
Abstract
Purpose
To support future dosing recommendations, the effect of food on the pharmacokinetics of adavosertib, a first-in-class, small-molecule reversible inhibitor of WEE1 kinase, was assessed in patients with advanced solid tumors.
Methods
In this Phase I, open-label, randomized, two-period, two-sequence crossover study, the pharmacokinetics of a single 300 mg adavosertib dose were investigated in fed versus fasted states.
Results
Compared with the fasted state, a high-fat, high-calorie meal (fed state) decreased adavosertib maximum plasma concentration (Cmax) by 16% and systemic exposure (area under the plasma concentration–time curve [AUC]) by 6%; AUC0–t decreased by 7% and time to maximum plasma concentration was delayed by 1.97 h (P = 0.0009). The 90% confidence interval of the geometric least-squares mean treatment ratio for AUC and AUC0–t was contained within the no-effect limits (0.8–1.25), while that of Cmax crossed the lower bound of the no-effect limits. Adverse events (AEs) related to adavosertib treatment were reported by 20 (64.5%) of the 31 patients treated in this study. Grade ≥ 3 AEs were reported by four (12.9%) patients (one in the fed state, three in the fasted state); two of these AEs were considered treatment-related by the investigator. Three serious AEs were reported in three (9.7%) patients; these were not considered treatment-related. No patients discontinued because of treatment-related AEs, and no new safety signals were reported.
Conclusion
A high-fat meal did not have a clinically relevant effect on the systemic exposure of adavosertib, suggesting that adavosertib can be administered without regard to meals.
Similar content being viewed by others

Introduction
Adavosertib (AZD1775) is an orally active, first-in-class, small-molecule reversible inhibitor of WEE1 kinase [1]. WEE1 is a protein tyrosine kinase that phosphorylates and inhibits cyclin-dependent kinase 1 (CDK1), which drives cells from the G2 phase into mitosis, and CDK2, which drives cells through the S phase of the cell cycle [1, 2]. In response to DNA damage, WEE1 inhibits both CDK1, to maintain the cell in an inactive state and prevent mitosis, and CDK2, to delay the replication process and allow time for DNA repair [3]. Through inhibition of WEE1, adavosertib prevents G2 cell cycle arrest, leading to premature mitosis, and enhances CDK2 activation in cells in the S phase; this results in uncontrolled DNA replication and replication stress [1, 4]. The majority of human cancers have abnormalities in the p53 G1/S checkpoint, making them more dependent on the S and G2 checkpoints [1, 5]. The S and G2 checkpoint abrogation induced by adavosertib could selectively enhance killing of p53-deficient tumor cells when administered in combination with other anticancer agents [6], while single-agent activity may be achieved in tumor cells by inducing high levels of replication stress and/or endogenous DNA damage, resulting in mitotic catastrophe. With this in mind, adavosertib is being developed for use as a chemosensitizing agent in combination with one or more cytotoxic agents, or with olaparib, or with durvalumab, and as monotherapy for the treatment of advanced solid tumors.
The mechanism of action of adavosertib was confirmed in a Phase I study, which provided the first direct evidence of a reduction in phosphorylated Tyr15-Cdk (a target of WEE1 kinase) levels in paired tumor biopsies [7]. This study also provided concurrent evidence for a DNA damage response (based on increased levels of phosphorylated histone H2AX) and, thus, demonstrated the single-agent activity of adavosertib [7]. Other Phase I studies have assessed the safety and tolerability of adavosertib as monotherapy [8, 9] and in combination with chemotherapy, olaparib, or durvalumab in patients with advanced or refractory solid tumors [10,11,12,13]. Multiple dosing schedules have been evaluated for adavosertib mono- and combination therapy to maximize clinical activity and minimize toxicity [8, 9, 14].
In addition to these Phase I studies, Phase II data have shown adavosertib to have promising antitumor activity in women with TP53-mutated ovarian cancer refractory or resistant to first-line platinum-based chemotherapy when administered in combination with carboplatin [15]. Preliminary results from an open-label, four-arm, Phase II study of adavosertib plus four different types of chemotherapy regimens in patients with platinum-resistant ovarian cancer also suggested that adavosertib plus carboplatin shows promise in terms of efficacy [16]. Furthermore, a randomized Phase II study, also in women with platinum-resistant ovarian cancer, has demonstrated that adavosertib and gemcitabine combination therapy improves treatment efficacy compared with gemcitabine alone [17].
The pharmacokinetic parameters of single-dose adavosertib are approximately linear and increase in a dose-proportional manner [10]. Adavosertib is absorbed at a modest rate, with a median time to peak plasma concentration (tmax) of 3–4 h and terminal half-life of 9.0–12.3 h following single doses of 325–1300 mg in patients with advanced solid tumors [10]. In vitro data indicate that the major pathway of adavosertib metabolism in humans involves CYP3A4, although FMO3 and FMO5 may also be involved [10], with no significant metabolites representing > 10% of unchanged adavosertib in human plasma.
