
Abstract
Metasilicic acid obtained from aqueous solution of sodium silicate was allowed to react with excess glycidol. The obtained semi-product was hydroxyalkylated with ethylene carbonate to obtain oligoetherol. The structure of the oligoetherol was determined by chemical and instrumental methods. Further the oligoetherol was used to obtain foamed polyurethane compositions. The properties of the foams were also studied.
Similar content being viewed by others
Introduction
Classic rigid polyurethane foams are flammable and have low thermal resistance, which restrict their use. The attempts to improve their properties are nowadays the matter of interest. One of the methods to improve their thermal stability is by introduction of oligoetherol with thermally resistant rings, like 1,3,5-triazine, purine, or perhydro-1,3,5-triazine ones to foamed composition [1]. The foams obtained with these substrates show thermal stability at 200 °C and can be exploited continuously within 140–150 °C temperature range [2]. In order to decrease their flammability it is reasonable to introduce phosphorus, chlorine, boron, or silicon elements into their structure [3,4,5]. This can be achieved both by chemical incorporation of these elements into the appropriate oligoetherols or polyurethane precursor (urethane oligomer) or addition of substances containing these elements into foaming composition. The oligoetherols with phosphorus and/or silicon elements are not common substrates for polyurethane foams. On the contrary, the addition of compounds with P and/or Si elements at foaming step to get polyurethane compositions were already studied [6]. The polyetherols with Si were synthesized from R(OH)3 triols obtained from propylene oxide (PO). PO was mixed with isophorone isocyanate at 1:3.05 molar ratio in presence of dibutyltin dilaurate and heated at 100 °C to get the product terminating with isocyanate groups [6]. The isocyanate groups were then blocked with 3-aminopropyltriethoxysilane and heated to 60 °C. The obtained product was hydrolyzed in water–alcohol mixture, pH = 5–6 to obtain a polymer useful to protect wool against felt.
Introduction of silicon-containing fillers into foaming compositions improves thermal stability of polyurethane foams (PUF). Yang et al. demonstrated that colloidal silica led to considerable increase of thermal stability of polyurethane foil in comparison with the non-modified foil [7]. When talc, mica, or montmorillonite are used as fillers they need to intercalate with amino acids, ammonium, or phosphonium salts [8], otherwise the fillers separate from compositions. Proper homogenization of silica nanoparticles with polyurethane led to materials of improved mechanical and thermal properties as well as the flame resistance, although these materials had diminished ability to form pores [9,10,11,12]. From other studies we have known that addition of small amount of reactive silicone compounds could improve thermal stability and flame resistance of PUFs [11, 12]. The effects can be attributed to decrease of flammable organic components in PUF and also that the silica appears as isolating layer on the surface of PUF inhibiting heat flow. Moreover, silicone components are ecologically friendly and combustion of such materials is not accompanied by corrosive smokes [13,14,15]. Siloxanes terminated with hydroxyl groups react with urethane prepolymers and incorporate into structure of polyurethane. The series of thermally resistant polyurethanes were obtained by polycondensation of bis(chloroformates) with aromatic diamines containing silicone or germanium as central atom [16]. Recently, silsesquioxanes were reported as polyurethane modifiers improving thermal resistance. The silsesquioxanes with many peripheral hydroxyl groups are able to facilely react with isocyanate groups of urethane prepolymers to give polyurethanes of improved thermal resistance [13,14,15,16]. They can also be modified by hydroxyalkylation, epoxidation, amination, etc., to give modified silsesquioxanes able to react with isocyanate groups. Hybrid foams formed of polyurethane and polysiloxanes were demonstrated as better heat insulators than PUFs [17].
The up-to-date methods on introduction of silicon to polyurethane foams required expensive silicon-containing substrates or sophisticated synthetic routes as that using silsesquioxanes. In our work we have attempted to obtain silicon-containing oligoetherols starting from metasilicic acid (MSA) for further application of silicon-containing oligoetherols to form polyurethane foams.
Experimental
Syntheses
Synthesis of metasilicic acid
40% water glass (DRAGON, Poland) was treated with concentrated hydrochloric acid (pure, POCH, Poland) to give a white precipitate, which was filtered off and washed with copious amount of water till a neutral filtrate was obtained. The precipitate was dried at ambient temperature, and then at 80 °C to constant mass.
