken with Pt electrode covered with the polyphenol films electrodeposited from the different solvents (a: bare electrode, b: acetonitrile, c: dimethyl formamide, d: dimethyl sulfoxide, e: acetone, f: tetrahydrofuran)
Performance of normal pulse voltammetry in the electrooxidation of phenol
By studying electrooxidation of phenol with cyclic voltammetry, the film formation is based on the application of anodic polarization for longer times resulting smaller and smaller voltammetric peaks by repeating the scans. The time of polarizing the working electrode to the potential appropriate for the electrooxidation of phenol is also a dominant factor in passivation of the electrode. The examination of techniques applying periodically interrupted potential program explores new alternatives also in electroanalysis. Normal pulse voltammetry (NPV) applies incrementally increased potential pulses where the pulse potential changes linearly from the base potential (where the scanning is started from) to the final potential (where the measurement ends). Electrode process occurs only during the pulses whose width is usually in the millisecond range. Between the pulses (this time is termed pulse interval), there is no electrode process as the electrode potential must be set to the base potential where the electroactive material does not react. If the pulse width is shortened significantly, it contributes to suppression of the fouling process and higher current signals can be obtained due to the large flux of electroactive material to the electrode. This technique was also tested in the electrooxidation of phenol in the selected aprotic non-aqueous solvents.
In the different solvents containing only 0.1 M TBuClO4, normal pulse voltammograms were recorded to see how the background current changes between 0 and 3 V by 0.1 V/s scan rate and 5 ms pulse width (insets of Fig. 7). This 5 ms pulse interval was a compromise as by setting smaller values the measured currents would interfere with the condenser current flowing in the first milliseconds and at higher values the possibility of the electrode passivation is significant. The anodic potential window was extended here to 3 V as the signal shifted to more positive potentials. Base potential was set to 0 V. By wider pulse widths, the electrode passivation was significant. Similarly to the cyclic voltammetric results, low currents could be obtained in acetonitrile and tetrahydrofuran in contrast to the other three solvents having high background current at potentials higher than 1 V.

Subsequent normal pulse voltammetric curves for 50 mM phenol in different non-aqueous solvents by setting 0.1 V/s scan rate and 5 ms pulse width in the presence of 0.1 M TBuClO4 (a acetonitrile, b acetone, c dimethyl formamide, d dimethyl sulfoxide, e tetrahydrofuran). Inset graphs show the normal pulse voltammograms in the pure solvents containing 0.1 M TBuClO4 supporting electrolyte
Five subsequent normal pulse voltammetric curves were recorded in the different solvents containing 50 mM phenol in the same conditions as in the pure solvents (Fig. 7). Generally, in an electrode process where fouling cannot take place the signal is a current plateau as function of potential. In contrary, when phenol was the electroactive compound current peaks appeared in the voltammograms taken in acetonitrile and acetone indicating that the electrode surface is also blocked by the formed polymer but speed of peak current decrease was sufficiently smaller than in the cyclic voltammetric experiments.
In dimethyl formamide, dimethyl sulfoxide and tetrahydrofuran, there were no anodic peaks and the curves recorded in the subsequent measurements were almost the same indicating that electrode passivation does not take place. On the other hand, currents increased linearly by increasing the pulse potential. Previously, in the cyclic voltammetric studies peaks of phenol oxidation could be observed in dimethyl formamide, but in NPV the current increased linearly from 1.5 V. In the potential range where phenol and solvent react, the slope of the NPV curves is smaller than in the pure solvents. It also indicates that these solvents are not good for electrochemical investigation of phenol, but the findings clearly show the absence of continuous electrode poisoning. Only the currents measured at the end of the short potential pulses are collected by the instrumentation, and during the potential pulses only smaller oligomers formed and they were still present at the electrode surface. During their formation, they hindered the diffusion of other molecules to the electrode surface and the subsequent diffusion into the bulk occurred in the pulse intervals where the potential was 0 V.