It is critical that the effect of food on drug exposure is investigated early in clinical development because food can alter systemic exposure of an orally administered drug and mitigate or exacerbate toxicities [18, 19]. FDA guidance stipulates that food effect studies should be conducted early in the drug development process (ideally Phase I) in oncology [18, 20]. Knowing whether food affects the bioavailability of a drug before the pivotal trial can help to optimize study design, increase patient comfort by avoiding unnecessary fasting, reduce cost and ultimately improve efficiency in oncology drug development [18, 19]. The data from several food effect studies for orally administered cancer drugs were the subject of a review published in 2018 [18]. Of the 16 drugs with available data, only two had completed food effect data collection before the start of the pivotal trial. Consequently, for four of the 14 drugs with a food effect study that was completed after the pivotal study, the food restrictions on the label were different to those imposed in the trial.
To support dosing recommendations in future clinical studies, the current study (NCT03315091) assessed the effect of a high-fat, high-calorie meal (representing the setting of maximum possible perturbation of oral bioavailability in the postprandial state) on the pharmacokinetics (PK) of a single daily oral dose of 300 mg adavosertib, which is the recommended Phase II dose when administered as monotherapy [9].
Materials and methods
Objectives
The primary objective of the study was to investigate the effect of food on the PK of a single oral dose of 300 mg adavosertib in patients with advanced solid tumors. The safety and tolerability of adavosertib were also assessed.
Study design
This Phase I, open-label, randomized study had a two-period, two-sequence crossover design (Fig. 1) and was conducted in patients with advanced solid tumors for which standard therapy did not exist or had proven ineffective or intolerable.

Study design. All test meals provided in the study were concordant with the recommendations of the FDA for a high-fat and high-calorie meal consisting of 50% calories from fat and 800–1000 calories in total [20]
Patients were screened within 28 days prior to day 1 of the first treatment period and randomly assigned to one of two treatment sequences before administration of the first dose of the study drug. In treatment sequence 1, patients received a single oral dose of 300 mg adavosertib after an overnight fast of at least 10 h and continued fasting for at least 4 h after administration (fasted state). After a washout period of at least 5 days, but no more than 14 days, patients received another single oral dose of 300 mg adavosertib within 30–45 min after having initiated consumption of a high-fat, high-calorie meal (fed state) [20]. In treatment sequence 2, patients received a single oral dose of 300 mg adavosertib first in the fed state, followed by the same washout period and another single oral dose of 300 mg adavosertib in the fasted state.
All test meals complied with FDA recommendations for a high-fat and high-calorie meal, where fat comprised approximately 50% of the total calorific content of the 800- to 1000-calorie meal [20]. An example test meal included 150 mL whole milk (3% fat), 45 g cereal (cornflakes), half a slice of fried bread, 60 g lean back bacon, one lightly fried egg, three slices of toast, 30 g butter, and 200 mL decaffeinated tea/coffee (with milk, which was part of the 150 mL allowance).
Patients stayed in the study center overnight to optimally manage fasting and food consumption, and all patients received intravenous anti-emetic medication (granisetron 1 mg or ondansetron 8 mg) within 30–40 min prior to adavosertib administration. In the fed state, patients were considered evaluable provided they consumed at least 75% of the high-fat meal within 45 min. If the patient did not consume at least 75% of the meal within 45 min, they were not dosed or sampled for PK in that treatment period. If a patient vomited within 3 h after adavosertib administration, all PK sampling ceased for that treatment period.
The study was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, applicable regulatory requirements and the AstraZeneca policy on bioethics [21]. Following the end-of-treatment assessment, patients could enroll in a continued-access study (NCT03313557) to receive adavosertib at the recommended Phase II dose of 300 mg per day on days 1–5 and 8–12 of a 21-day treatment cycle.
Patients
Both male and female patients of at least 18 years of age were eligible for inclusion in this study. Key inclusion criteria were as follows: patients had to have a histologically or cytologically documented locally advanced or metastatic solid tumor, excluding lymphoma, for which standard therapy did not exist or had proven ineffective or intolerable; an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; any prior palliative radiation had to have been completed at least 7 days before the start of study treatment, and patients had to have recovered from any acute adverse effects. Required thresholds for baseline laboratory values (within 7 days of study treatment initiation) were: absolute neutrophil count ≥ 1500/μL; hemoglobin ≥ 9 g/dL; platelets ≥ 100,000/μL; alanine aminotransferase and aspartate aminotransferase ≤ 3 × upper limit of normal (ULN), or ≤ 5 × ULN if the patient had documented hepatic metastases; serum bilirubin within normal limits, or ≤ 1.5 × ULN in patients with hepatic metastases; and serum creatinine ≤ 1.5 × ULN. Patients had to be able to consume a high-fat meal. Key exclusion criteria were as follows: known malignant central nervous system disease other than neurologically stable, treated brain metastases; use of any anticancer treatment drug ≤ 21 days or ≤ 5 half-lives (whichever was shorter) prior to first administration of the study drug; patients suffering from conditions that were likely to adversely affect gastrointestinal motility and/or transit (including diarrhea, vomiting and nausea), or with gastrointestinal resection likely to interfere with absorption of study treatment; and patients who were dependent on a medication that could adversely affect gastrointestinal motility or transit.