Reaction of metasilicic acid with glycidol
7.8 g (0.1 mol) of metasilicic acid (MSA) and 29.6 g (0.4 mol) of glycidol (GL, pure, SIGMA-ALDRICH, Germany) were placed in a three-necked 250-cm3 flask equipped with mechanical stirrer, reflux condenser and thermometer and heated to 120 °C. An exothermic process occurred causing an increase in the reaction temperature to 170 °C. The reaction was carried out for 1.5 h at 180 °C. A semi-product was obtained wherein the molar ratio MSA:GL = 1:4.
Reactions of semi-product MSA:GL = 1:4 with ethylene carbonate
37.4 g (0.1 mol) of semi-product MSA:GL = 1:4, ethylene carbonate (EC, 26.3 g, 0.3 mol; pure, SIGMA-ALDRICH, Germany), and 0.35 g (0.0025 mol) of potassium carbonate as catalyst (pure, POCH, Poland) were placed in a three-necked 250-cm3 flask equipped with mechanical stirrer, reflux condenser and thermometer and heated 145 °C for 14 h. The isolated product with MSA:GL:EC = 1:4:2.7 molar ratio was obtained (based on mass balance).
Analytical methods
The course of reaction between MSA and GL was followed by measuring the content of epoxide groups [18]. The progress of reaction between semi-product and EC was monitored using barium hydroxide method described in [19]. Acidic number (AN) of obtained oligoetherol was determined by titration with 0.1 M NaOH in the presence of phenolphthalein. Elemental analysis for C, H, N, were done with EA 1108, Carlo-Erba analyzer. Silicon was determined spectrophotometrically in the form of SiO2 after previous mineralization of oligoetherol. The 1H-NMR spectra of products were recorded at 500 MHz Bruker UltraShield in DMSO-d6 with hexamethyldisiloxane as internal standard. IR spectra were registered on ALPHA FT-IR BRUKER spectrometer in KBr pellets or ATR technique. MALDI-ToF spectra (matrix-associated laser desorption ionization time of flight) of oligoetherols were obtained on Voyager-Elite Perceptive Biosystems (US) mass spectrometer working at linear mode with delayed ion extraction, equipped with nitrogen laser working at 352 nm. The method of laser desorption from matrix gold was used. Therefore, the observed peaks corresponded to the molecular ions plus Au and K+ (from catalyst) ions. The samples with diluted with water to 0.5 mg/cm3 concentration. Molecular mass (number-averaged and weight-averaged) and polydispersity of oligoetherol were determined by gel permeation chromatography using the following parameters: 25 ± 0.1 °C temperature, 1 cm3/min volume flow of eluent, 20 μdm3, volume of inlet chamber, 4–5 mg/cm3 polymer concentration, 30 min analysis time, eluent: N,N-dimethylformamide, calibration reference: polystyrene. In order to identify side products (glycols and polyglycols) formed in the reaction of semi-product MSA:GL = 1:4 with EC and the compounds formed in consecutive reactions with EC, the oligoetherols were separated chromatographically using cyclohexanone (analytical grade S.A. POCH, Gliwice, Poland) as internal standard. The gas chromatograph HP 4890A was used, equipped with HP-FFAP column of 30 m length, 0,53 mm diameter, 1,5 μm film thickness and 220 °C port temperature and temperature profile: 50–220 °C, with 20 deg/min heating rate, the helium flow 18.3 cm3/min, and 0.2 μdm3 sample volume. Series of reference substances were used: ethylene glycol (ED), diethylene glycol (DEG), triethylene glycol (TEG) tetraethylene glycol (TeEG) (pure Aldrich, UK). The percentage of diols in products was determined based on calibration curves with the same internal standard using formula (1):
where m cd, m t diol mass or consecutive product of its reaction with alkylene carbonate and mass of standard, respectively, S cd, S t integrated peak area of diol or consecutive product and standard, respectively, a, b experimental coefficients of calibration curves.
The calibration coefficients and retention times of diols are collected in Table 1. Mass of products obtained from EC or PC and water and mass of products of consecutive reactions between diols and alkylene carbonates were calculated from formula (3) (m cd). The percentage of side products were calculated considering total sample mass (m p) according to formula 2:
Physical properties of oligoetherols
Refraction index, density, viscosity, and surface tension of oligoetherols were determined with Abbe refractometer, pycnometer, Höppler viscometer (type BHZ, prod. Prüfgeratewerk, Germany) [20] and by the detaching ring method [21], respectively.