Conclusions
The results obtained with phenol electrooxidation in different aprotic non-aqueous solvents showed that polyphenol film formation can also take place at the electrode. Fouling effect was less significant by using normal pulse voltammetry compared with cyclic voltammetry in all of the solvents. Dimethyl sulfoxide can provide an excellent solvent for electrolysis of phenols and other organic compounds, and it is a good solvent for electrochemical in situ synthesis of polyphenol-based membranes which is a subject of our future investigations. The permeability tests showed that the size exclusion was the most significant in case of film deposited from acetone. Acetonitrile is the most promising of the selected solvents in respect of electrode reactivation as it can provide an appropriate medium polarizing the electrode to potentials above 2 V for the removing of the formed polymer film. Further investigations are needed to clarify its usefulness in electrode reactivation after electrodeposition from different media.
Although dimethyl sulfoxide, dimethyl formamide and tetrahydrofuran are not good in respect of investigation of phenol due to the significant overlapping of their electrooxidation, films having specific properties can be built with the use of these solvents. Especially, the layer formed in dimethyl sulfoxide might be useful in future applications.
References
Ureta-Zanartu MS, Bustos P, Berríos C, Diez MC, Mora ML, Gutiérrez C (2002) Electrooxidation of 2,4-dichlorophenol and other polychlorinated phenols at a glassy carbon electrode. Electrochim Acta 47:2399–2406
Sistiaga M, Pierna AR, Marzo FF, Altube A, Lorenzo A (1998) Electrooxidation of phenol on amorphous Ni–40Nb–_1yx/Pt–xSn alloys. Appl Surf Sci 133:124–128
Ahmed S, Ahmad M, Butt SB (2012) Electrooxidation of chloro, nitro, and amino substituted phenols in aqueous medium and their heterogeneous kinetics. Res Chem Intermed 38:705–722. https://doi.org/10.1007/s11164-011-0410-z
Fino D, Jara CC, Saracco G, Specchia V, Spinelli P (2005) Deactivation and regeneration of Pt anodes for the electro-oxidation of phenol. J Appl Electrochem 35:405–411. https://doi.org/10.1007/s10800-005-0799-4
Skowronski JM, Krawczyk P (2004) Electrooxidation of phenol at exfoliated graphite electrode in alkaline solution. J Solid State Electrochem 8:442–447. https://doi.org/10.1007/s10008-003-0483-8
Dulal SMSI, Won M, Shim Y (2010) Carbon fiber supported platinum nanoparticles for electrooxidation of methanol and phenol. J Alloy Compd 494:463–467
Tahar NB, Savall A (2009) Electropolymerization of phenol on a vitreous carbon electrode in alkaline aqueous solution at different temperatures. Electrochim Acta 55:465–469
Uskova IK, Bulgakova ON (2014) Cyclic voltammetry of phenol. J Anal Chem 69:542–547. https://doi.org/10.1134/s1061934814060148
Tessensohn ME, Hirao H, Webster RD (2013) Electrochemical properties of phenols and quinones in organic solvents are strongly influenced by hydrogen-bonding with water. J Phys Chem C 117:1081–1090. https://doi.org/10.1021/jp311007m
Richards JA, Whitson PE, Evans DH (1975) Electrochemical oxidation of 2,4,6-tri-tert-butylphenol. J Electroanal Chem 63:311–327
McCarley RL, Irene EA, Murray RW (1991) Permeant molecular sieving with electrochemically prepared 6-nm films of poly(phenylene oxide). J Phys Chem 95:2492–2498. https://doi.org/10.1021/j100159a071
Rhodes CP, Long JW, Doescher MS, Fontanella JJ, Rolison DR (2004) Nanoscale polymer electrolytes: ultrathin electrodeposited poly(phenylene oxide) with solid-state ionic conductivity. J Phys Chem 108:13079–13087. https://doi.org/10.1021/jp047671u