Patients were not permitted to use any other anticancer therapy, biologic therapy or novel investigational agent during the study treatment. All patients had to avoid concomitant use of medications, herbal supplements, and ingestion of foods with known inducer/inhibitory effects on CYP3A4. Loperamide was permitted during the study for treatment of diarrhea.
Assessments
Pharmacokinetic samples and safety assessments were obtained pre-dose and up to 72 h post-dose (every 15 min up to 1 h post-dose, hourly from 1 to 4 h post-dose, bi-hourly from 4 to 12 h post-dose, every 12 h from 12 to 48 h post-dose, and at 72 h post-dose) in each treatment period. The concentration of adavosertib in human plasma, containing dipotassium ethylenediaminetetraacetic acid as an anticoagulant, was determined using protein precipitation extraction followed by analysis with high-performance liquid chromatography and tandem mass spectrometric detection. The sample analysis was performed by Covance Laboratories (Madison, WI, USA) using the bioanalytical method initially developed and validated by Merck (described previously [22]), which has been transferred and cross-validated by Covance Laboratories for sample analysis in clinical trials. All samples were stored at − 70 °C prior to analysis and analyzed within the validated time frame. A total of 939 samples were analyzed for this study. The adavosertib concentration in human plasma ranged from 4 to 2000 nM. The precision (coefficient of variation [CV]) was ≤ 4.7%, and the accuracy (percentage bias) of the quality control samples was within − 2.0% to − 0.4%. Statistical analysis was performed to determine the reliability of the analysis. Reproducibility was confirmed by re-analysis of 102 samples (with adavosertib concentrations above the lower limit of quantification) selected at random: 99.0% of the results obtained following the initial and repeat analysis were within 20.0% of the mean of the two values, thus meeting the acceptance criteria [23].
Pharmacokinetic assessments
The following PK variables were measured: area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration (AUC0–t), calculated using the linear up, log down trapezoidal rule; area under the plasma concentration–time curve from time zero to infinity (AUC), obtained by extrapolating AUC to infinity using the terminal elimination rate constant; and maximum plasma drug concentration (Cmax). To characterize the response, the following PK variables were measured: tmax; elimination rate constant (λz); terminal half-life (t½); apparent clearance (CL/F); and apparent volume of distribution (Vz/F).
Safety and tolerability assessments
Adverse events (AEs) were defined by the Medical Dictionary for Regulatory Activities (version 20.1) and graded by Common Terminology Criteria for Adverse Events (CTCAE; version 4.03). AEs were assessed by physical examination, monitoring of vital signs (blood pressure, pulse rate and body temperature), and evaluation of laboratory parameters (clinical chemistry and hematology). Safety assessments were performed up to 72 h post-dose in each treatment period. Upon completion of the 72-h safety assessment in treatment period 2, patients entered a 4-day washout period (relative to the last dose of adavosertib) and were required to attend an end-of-treatment assessment within 3 days of the washout period. AEs were captured throughout the study, starting 28 days prior to day 1 of the first treatment period until the end-of-treatment assessment. For patients who enrolled in the continued-access study, the end-of-treatment assessment was carried out within 3 days after the 4-day washout period relative to the last dose of adavosertib in treatment period 2; for patients who did not enroll in the continued-access study, this was within 30 days (permitted minus 7-day window) after the last dose of adavosertib.
Statistical analyses
Determination of sample size
The size of this study was based on careful clinical consideration of the need to gain adequate PK data on the effect of food on adavosertib while exposing as few patients as possible to study procedures. No formal hypothesis testing was planned. Interpretation of the results was based on the size of the treatment ratio and associated 90% confidence interval (CI). The intended enrollment was 24 patients in total, with the aim of ensuring 18 PK-evaluable patients; however, the study protocol allowed for enrollment of additional patients to ensure that there were at least 18 evaluable patients.
Analysis sets
The PK analysis set included all patients who received at least one dose of adavosertib in one treatment period and who had at least one quantifiable plasma concentration recorded post-dose without protocol deviations or events that would affect the PK analysis. The safety analysis set included all patients who received at least one dose of adavosertib.