Obtaining of polyurethane foams
Foaming of oligoetherols was performed in 500-cm3 cups at room temperature. The foams were prepared from 10 g of oligoetherol, to which 0.23–0.31 g of surfactant (Silicon 5340, pure, Houdry Hülls, USA) and 0.05–0.32 g of triethylamine (pure, Fluka, Switzerland) as catalyst and water (2–4%) as blowing agent were added. After the homogenization the polymeric diphenylmethane 4,4′-diisocyanate was added. The commercial isocyanate containing 30% of tri-functional isocyanates was used (Merck, Germany). The mixture was vigorously stirred until creaming began. The samples for further studies were cut from the obtained foams.
Properties of foams
The apparent density [22], water uptake [23], dimensional stability in temperature 150 °C within 24 h [24], heat conductance coefficient (IZOMET 2104, Slovakia) and compressive strength [25] of PUFs were measured. Thermal resistance of modified foams was determined both by static and dynamic methods. In static method the foams were heated at 150 and 175 °C with continuous measurement of mass loss and determination of mechanical properties before and after heat exposure. In a dynamic method thermal analyses of foams were performed in ceramic crucible at 20–600 °C temperature range, with ca 100 mg sample, under air atmosphere with Thermobalance TGA/DSC 1 derivatograph, Mettler, with 10 °C/min heating rate. Flammability of foams was determined by oxygen index and horizontal test according to norm [26] as follows: the foam samples (150 × 50 × 13 mm) were weighed, located on horizontal support (wire net of 200 × 80 mm dimensions) and the line was marked at the distance of 25 mm from edge. The sample was set on fire from the opposite edge using Bunsen burner with the blue flame of 38 mm height for 60 s. Then the burner was removed and time of free burning of foam reaching marked line or cease of flame was measured by stopwatch. After that the samples were weighed again. The rate of burning was calculated according to the expression:
if the sample was burned totally, or using equation:
if the sample ceased burning, where L e the length of burned fragment, measured as the difference 150 minus the length of unburned fragment (in mm). According to norms, if the burned fragment has 125 mm length, the foam is considered as flammable.
t b , t e the time of propagation of flame measured at the distance between starting mark up to the end mark or as the time of flame cease.
The mass loss Δm after burning was calculated from the formula:
where m o and m mean the sample mass before and after burning, respectively.
The morphology of PUFs was investigated by an optical microscope Nikon Eclipse LV100 POL, using a digital camera Sight DS-5Mc at 5.0 or tenfold magnification.
Results and discussion
Synthesis of oligoetherol
MSA was obtained straight by acidifying aqueous sodium silicate solution with hydrochloric acid:
Silicic acid is soluble in neither water nor in organic solvents, thus its reactivity is quite low. It does not react with alkylene carbonates, which are strong hydroxyalkylating agent for organic compounds slightly soluble in organic solvents like isocyanuric acid, melamine, or uric acid [27]. Previously we have demonstrated [28] that glycidol (GL) is comparably reactive hydroxyalkylating agent, therefore we have applied it for MSA hydroxyalkylation. Preliminary experiments have indicated that MSA partially dissolved and reacted with GL. When heated with GL up to 120 °C the MSA has reacted exothermally causing temperature jump to 180 °C. The 1:4 MSA:GL system was kept at this temperature for 1.5 h. The isolated resin product indicated acidic number equal to zero; neither did it contain unreacted GL, as was determined by epoxide number equal zero. The product was contaminated with poly(silicic acid). Nonetheless, this semi-product was mixed with EC at 1:3 molar ratio. It dissolved slowly in EC and reacted to form straw colored liquid oligoetherol, still with admixture of polysilicic acid, according to the reaction scheme below:

where x + y = p; a + b + c + d = n.
Poly(silicic acid) was filtered off. Its percentage in post-reaction mixture was 9.5 mass %. Taking into account the mass balance the ratio or MSA:GL:oxyethylene group (formed from EC) was 1:10:6.8 in the product. The C, H elemental analysis (Table 2) corroborated well with the calculated values, considering that about 0.3 mol of ethylene carbonate per mole of acid decomposed thermally.
