
Abstract
Purpose
Afatinib, an irreversible ErbB family blocker, has demonstrated preclinical antitumor activity with chemotherapy.
Methods
As part of a phase I trial in patients with advanced solid tumors (NCT00809133; 3 + 3 dose-escalation design), we determined the maximum tolerated dose (MTD) of afatinib with carboplatin (A/C) or with carboplatin plus paclitaxel (A/C/P). Starting doses: afatinib 20 mg/day, carboplatin AUC6 (A/C) with paclitaxel 175 mg/m2 (A/C/P) (chemotherapy: Day 1 of 21-day cycles). The primary objective was to determine the MTDs; safety, pharmacokinetics and antitumor activity were also evaluated.
Results
Thirty-eight patients received A/C (n = 12) or A/C/P (n = 26). No dose-limiting toxicities (DLTs) were reported with A(20 mg)/C(AUC6). One patient experienced DLT in the A(40 mg)/C(AUC6) cohort (grade 3 acneiform rash); A(40 mg)/C(AUC6) was determined as the recommended phase II dose (RP2D) for A/C. Two patients each had DLTs with A(20 mg/day)/C(AUC6)/P(175 mg/m2): fatigue, infection, diarrhea, small intestine hemorrhage, dehydration, renal impairment, neutropenic sepsis (n = 1), mucositis (n = 1); A(40 mg)/C(AUC5)/P(175 mg/m2): febrile neutropenia (n = 1), mucositis, fatigue (n = 1); and A(30 mg)/C(AUC5)/P(175 mg/m2): stomatitis (n = 1), mucositis (n = 1). No DLT was observed with A(20 mg)/C(AUC5)/P(175 mg/m2), determined as the RP2D for A/C/P. The most frequent drug-related adverse events were (A/C; A/C/P): rash (75%; 73%), fatigue (67%; 69%), and diarrhea (58%; 88%). Drug plasma concentrations were similar between cycles, suggesting no drug–drug interactions. Objective response rates in these heavily pretreated patients were A/C, 3/12 (25%); A/C/P, 5/26 (19%).
Conclusions
Afatinib 40 mg/day (approved monotherapy dose) with carboplatin AUC6, and afatinib 20 mg/day with carboplatin AUC5 and paclitaxel 175 mg/m2 demonstrated manageable safety and antitumor activity. Afatinib > 20 mg/day in the triple combination was not well tolerated.
Similar content being viewed by others


Introduction
Afatinib is an irreversible ErbB family blocker, which binds covalently to the intracellular kinase domains of, and prevents signaling from, all kinase-active members of the ErbB family. It is, therefore, active against epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2) and ErbB4, and indirectly inhibits transphosphorylation of the kinase-inactive ErbB3 [1]. As a single agent, afatinib is approved for the first-line treatment of advanced EGFR mutation-positive lung adenocarcinoma, having demonstrated improved progression-free survival (PFS) versus platinum-based chemotherapy in these patients [2, 3]. In addition, afatinib was recently approved for the treatment of patients unselected for EGFR mutations with advanced squamous cell carcinoma of the lung following first-line chemotherapy, with activity superior to erlotinib in this setting [4].
Given their non-overlapping mechanisms of action, the addition of targeted therapies such as afatinib to existing chemotherapy regimens may improve outcomes in patients with advanced solid tumors. In vitro cell-based assays have shown greater efficacy with afatinib combined with paclitaxel than with single-agent treatment [5]. The combination of afatinib with paclitaxel or docetaxel in xenograft animal models has also demonstrated improved efficacy compared with single-agent therapy [6].
The clinical feasibility and tolerability of combining afatinib with paclitaxel, and with paclitaxel plus bevacizumab, were previously explored in earlier parts of this trial (a total of four drug combinations were assessed in patients with advanced solid tumors in this trial [NCT00809133]) [5, 7]. Both of these earlier combination regimens demonstrated manageable safety profiles and antitumor activity at the maximum tolerated doses (MTDs). Here, we evaluated the combination of afatinib with carboplatin, and with carboplatin and paclitaxel, in patients with advanced solid tumors.
Materials and methods
Patients
Patients eligible for treatment had advanced, non-resectable and/or metastatic solid tumors suitable for treatment with either carboplatin or carboplatin and paclitaxel, and were recruited at two centers in the United Kingdom. Key eligibility criteria included age ≥ 18 years; Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1; life expectancy of at least 3 months and adequate organ function (cardiac left ventricular function with resting ejection fraction [LVEF] ≥ 50%; absolute neutrophil count ≥ 1.5 × 109/L [> 2.0 × 109/L for patients allocated to carboplatin]; platelets ≥ 100,000/µL; total bilirubin ≤ 26 µmol/L; aspartate aminotransferase and/or alanine aminotransferase ≤ 2.5 times the upper limit of normal; creatinine ≤ 132 µmol/L and creatinine clearance > 60 mL/min by Cockcroft–Gault equation).