Statistical methods
Statistical analysis was performed by IQVIA Early Clinical Development (ECD) under the direction of the AstraZeneca Biostatistics group and employed SAS® version 9.4. Calculation of the PK parameters was the responsibility of IQVIA ECD Pharmacokinetics/Pharmacodynamics Department using non-compartmental methods with Phoenix® WinNonlin® version 6.4 (Certara, LP, Princeton, NJ, USA) and/or SAS version 9.4. For qualitative variables, the population size (N for sample size and n for available data) and the percentage (of available data) for each class of variable were reported. Quantitative variables were summarized using descriptive statistics, including n, arithmetic mean, standard deviation (SD), median, and minimum and maximum values. To assess the primary objective of this study, geometric mean ratios and 90% CIs of PK parameters were calculated based on a mixed-effects model, with fixed effects for sequence, period, and treatment, and subject nested within sequence as a random effect.
Results
Patients
A total of 37 patients were enrolled in seven study centers in France, the UK, and The Netherlands. There were six screening failures; therefore, 31 patients were randomized to one of the two treatment sequences. A total of 15 patients were assigned to treatment sequence 1 and 16 patients to treatment sequence 2. Three patients discontinued treatment because of worsening of the disease, resulting in 28 patients (14 per treatment sequence) who completed the study. As the three patients discontinued treatment after having received one dose of adavosertib (one in the fasted state and two in the fed state), data for the study safety and PK analyses were available for 29 and 30 patients in the fasted and fed states, respectively. Thus, data for all 31 randomized patients were included (Supplementary Fig. 1).
Fourteen patients had important protocol deviations. Of these, six were excluded from the PK analysis: one patient in the fasted state was excluded because it was uncertain whether movicolon had been withheld for 3 h before and after adavosertib dosing in accordance with the protocol; and five patients in the fed state were excluded, one because they received metoclopramide, potentially affecting the absorption of adavosertib, and four because they received adavosertib more than 45 min after the start of the meal. By accounting for these six excluded patients and the three patients who had discontinued because of worsening of the disease after the first treatment period, data were available for the final PK analysis for 28 and 25 patients in the fasted and fed states, respectively. A total of 22 patients had evaluable PK results for both the fed and fasted states.
Patient demographics and baseline characteristics were representative of the intended patient population and were generally balanced between treatment sequences (Table 1). A total of 19 (61.3%) of the 31 patients were female. The primary tumor location was varied; however, the most common primary tumor sites (in at least 10% of patients overall) were the ovaries in 6 (19.4%) patients and the rectum in 4 (12.9%) patients.
All patients participating in the study used concomitant medication (in addition to the mandatory pre-medication) during both treatment periods. Anti-emetic agents, including serotonin antagonists, were the most commonly used concomitant medication (used by 18 [62.1%] and 21 [70.0%] patients in the fasted and fed states, respectively), followed by pain medication, including opiates (used by 11 [37.9%] and 12 [40.0%] patients in the fasted and fed states, respectively) and non-steroidal anti-inflammatory drugs (5 [17.2%] and 6 [20.0%] patients in the fasted and fed states, respectively).
In accordance with the study protocol, all medications that could affect gastrointestinal motility or stomach acidity were prohibited to avoid alterations in the absorption of adavosertib. A total of 17 patients received one or more prohibited medications; of these, 12 patients were in both treatment groups. Most patients taking prohibited medications received laxatives for the treatment of constipation or other non-parenteral medications for the treatment of diarrhea or other gastrointestinal symptoms. However, the study centers dosed these medications at least 3 h before or after each adavosertib administration, so their use was not expected to affect adavosertib PK results.
Pharmacokinetic assessments
The adavosertib PK parameters are summarized by treatment group (PK analysis set) in Table 2. The geometric mean AUC and AUC0–t values were similar whether adavosertib was administered in the fasted or fed state. The geometric mean Cmax was 19% lower in the fed versus fasted state (965.9 vs 1192 nM, respectively).
Systemic exposure was lower in the fed state compared with the fasted state, as shown for AUC0–t in Fig. 2a. Two patients showed pronounced higher systemic exposure in the fed state compared with the fasted state. Furthermore, the systemic exposure for one patient in the fed state was lower than for other patients under the same conditions (Fig. 2a). These outliers may account for the approximate tenfold difference in the median value ranges of AUC and AUC0–t in the fed state compared with the approximate threefold difference in median value ranges seen in the fasted state reported in Table 2. No events or patient characteristics were identified that may explain these observations.

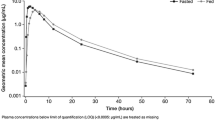
a Individual and geometric mean AUC0–t of adavosertib by treatment group (PK analysis set). b Geometric mean (± geometric SD) plasma concentration of adavosertib by treatment group over time (PK analysis set)
Geometric mean adavosertib plasma concentration over time is shown in Fig. 2b. Overall, the presence of food resulted in slower initial absorption in the fed state versus the fasted state, leading to lower initial plasma concentrations. By 6 h after administration of adavosertib, geometric mean concentration–time profiles were comparable between groups for the remaining time course. Geometric mean plasma concentrations peaked at 6 h in the fed state and 4 h in the fasted state.