The structures of products were determined also by IR and H-NMR spectra. There are two valence stretching vibration bands of O–H and C–OH groups at 3404 and 1035 cm−1, respectively, in the IR spectrum of GL. The band at 1260 cm−1 is attributed to GL oxirane ring band (Supplementary materials, Fig. S.1). In the IR spectrum of MSA (Fig. S.2) there are two broad bands at 3600–3000 cm−1 from hydroxyl stretching vibrations and strong, broad band from Si–OH overlapping with Si–O–Si band at 1100 cm−1, characteristic of poly(silicic acid). In the IR spectrum of the product of reaction between MSA and GL (Fig. S.3), the broad band within 3500–3000 cm−1 is replaced by lower intensity band attributed to OH groups in hydroxyalkylated acid. Also the band due to Si–OH at 1100 cm−1 disappears; instead the C–OH band at 1034 cm−1 is present. Additional strong band at 2868 cm−1 from C–H stretching vibrations is present. In the IR spectrum of the obtained oligoetherol (Fig. S. 4) there is broad valence vibration band from OH groups within 3250–3600 cm−1, while C–OH bands is observed at 1117 cm−1. The high-intensity C–O–C band at 1042 cm−1 is overlapped by the C–O–Si one. The presence of C–O–Si bonds is confirmed by the band at 934 cm−1 [29].
The 1H–NMR spectrum of GL (Fig. S. 5) is complicated due to the presence of chiral center at C-2 imposing magnetic non-equivalence on both flanking methylene CH2 groups attached to C-2. The assignment of multiplets is in accordance with data in Spectra Database for Organic Compounds (SDBS) [30]:

In the 1H-NMR spectrum of the product of reaction between MSA and GL (Fig. S.6) the absence of resonances around 3 ppm and at lower chemical shift clearly indicates the absence of methine proton in the epoxide ring. Instead, all methine and methylene proton resonances of hydroxyalkyl groups occur within 3.2–3.6 ppm. The hydroxyl proton broad signal at 4.4–4.9 ppm was identified by selective deuteration with D2O. In the spectrum of oligoetherol (Fig. S. 7), the resonances from protons of methylene and methine groups formed by ring opening of GL and EC are present within 3.2–3.6 ppm, while hydroxyl group resonance is centered at 4.5 ppm.
The composition of oligoetherols can be determined by MALDI-ToF spectrometry method. The spectra showed trace amounts of substrates identified by the appropriate low m/z, and also corresponding to oligomeric forms of MSA of the general formula (H2SiO3) n , where n = 2, 3 or 4. From detailed analysis of MALDI-ToF spectra it can be concluded that even at the appropriate molar ratio of reagents the mixtures are obtained, in which MSA is hydroxyalkylated with GL at various levels and the product of reaction between the semi-product and EC at various levels of hydroxyalkylation. Thus, the satellite peaks with masses increased by m/z = 74 or by masses of attached ions from matrix, and also the peaks of masses diminished by m/z = 18 due to elimination of water in self-condensation of MSA are present. The series of peaks differing by m/z = 44 derived from consecutive addition of EC are also present.
The obtained oligoetherols were analyzed by gas chromatography in order to determine the amount of side products formed in reaction of EC with water released from products of MSA hydroxyalkylation or with diols. In separate control experiments the products of EC with reference diols like glycol and polyglycols were analyzed chromatographically (Table 1). It has been found that the percentage of glycols and their consecutive products did not exceed 20 mass % in the products (Table 3), which is comparable to that found previously in case of melamine–ethylene carbonate system [31]. The presence of low molecular products in oligoetherol corroborates well with hydroxyl number (Table 2) which is 806.8 mg KOH/g, i.e., higher than the theoretical (602.6 mg KOH/g). The number-averaged molecular mass was determined by GPC as 314 g/mol, due to the presence of low molecular weight admixtures. Weight-averaged molecular mass was 899 g/mol with polydispersity 2.86. The result on M n and M w are in good accordance with the masses calculated from assumed oligoetherol and side products (Table 3), which were M n = 333 g/mol and M w = 917 g/mol, respectively.
The physical properties of oligoetherol, namely refraction index, density, viscosity, and surface tension were determined (Table 4). It has been found that these parameters are within the limits for oligoetherols used for obtaining polyurethane foams (PUF).