Mengoli G, Musiani MM (1994) Phenol electropolymerization: a straight route from monomers to polymer coatings. Prog Org Coat 24:237–251
Prabakaran M, Kim S, Hemapriya V, Chung I (2016) Evaluation of polyphenol composition and anti-corrosion properties of Cryptostegia grandiflora plant extract on mild steel in acidic medium. J Ind Eng Chem 37:47–56
Lapuente R, Cases F, Garcés P, Morallón E, Vázquez JL (1998) A voltammetric and FTIR–ATR study of the electropolymerization of phenol on platinum electrodes in carbonate medium: influence of sulfide. J Electroanal Chem 451:163–171
Tahar NB, Savall A (2012) Effect of electropolymerisation conditions on the permeability of polyphenol films deposited on a vitreous carbon electrode. Electrochim Acta 82:427–433
Cihaner A, Önal AM (2000) Electroinitiated polymerization of 2-allylphenol. Polym Bull 45:45–52. https://doi.org/10.1007/s002890070055
Sen S, Usanmaz A, Önal AM (1995) Electroinitiated polymerization of allylphenylether. J Polym Sci Polym Chem 33:1817–1821. https://doi.org/10.1002/pola.1995.080331108
Sacak M, Akbulut U, Kisakurek D, Turker L, Toppare L (1989) Electroninitiated polymerization of bis(trichlorophenoxo)-tetramethylethylene diamine copper(II). J Polym Sci Polym Chem 27:1599–1608. https://doi.org/10.1002/pola.1989.080270511
Kisakurek D, Sen S, Aras L, Turker L, Toppare L (1991) Polymerization of bis(2-bromo-4,6-dichlorophenoxo)bis(pyridine) copper (11) complex in DMF by electroinitiation. Polymer 32:1323–1328
Sacak M, Akbulut U, Kisakurek D, Toppare L (1989) Electroinitiated polymerization of bis(4-bromo-2,6-dichIorophenoxo)N, N, Nʹ, Nʹ-tetramethylethylenediamine copper(ll) complex. Polymer 30:928–932
Aydeniz K, Önal AM, Kisakurek D (2001) Electrochemical polymerization of (2,4,6-trihalophenolato)nickel(II) complexes in solution. Eur Polym J 37:2017–2023
Sen S, Kisakurek D (1993) Synthesis and characterization of poly(dichlorophenylene oxide)s based on the electro-oxidation of bis(2,4,6-trichIorophenoxo)bis(pyridine)copper(II) complex. Polymer 34:4146–4149
Özalp-Yaman S, Bastürkmen M, Kisakurek D (2005) Simultaneous novel synthesis of conducting and non-conducting halogenated polymers by electroinitiation of (2,4,6-trichloro- or 2,6-dichlorophenolato)Ni(II) complexes. Polymer 46:6786–6796
Kesici N, Kisakurek D (2001) Polymerization and characterization of various di(pyridine)bis(trihalophenolato)cobalt(II) complexes in solid and melt states. Polym Int 50:1061–1067. https://doi.org/10.1002/pi.744
Akbulut U, Sacak M, Kisakurek D, Toppare L (1990) Electroinitiated polymerization of bis(tribromophenoxo)-N, N, Nʹ, Nʹ-tetramethylethylenediamine copper(II) complex. Br Polym J 22:65–71. https://doi.org/10.1002/pi.4980220110
Abidi M, Derbel N, Hkiri R, Sbihi HM, Said H, Morallon E, Besbes-Hentati S (2017) Electrodeposition of 4,4ʹ-di-tert-butylbiphenyl peroxide from the anodic oxidation of p-tert-butylphenol in an alkaline acetonitrile solution. J Appl Electrochem 47:507–516. https://doi.org/10.1007/s10800-016-1041-2
Chan YY, Yue Y, Li Y, Webster RD (2013) Electrochemical/chemical oxidation of bisphenol A in a four-electron/two-proton process in aprotic organic solvents. Electrochim Acta 112:287–294
Loyson P, Imrie C, Gouws S, Barton B, Kruger E (2009) Bmim ionic liquids as media for the electrochemical oxidation of 2,6-di-t-butylphenol. J Appl Electrochem 39:1087–1095. https://doi.org/10.1007/s10800-008-9760-7