Statistical comparisons of adavosertib primary exposure parameters (AUC, AUC0–t, and Cmax), as well as tmax, are summarized in Table 3. Statistical analysis confirmed that systemic exposures to adavosertib for AUC, AUC0–t, and Cmax were similar in the fed and fasted states. Co-administration of adavosertib with food decreased adavosertib geometric least-squares (LS) mean systemic exposure by 6% (AUC) and 7% (AUC0–t), and decreased geometric LS mean Cmax by approximately 16%, compared with adavosertib administered in the fasted state. The 90% CIs of the geometric LS mean treatment ratios for AUC and AUC0–t were fully contained within the no-effect limits of 0.8 and 1.25, while that of Cmax crossed the lower bound of the no-effect limits. Administration of adavosertib in the fed state delayed median tmax by 1.97 h compared with the fasted state, which was statistically significant (P = 0.0009).
Median (range) λz was 0.595 (0.0247–0.1013) for patients in the fed state and 0.0612 (0.0270–0.0919) for patients in the fasted state. Other PK parameters of interest are summarized by treatment group in Table 2. Median tmax was 5.95 h in the fed state and 3.01 h in the fasted state. Arithmetic mean t½ of adavosertib was similar whether administered in the fed or fasted state: 12.29 and 12.28 h, respectively. Arithmetic mean CL/F was 23% (46.05 vs 37.40 L/h) higher, and arithmetic mean Vz/F was 37% (916.5 vs 670.3 L) higher, when adavosertib was administered in the fed versus fasted state, respectively.
Safety
Adverse events by category are summarized in Table 4. At least one AE of any cause and grade occurred in 30 (96.8%) of the 31 patients in the safety analysis set. In the fed treatment group, 25 (83.3%) patients experienced AEs, while 23 (79.3%) patients experienced AEs in the fasted treatment group. One patient enrolled in the continued-access study 9 days after the end-of-treatment visit; for this patient, AEs were reported up to and including the date of enrollment in the continued-access study.
The AEs that occurred most frequently (in at least two patients overall) are summarized in Supplementary Table 1 and included nausea and vomiting (both 38.7%), diarrhea and headache (both 19.4%), constipation (16.1%), and abdominal pain, anemia, cough, and fatigue (all 12.9%). Of the AEs that occurred in at least two patients, the following were more common in the fasted state than the fed state: vomiting (34.5% vs 20.0%); headache (20.7% vs 6.7%); constipation (17.2% vs 6.7%); abdominal pain (13.8% vs 0%); back pain (10.3% vs 3.3%); and pyrexia (10.3% vs 0%). AEs that were more common in the fed state than the fasted state included: nausea (33.3% vs 24.1%); diarrhea (16.7% vs 10.3%); and decreased appetite (10.0% vs 0%). There were no AEs that led to the discontinuation of adavosertib treatment.
AEs that were considered to be related to adavosertib treatment by the investigator were reported in 16 (53.3%) patients in the fed state and 14 (48.3%) patients in the fasted state (Supplementary Table 2). Three AEs considered by the investigator to be treatment-related were reported in at least two patients in both the fed and fasted states; these were nausea (38.7%), vomiting (35.5%), and diarrhea (16.1%). The following treatment-related AEs were more common in the fasted state than the fed state: vomiting (27.6% vs 20.0%), headache (10.3% vs 3.3%), fatigue (6.9% vs 3.3%), and abdominal pain (6.9% vs 0%). Treatment-related nausea (33.3% vs 24.1%) and diarrhea (13.3% vs 6.9%) were more common in the fed state than the fasted state.
No grade ≥ 4 AEs were reported. Four (12.9%) patients experienced a grade-3 AE: one patient in the fed state and three patients in the fasted state. In two of these cases, the AE was deemed to be treatment-related: diarrhea and headache in one patient in the fasted state, and hypokalemia in one patient in the fed state.
Three serious AEs (SAEs), which all led to hospitalization, were reported in 3 (9.7%) patients; none were considered treatment-related. Two patients experienced grade-1 SAEs, pyrexia and hydronephrosis, respectively, and one patient experienced a grade-3 SAE, urosepsis. There were no SAEs that led to the discontinuation of adavosertib treatment. No deaths were reported in the study.
No new clinically important findings or trends for safety laboratory parameters or vital signs were observed; however, some abnormal laboratory results were clinically significant and were reported as AEs. Notably, 1 (3.4%) patient in the fasted state (n = 29) experienced a single event of prolonged QTc (CTCAE grade 1), which was deemed treatment-related but was potentially confounded by hypomagnesemia (CTCAE grade 3). The event did not recur when the patient was dosed in the fed state.
Discussion
The purpose of this Phase I, open-label, randomized, two-period, two-sequence crossover study was to assess the effect of food on the PK of a single oral dose of 300 mg adavosertib in patients with advanced solid tumors. Food is an important extrinsic factor that may influence the PK profile of a drug because it can change the extent of absorption, or delay absorption by decreasing the rate of gastric emptying [18].