Obtaining polyurethane foams and their properties
The obtained oligoetherols were tested to obtain PUFs. The crude oligoetherol with admixed suspended metasilicic acid was used intentionally. We have assumed that metasilicic acid could be the additive component playing the role of flammability quencher in foamed compositions. We have optimized the amount of catalyst, isocyanate, and water as foaming agent. We have found that the best PUFs were obtained with isocyanate coefficient (the molar ratio of isocyanate to hydroxyl groups) within 0.8–1.1 (Table 5). The optimized amount of water as foaming agent was 2–4% related to the mass of oligoetherol. For lower water percentage, lower foaming was observed, while more than 4% water caused decrease of mechanical resistance and fragile PUFs. The amount of catalyst depended on water percentage and for higher water percentage, less catalyst was necessary (Table 5, compositions 6 and 10). Cream time and rise time depended on amount of water; in case of optimal composition these parameters were 30 and 58 s and 25 and 63 s for water at the level of 2 and 4%, respectively. Tack-free time was very short.
Physical properties of optimized PUFs were studied. The apparent density was quite high 95.5–96.8 kg/m3 (Table 6, column 3). At elevated temperature (150 °C) the PUF showed up to 6% shrinkage, while water uptake after 24 h exposure at RT was up to 8%. Heat conductance coefficient was within typical value for rigid PUFs [32] used for thermal isolation. It has been noticed that heat conductance coefficient increased considerably upon heat exposure, presumably due to partial carbonization of PUF.
Thermal resistance of obtained PUFs was studied by static method, i.e., by measuring mass loss upon thermal exposure of PUF sample at 150, 175, and 200 °C with concomitant determination of physical properties. It has been noticed that the highest mass loss took place within first day of thermal exposure (Fig. 1). The mass loss of obtained foams after one month thermal exposure at 150 and 175 °C was equal to 16.7–24.0 and 36.5%, respectively (Table 7), while classic PUFs obtained from polymeric diphenylmethane 4,4′-diisocyanate and oligoetherol obtained by oxyethylenation of glycerol (the hydroxyl number was 380 mg KOH/g) showed 21 and 37% mass loss already after 1 day of the exposure, respectively.

Thermal stability of the polyurethane foam as the mass loss after heating at high temperature (the composition number from Table 5 is given in inset)
The PUFs showed improved compressive strength after thermal exposure at 150 and 175 °C (Table 7, column 4). All PUFs were not resistant to heating at 200 °C temperature, at which they deformed. Dynamic heating test was performed for the sample obtained at foaming composition with 4 weight % water. This PUF started to decompose at 167 °C (5% mass loss, Table 8). Fast decomposition was observed at 260 °C (Fig. 2) presumably related to degradation of oxyalkylene chains. At 390 °C the second maximum of mass loss rate was found, probably related to degradation of isocyanuric ring decomposition, which are formed in trimerization of isocyanate. The samples which were pre-annealed at lower temperatures generally decomposed at higher temperatures. This can be attributed to some changes in chemical structure of polymers. Considering all the observations, it can be concluded that some additional crosslinking occurred during annealing PUFs at 150 °C resulting in increase of compressive strength. Another possibility is that the spatial structure of annealed polymer changed due carbonization and such modification led to PUF of different mechanical properties.

Thermal analysis of foam No. 10 according to the Table 5: weight change as a function of temperature (a) differential mass change as a function of temperature (b)
Flaming properties of obtained PUFs depended on amount of water used for foaming. Thus PUF obtained from foaming composition with 2% water was flammable (Table 7). In horizontal test it burned completely with flaming rate 2 mm/s. The PUF obtained from composition with 4% water was self-extinguishing; flame zone was only 21 mm, and flaming rate was 0.7 mm/s. The PUFs after thermal exposure flamed only upon flame contact and self-extinguished after flame source removal.
The oxygen index (OI) of PUFs before and after thermal exposure was determined. It has been noticed that the higher heat exposure temperature the higher values of IO were found both for compositions obtained from 2 and 4% foaming water compositions. The PUFs after thermal exposure did not release smoke upon combustion contrary to untreated ones. The PUFs annealed for one month at 175 °C did not burn with flame at IO 36–38%. Thus it has been concluded that PUFs annealed for a long time lost their flammability. From elemental analysis (Table 9) it could be seen that these materials lost hydrogen, and resulted in increase of nitrogen percentage, which is a flammability reducing element contrary to hydrogen. The IR spectral control of the samples after thermal exposure (Fig. S. 8) showed the disappearance of carbodiimide groups (by decrease of the band at 2150 cm−1), probably due to their oxidation and residual isocyanate groups (by decrease of the band at 2275 cm−1) due to their involvement in additional crosslinking reaction. The oxidation of the former was confirmed by the increase of intensity of the bands at 3400 and 1150 cm−1 attributed to valence bands from O–H and C–OH groups.