Saravanan KR, Sathyamoorthi S, Velayutham D, Suryanarayanan V (2012) Voltammetric investigations on the relative deactivation of boron-doped diamond, glassy carbon and platinum electrodes during the anodic oxidation of substituted phenols in room temperature ionic liquids. Electrochim Acta 69:71–78
Li H, Nie JC, Kunsági-Máté S (2010) Modified dispersion of functionalized multi-walled carbon nanotubes in acetonitrile. Chem Phys Lett 492:258–262
Li H, Petz A, Yan H, Nie JC, Kunsági-Máté S (2011) Morphology dependence of Raman properties of carbon nanotube layers formed on nanostructured CeO2 films. J Phys Chem C 115:1480–1483. https://doi.org/10.1021/jp108023f
Rizvi MA, Akhoon SA, Maqsood SR, Peerzada GM (2015) Synergistic effect of perchlorate ions and acetonitrile medium explored for extension in copper redoximetry. J Anal Chem 70:633–638. https://doi.org/10.1134/S1061934815050093
Cassidy J, Khoo SB, Pons S, Fleischmann M (1985) Electrochemistry at very high potentials: the use of ultramicroelectrodes in the anodic oxidation of short-chain alkanes. J Phys Chem 89:3933–3935. https://doi.org/10.1021/j100264a034
Dibble T, Bandyopadyay S, Ghoroghcian J, Smith JJ, Sarfarazi F, Fleischmann M, Pons S (1986) Electrochemistry at very high potentials: oxidation of rare gases and other gases in nonaqueous solvents at ultramicroelectrodes. J Phys Chem 90:5275–5277. https://doi.org/10.1021/j100412a075
Heras MA, Lupu S, Pigani L, Pirvu C, Seeber R, Terzi F, Zanardi C (2005) A poly(3,4-ethylenedioxythiophene)-poly(styrene sulphonate) composite electrode coating in the electrooxidation of phenol. Electrochim Acta 50:1685–1690
Kennedy B, Glidle A, Cunnane VJ (2007) A study of the oxidation and polymerisation of meta substituted phenol and aniline derivatives. J Electroanal Chem 608:22–30. https://doi.org/10.1016/j.jelechem.2007.05.006
Bankovic P, Mojovic Z, Milutinovic-Nikolic A, Jovic-Jovicic N, Marinovic S, Jovanovic S (2010) Mixed pillared bentonite for electrooxidation of phenol. Appl Clay Sci 49:84–89. https://doi.org/10.1016/j.clay.2010.04.012
Tripathi GNR, Schuler RH (1987) Resonance Raman spectra of p-benzosemiquinone radical and hydroquinone radical cation. J Phys Chem 91:5881–5885. https://doi.org/10.1021/j100307a014
Larsen KL, Barsberg S (2010) Theoretical and Raman spectroscopic studies of phenolic lignin model monomers. J Phys Chem B 114:8009–8021. https://doi.org/10.1021/jp1028239
Perera PN, Schmidt M, Chiang VL, Schuck PJ, Adams PD (2012) Raman-spectroscopy-based noninvasive microanalysis of native lignin structure. Anal Bioanal Chem 402:983–987
Pompeu DR, Larondelle Y, Rogez H, Abbas O, Pierna JAF, Baeten V (2018) Characterization and discrimination of phenolic compounds using Fourier transform Raman spectroscopy and chemometric tools. Biotechnol Agron Soc 22:13–28
Abbas O, Compére G, Larondelle Y, Pompeu D, Rogez H, Baeten V (2017) Phenolic compound explorer: a mid-infrared spectroscopy database. Vib Spectrosc 92:111–118. https://doi.org/10.1016/j.vibspec.2017.05.0080924-2031
Acknowledgements
Open access funding provided by University of Pécs (PTE). Financial support of the GINOP 2.3.2-15-2016-00022 and the work was also supported by Fundamental Research Funds for the Central Universities (20720170084) of China. The present scientific contribution is dedicated to the 650th anniversary of the foundation of the University of Pécs, Hungary.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kiss, L., Bősz, D., Kovács, F. et al. Investigation of phenol electrooxidation in aprotic non-aqueous solvents by using cyclic and normal pulse voltammetry. Polym. Bull. 76, 5849–5864 (2019). https://doi.org/10.1007/s00289-019-02678-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-019-02678-2