The current study was designed to meet key FDA recommendations for a food effect study [18, 20]. Specifically, the FDA recommends a two-period, two-sequence crossover design testing a fed and a fasting condition to support the clinical development of a new agent. Furthermore, to generate the maximum possible effect on systemic drug availability, the fed group should receive a high-fat, high-calorie meal consisting of 50% calories from fat and 800–1000 calories [20]. The current study was not, however, conducted in healthy volunteers because adavosertib is a DNA damage response inhibitor, and it is neither safe nor ethical to administer this anticancer agent to healthy individuals. Therefore, the study was conducted in patients with advanced solid tumors for which standard therapy did not exist or had proven ineffective or intolerable.
The primary objective of this study was to investigate the effect of food on the PK of a single dose of adavosertib following oral dosing. Co-administration of adavosertib with food did not meaningfully affect its systemic exposure (AUC, AUC0–t, and Cmax) compared with administration in the fasted state. Co-administration of adavosertib with food decreased geometric LS mean systemic exposure by 6% (AUC) and 7% (AUC0–t), and decreased geometric LS mean Cmax by approximately 16%, compared with the fasted state. The decrease in adavosertib exposure was not considered to be clinically relevant.
The secondary objective of this study was to characterize the PK of a single dose of adavosertib following oral administration. Adavosertib taken with food delayed the rate of absorption (median tmax) by approximately 2 h compared with administration in the fasted state. The study also assessed the safety and tolerability of a single dose of adavosertib following oral administration. The safety profile of single-dose adavosertib in this study was consistent with its known safety profile as monotherapy [7, 8, 10]. The most prevalent AEs were gastrointestinal toxicities (nausea, vomiting and diarrhea). Four patients experienced AEs of grade 3 or worse; these were considered to be treatment-related in two patients (one in the fed state and one in the fasted state). Of the three SAEs reported in three patients, none were considered to be treatment-related. No significant AEs of clinical importance were observed in this study, nor were new safety signals identified.
The results of this study have several important clinical implications; for example, a lack of food effect minimizes the risk of unintended alterations to drug exposure, toxicity, and efficacy [24]. Furthermore, food restrictions associated with a dosing regimen can have a negative impact on patient adherence [25], especially when cancer patients are taking multiple agents with different and confusing instructions relating to dosing with food [19]. For cancer treatments that are subject to positive food effects, resources may subsequently be used in the development of alternative formulations that negate the restriction of a food label [26].
The study also has a number of limitations. Patients only received single doses of adavosertib; thus, although this is sufficient for PK assessments, no comparison could be made between the fed and fasted states in relation to toxicities occurring with the full treatment schedules, such as myelosuppression [7]. To that end, the AEs reported herein occurring in the fed and fasted states were observational; no formal analyses were performed, and it is not possible to conclude whether adavosertib is better tolerated when administered with or without food. The high-fat, high-calorie meal consumed by patients in this study was based on FDA guidance; therefore, the assumption was made that it would have the greatest effect on gastrointestinal physiology and would thus have the maximum effect on systemic drug availability. All patients in the present study received intravenous anti-emetic medication with granisetron or ondansetron prior to adavosertib administration; while no formal clinical drug interaction studies have been performed with adavosertib, a potential drug–drug interaction seems unlikely based on the low potential of these agents to significantly induce or inhibit CYP450 enzymes. Finally, the small sample size was chosen to balance the need to gain adequate PK data against exposing as few patients as possible to study procedures. No formal hypothesis testing was planned, and no conclusions can be drawn in relation to the efficacy of adavosertib.
In summary, administration of adavosertib 300 mg as a single oral dose following a high-fat, high-calorie meal did not have a clinically relevant effect on its systemic exposure, and the data from this study suggest that adavosertib can be administered in a fasted or fed state. Excluding SAEs and events leading to discontinuation of study treatment, no other significant AEs of clinical importance were observed in this study. Furthermore, no new safety signals were identified in this study.