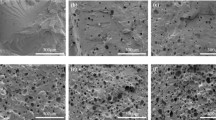
Based on microscopic observation (Fig. 3) it was found that foam pore diameters are dependent on water percentage in foaming composition. When water content increased from 2 to 4% the average pore diameter increased from ca 110 µm to 580–600 µm (Fig. 3a, c). This observation is consistent with higher compression strength of PUF obtained with 2% water than that of PUF obtained with 4% of water in foaming composition. Generally the heating caused decrease of pore size, especially observed at temperature 175C and higher, and also tearing of pores. For example, the PUF obtained from composition with 2% water before thermal exposure for one month had the pores of 110 µm diameter, while this PUF exposed at 175 °C and higher showed the pores of 70–80 µm.

Morphology of foam frothed with 2% of water before exposure (a), after exposure in temperature 175 °C (b), and frothed with 4% of water before exposure (c)
We have elaborated a simple synthesis of oligoetherols with incorporated silicon atoms starting from cheep and easily available metasilicic acid. There were not many efforts on obtaining silicon-modified PUFs reported, except those with organosilicon compounds [6] or silsesquioxanes [13,14,15,16] as silicon source. Using the silicon-containing oligoetherols described in our work we have obtained PUFs of increased thermal resistance and mechanical properties, as well as less flammable in comparison with classic rigid PUFs.
Conclusions
MSA reacts with GL without catalyst at 180 °C, and with EC in presence of potassium carbonate at 145 °C. The method of hydroxyalkylation of MSA with GL and EC leads to oligoetherol useful in obtaining PUFs containing 9.5 mass % of poly(metasilicic acid) which is an additive flame retardant. The obtained PUFs are similar to the classic rigid PUFs except in having advantageous thermal and mechanical resistance. They stand long-term heating at 175 °C; additionally, after thermal exposure they gain higher compressive strength. The PUF obtained from composition with 4% water was self-extinguishing. The PUFs after thermal exposure flamed only upon flame contact and self-extinguished after flame source removal.
References
Lubczak J (2011) Polyhydroxyalkyl derivatives and polyetherols obtained from azacyclic compounds. Part I. Reactions with oxiranes. Polimery 56:360–368
Wirpsza Z (1991) Polyurethanes. WNT, Warsaw
Janowska G, Przygocki W, Włochowicz A (2007) Flammability of polymers and plastics. WNT, Warsaw
Czupryński B, Paciorek-Sadowska J, Liszkowska J (2006) Studies on effect of tri(2-hydroxypropyl), tri(2-hydroxybutyl) and tri(hydroxythiodiethylene) borates on thermal and heat properties of rigid polyurethane-polyisocyanurate foams. Chin J Chem 24:1796–1799. doi:10.1002/cjoc.200690336
Thirumal M, Khastgir D, Singha N, Manjunath B, Naik Y (2008) Effect of expandable graphite on the properties of intumescent flame-retardant polyurethane foam. J Appl Polym Sci 110:2586–2594. doi:10.1002/app.28763
Yi H, Yan K (2008) Polyurethane modified with 3-aminopropyltriethoxysilane as wool antifelting agent. J Appl Polym Sci 109:2169–2175. doi:10.1002/app.28012
Yang CH, Liu FJ, Liu YP, Liao WT (2006) Hybrids of colloidal silica and waterborne polyurethane. Colloid Int Sci 302:123–132. doi:10.1016/j.jcis.2006.06.001
Kim BS, Park SH, Kim BK (2006) Nanosilica-reinforced UV-cured polyurethane dispersion. Colloid Polym Sci 284:1067–1072. doi:10.1007/s00396-006-1488-5
Chen S, Sui J, Chen L (2004) Positional assembly of hybrid polyurethane nanocomposites via incorporation of inorganic building blocks into organic polymer. Colloid Polym Sci 283:66–73. doi:10.1007/s00396-004-1093-4