References
Do K, Doroshow JH, Kummar S (2013) WEE1 kinase as a target for cancer therapy. Cell Cycle 12(19):3159–3164. https://doi.org/10.4161/cc.26062
Gerard C, Goldbeter A (2014) The balance between cell cycle arrest and cell proliferation: control by the extracellular matrix and by contact inhibition. Interface Focus 4(3):20130075. https://doi.org/10.1098/rsfs.2013.0075
O’Connor MJ (2015) Targeting the DNA damage response in cancer. Mol Cell 60(4):547–560. https://doi.org/10.1016/j.molcel.2015.10.040
Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuasen RG, Sorensen CS (2012) Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol 32(20):4226–4236. https://doi.org/10.1128/MCB.00412-12
Sherr CJ (1996) Cancer cell cycles. Science 274(5293):1672–1677. https://doi.org/10.1126/science.274.5293.1672
Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y (2001) Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res 61(22):8211–8217
Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S (2015) Phase I study of single-agent AZD1775 (MK-1775), a WEE1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol 33(30):3409–3415. https://doi.org/10.1200/JCO.2014.60.4009
Bauer TM, Moore K, Rader JS, Simpkins F, Mita A, Beck JT, Hart L, Chu Q, Oza A, Tinker AV, So K, Imedio E, Kumar S, Mugundu G, Jenkins S, Chmielecki J, Jones S, Spigel D, Fu S (2019) Open-label, multicenter, Phase Ib study to assess safety, tolerability and efficacy of adavosertib monotherapy in patients with advanced solid tumors: expansion cohorts. Cancer Res 79(13 Suppl). Abstract CT012
Falchook GS, Sachdev J, Imedio ER, Kumar S, Mugundu G, Chmielecki J, Jones S, Spigel DR, Johnson M (2019) A Phase Ib study of WEE1 inhibitor adavosertib in patients with advanced solid tumors. Cancer Res 79(13 Suppl). Abstract CT022
Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, Lam R, Demuth T, Rose S, Lee MA, Freshwater T, Shumway S, Liang LW, Oza AM, Schellens JH, Shapiro GI (2016) Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol 34(36):4371–4380. https://doi.org/10.1200/JCO.2016.67.5991
Hamilton E, Falchook GS, Wang JS, Fu S, Oza AM, So K, Imedio ER, Kumar S, Ottesen L, Mugundu GM, Chmielecki J, Jones S, Spigel D, Li B (2019) Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: dose escalation. Cancer Res 79(13 Suppl). Abstract CT025
Patel MR, Falchook GS, Wang JS-Z, Imedio ER, Kumar S, Motlagh P, Miah K, Mugundu G, Fields Jones S, Spigel D, Hamilton E (2019) Open-label, multicenter, Phase I study to assess safety and tolerability of adavosertib plus durvalumab in patients with advanced solid tumors. J Clin Oncol 37(Suppl). Abstract 2562
Kato H, de Souza P, Kim SW, Lickliter JD, Naito Y, Park K, Kumar S, Mugundu GM, Bang YJ (2020) Safety, pharmacokinetics, and clinical activity of adavosertib in combination with chemotherapy in asian patients with advanced solid tumors: Phase Ib study. Target Oncol 15(1):75–84. https://doi.org/10.1007/s11523-020-00701-5
Geenen JJJ, Schellens JHM (2017) Molecular pathways: targeting the protein kinase WEE1 in cancer. Clin Cancer Res 23(16):4540–4544. https://doi.org/10.1158/1078-0432.CCR-17-0520
Leijen S, van Geel R, Sonke G, de Jong D, Rosenberg E, Marchetti S, Pluim D, van Werkhoven E, Rose S, Lee M, Freshwater T, Beijnen J, Schellens J (2016) Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol 34(36):4354–4361. https://doi.org/10.1200/JCO.2016.67.5942
Moore K, Chambers S, Hamilton E, Chen L-M, Oza AM, Ghamande S, Konecny G, Plaxe S, Lewis Spitz D, Geenen J, Troso-Sandoval T, Cragun J, Imedio E, Kumar S, Mugundu G, Lai Z, Chmielecki J, Fields Jones S, Spigel D, Cadoo K (2019) Adavosertib with chemotherapy (CT) in patients (pts) with platinum-resistant ovarian cancer (PPROC): an open label, four-arm, Phase II study. J Clin Oncol 37(Suppl). Abstract 5513
Lheureux S, Cabanero M, Cristera M, Mantia-Smaldone G, Olwaiye A, Ellard S, Weberpals J, Wahner Hendrickson A, Fleming G, Welch S, Dhani N, Piskorz A, Tan S, Chang K, Wang L, Kunos C, Pugh T, Brenton J, Oza A (2019) A randomized, double-blind, placebo-controlled, Phase II trial comparing gemcitabine monotherapy to gemcitabine in combination with adavosertib in women with recurrent, platinum-resistant epithelial ovarian cancer: a trial of the Princess Margaret, California, Chicago, and Mayo Phase II consortia. J Clin Oncol 37(Suppl). Abstract 5518
Farha M, Masson E, Tomkinson H, Mugundu G (2018) Food effect study design with oral drugs: lessons learned from recently approved drugs in oncology. J Clin Pharmacol 59(4):463–471. https://doi.org/10.1002/jcph.1351
Yu G, Wu DN, Gong Y, Li G, Zhou HH (2018) Conflicting meal recommendations for oral oncology drugs: pose risks to patient care? Eur J Clin Pharmacol 74(6):833–842. https://doi.org/10.1007/s00228-018-2439-z
US Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER) (2002) Guidance for industry. Food-effect bioavailability and fed bioequivalence studies
AstraZeneca (2019) Code of ethics. Available at: https://www.astrazeneca.com/content/dam/az/PDF/Sustainability/code-of-ethics-2019/Code%20of%20Ethics%20in%20English%202019.pdf. Accessed June 2020
Xu Y, Fang W, Zeng W, Leijen S, Woolf EJ (2012) Evaluation of dried blood spot (DBS) technology versus plasma analysis for the determination of MK-1775 by HILIC-MS/MS in support of clinical studies. Anal Bioanal Chem 404(10):3037–3048. https://doi.org/10.1007/s00216-012-6440-6
US Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER) (2018) Guidance for industry. Bioanalytical method validation
Bullock JM, Lin T, Bilic S (2017) Clinical pharmacology tools and evaluations to facilitate comprehensive dose finding in oncology: a continuous risk-benefit approach. J Clin Pharmacol 57(Suppl 10):S105–S115. https://doi.org/10.1002/jcph.908
Ingersoll KS, Cohen J (2008) The impact of medication regimen factors on adherence to chronic treatment: a review of literature. J Behav Med 31(3):213–224. https://doi.org/10.1007/s10865-007-9147-y
Solymosi T, Otvos Z, Angi R, Ordasi B, Jordan T, Molnar L, McDermott J, Zann V, Church A, Mair S, Filipcsei G, Heltovics G, Glavinas H (2017) Novel formulation of abiraterone acetate might allow significant dose reduction and eliminates substantial positive food effect. Cancer Chemother Pharmacol 80(4):723–728. https://doi.org/10.1007/s00280-017-3406-6
Acknowledgments
We thank Carol Wareham, MRes, PhD, and Laura Atkins-Schmidt, MRes, MPhil, of AMICULUM Ltd, for medical writing support, funded by AstraZeneca. We thank Covance Laboratories, Madison, WI, USA, for conducting the PK sample analysis, funded by AstraZeneca. We also thank IQVIA, Durham, NC, USA, for operational support and statistical analyses. Finally, we thank the patients who participated in this study and their families, as well as the research nurses, data managers, and trial coordinators for their support with conducting the trial.
Funding
This study was funded by AstraZeneca. The UK-based centers acknowledge support from the Experimental Cancer Medicine Centre initiative jointly funded by Cancer Research UK, the National Institute for Health Research in England, and the Departments of Health for Scotland, Wales, and Northern Ireland. Patients from The Christie NHS Foundation Trust were treated in the NIHR Manchester Clinical Research Facility.
Author information
Authors and Affiliations
Contributions
Conception and design by G Mugundu, M Någård and L Ottesen; development of methodology by Y Li and G Mugundu; acquisition of data by M Campone, M Labots, Y Li, A Ravaud, P Roxburgh, F Thistlethwaite, L Valkenburg-van Iersel, and M Vermunt; analyses and interpretation of data by M-L Ah-See, M Någård, G Mugundu, and K So; administrative, technical, or material support of the study provided by Y Li, G Mugundu, and K So; study supervision provided by M-L Ah-See and G Mugundu; all authors reviewed the manuscript at least once for important content and approved the final version prior to submission.
Corresponding author
Ethics declarations
Conflict of interest
M Labots, A Ravaud, L Valkenburg-van Iersel and M Vermunt have nothing to disclose in relation to this study. M-L Ah-See, L Ottesen and K So are employees and shareholders of AstraZeneca UK Limited. Y Li, G Mugundu and M Någård are employees and shareholders of AstraZeneca US Limited. M Campone has participated in advisory boards (fees paid to institution) for Abbvie, Accord, AstraZeneca, Novartis, Pfizer, Sandoz, Sanofi and (personal fees paid) for G1 Therapeutics and Lilly, acted as a consultant (fees paid to institution) for Novartis, Pierre Fabre Oncology, Sanofi and Servier, is a member of a speaker bureau (personal fees paid) for Lilly and Novartis, and has received travel reimbursement from AstraZeneca, Novartis, Pfizer and Roche. P Roxburgh has received honoraria from AstraZeneca and travel reimbursement and research funding from AstraZeneca and Tesaro. F Thistlethwaite has consultancy agreements with Novartis, Pfizer, Bristol-Myers Squibb, Achilles Therapeutics, GlaxoSmithKline, Kite and Gilead.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The institutional review board at each study center approved the protocol, and the study was conducted in accordance with the principles expressed in the Declaration of Helsinki, Good Clinical Practice, applicable regulatory requirements and the AstraZeneca policy on bioethics [21].
Informed consent
All patients provided written informed consent prior to enrollment.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Någård, M., Ah-See, ML., So, K. et al. Effect of food on the pharmacokinetics of the WEE1 inhibitor adavosertib (AZD1775) in patients with advanced solid tumors. Cancer Chemother Pharmacol 86, 97–108 (2020). https://doi.org/10.1007/s00280-020-04101-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-020-04101-4