Chen T-K, Tien Y-I, Wei K-H (2000) Synthesis and characterization of novel segmented poly-urethane/clay nanocomposites. Polymer 41:1345–1353. doi:10.1016/S0032-3861(99)00280-3
Nikje MMA, Tehrani ZM (2010) Thermal and mechanical properties of polyurethane rigid foam/modified nanosilica composite. Polym Eng Sci 50:468–473. doi:10.1002/pen.21559
Francés AB, Navarro Bañón NV (2014) Effect of silica nanoparticles on polyurethane foaming process and foam properties. Con Ser Mat Sci Eng 64:1–6. doi:10.1088/1757-899X/64/1/012020
Levchik SV, Weil ED (2004) Thermal decomposition, combustion and fire-retardancy of polyurethanes—a review of the recent literature. Polym Int 53:1901–1929. doi:10.1002/pi.1314
Zhang S, Horrocks AR (2003) A review of flame retardant polypropylene fibres. Prog Polym Sci 28:1517–1538. doi:10.1016/j.progpolymsci.2003.09.001
Mercado LA, Galia M, Reina JA (2006) Silicon-containing flame retardant epoxy resins. Polym Degrad Stabil 91:2588–2594. doi:10.1016/j.polymdegradstab.2006.05.007
Terraza CA, Tagle LH, Leiva A, Poblete L, Concha F (2008) Poly(urethanes) containing silarylene and/or germarylene units. J Appl Polym Sci 109:303–308. doi:10.1002/app.28170
Verdolotti L, Lavorgna M, Lamanna R, Di Maio E, Iannace S (2015) Polyurethane-silica hybrid foam by sol–gel approach: chemical and functional properties. Polymer 56:20–28. doi:10.1016/j.polymer.2014.10.017
Brojer Z, Penczek P (1972) Epoxide resins. WNT, Warsaw
Kijowska D, Wołowiec S, Lubczak J (2004) Kinetics and mechanism of initial steps of syn-thesis of polyetherols from melamine and ethylene carbonate. J Appl Polym Sci 93:294–300. doi:10.1002/app.20453
Broniewski T, Iwasiewicz A, Kapko J, Płaczek W (1967) Testing and evaluation of properties of plastics. WNT, Warsaw
Dryński T (1967) Laboratory of physics. PWN, Warsaw
Cellular Plastics and Rubbers. Determination of apparent (bulk) Density, Polish (European) Standards PN-EN ISO 845-2000. Ed. Polish Committee for Standardization
Cellular Plastics, rigid. Determination of Water Absorption. Polish (European) Standards PN-EN ISO 2896-1986. Ed. Polish Committee for Standardization
Cellular Plastics, rigid. Test of dimensional Stability. Polish (European) Standards PN-EN ISO 2796-1986. Ed. Polish Committee for Standardization
Cellular Plastics, Compression Test for rigid Materials. Polish (European) Standards PN- EN ISO 844-1978, Ed. Polish Committee for Standardization
Flexible Cellular polymeric Materials—Laboratory Characteristics of small specimens Subject to a small Flame. Polish (European) Standards PN-EN ISO 3582-2002. Ed. Polish Committee for Standardization
Lubczak J (2011) Polyhydroxyalkyl derivatives and polyetherols obtained from azacyclic compounds. Part II. Reaction with formaldehyde and alkylene carbonates. Polimery 56:452–460
Cyzio K, Lubczak J (2011) New possibilities of synthesis of oligoetherols with azacyclic com-pounds. Polimery 56:856–860
Listoś T, Kuran W (2000) Studies on the synthesis of oligomeric sililene carbonate. Polimery 45:603–607
Kucharski M, Kijowska D (2001) Synthesis of polyetherols from melamine and ethylene car-bonate. J Appl Polym Sci 80:1776–1784. doi:10.1002/app.1273
Czupryński B (2004) Topics on chemistry and technology of polyurethanes. Casimir the Great Academy of Bydgoszcz, Bydgoszcz
Acknowledgements
The NMR spectra were recorded within U-8689/DS Grant placed at Rzeszow University of Technology.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chmiel, E., Lubczak, J. Oligoetherols and polyurethane foams obtained from metasilicic acid. Polym. Bull. 75, 1579–1596 (2018). https://doi.org/10.1007/s00289-017-2109-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-017-2109-9