Exclusion criteria included gastrointestinal tract disease that could impair drug absorption; significant cardiovascular disease; active infectious disease; known interstitial lung disease; untreated or symptomatic brain metastases; persistent grade ≥ 2 neuropathy or neurotoxicity from any cause; and treatment with chemotherapy, immunotherapy, radiotherapy, biologic therapy, hormone therapy, EGFR- or HER2-targeting drugs or other investigational drugs within 4 weeks prior to starting trial medication.
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice, and ethical approval was provided by the UK Integrated Research Application System. All patients provided written informed consent.
Study design and treatment
A phase I open-label trial assessed four different drug combinations using a 3 + 3 dose-escalation design (NCT00809133); previous analyses of other combination regimens included in the trial have been reported [5, 7]. In this report, afatinib was evaluated in combination with carboplatin (A/C) or carboplatin plus paclitaxel (A/C/P). In both combinations, oral afatinib was administered once daily, beginning on Day 2 of Cycle 1 in 21-day cycles, at a starting dose of 20 mg/day, and escalated to 40 mg (the approved monotherapy starting dose) and then 50 mg in subsequent dose cohorts. Chemotherapy was administered intravenously on Day 1 of each 21-day cycle: carboplatin at a dose targeting an area under the concentration–time curve of 6 mg/mL.min (AUC6), and paclitaxel at 175 mg/m2 (carboplatin was administered after paclitaxel, consistent with standard medical practice in the United Kingdom) [8]. Target doses for carboplatin were calculated using the Calvert formula (dose [mg] = target AUC [mg/mL.min] × (glomerular filtration rate [GFR; mL/min] + 25 mL/min)). Based on the American Society for Clinical Oncology guidelines, the GFR used in the Calvert formula was capped at 125 mL/min [9]. In the event of toxicity with carboplatin AUC6, a dose reduction to AUC5 was allowed. Patients continued combination treatment for six cycles or until tumor progression, intolerable adverse events (AEs) or withdrawal of consent. Patients with clinical benefit after six cycles could continue either with combination treatment or single-agent afatinib.
The primary endpoint was safety, assessed as dose-limiting toxicities (DLTs) to define the MTD of the two afatinib–chemotherapy combinations. MTD was defined as the highest dose of afatinib in combination with carboplatin, or with carboplatin and paclitaxel, at which no more than one of six patients experienced a DLT during Cycle 1. To be evaluable for determination of the MTD, patients must have completed the first 2 weeks of therapy or have experienced a DLT; patients who did not meet either of these criteria were replaced within the respective dose cohort. Secondary endpoints included pharmacokinetic parameters and antitumor activity (objective tumor responses [OR]).
Assessments
All patients were assessed for AEs according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, version 3.0); relationship to study treatment was assessed by the investigators.
DLTs were defined as any of the following drug-related AEs: grade 4 uncomplicated neutropenia (i.e., fever ≤ 38.3 °C) for > 7 days; neutropenia associated with fever > 38.5 °C; platelets < 25 × 109/L or grade 3 thrombocytopenia associated with bleeding requiring transfusion; grade ≥ 2 decrease in LVEF; uncontrolled hypertension despite multiple anti-hypertension therapies; grade ≥ 2 worsening of renal function; grade > 2 diarrhea despite anti-diarrheal treatment; persistent grade ≥ 2 diarrhea for ≥ 7 days despite supportive care; grade > 2 nausea and/or vomiting despite antiemetic treatment; persistent grade ≥ 2 vomiting for ≥ 7 days despite supportive care; and all other non-hematologic toxicities of grade ≥ 3, except incompletely treated nausea, vomiting or diarrhea.
For the assessment of pharmacokinetic parameters of afatinib (AUCτ,ss and Cmax,ss), total platinum (AUC0−24 and Cmax) and paclitaxel (AUC0−23 and Cmax), blood was collected on Days 1 and 2 of Cycles 1 and 2. Samples were taken pre- and post- (1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0 and 24.0 h) carboplatin infusion (in the A/C arm) and pre- and post- (1.0, 2.0, 3.0, 3.5, 4.0, 4.5, 5.0, 6.0, 8.0, 9.0 and 24.0 h) paclitaxel infusion (in the A/C/P arm). Of note, the 1.0- and 2.0-h time points post-paclitaxel infusion were conducted only during Cycle 2. A sample was also collected on Day 15 of Cycle 1 before the administration of afatinib in both treatment combinations. Plasma concentrations of afatinib and paclitaxel were analyzed by a validated high-performance liquid chromatography–tandem mass spectrometry method at Boehringer Ingelheim (Biberach, Germany) and Nuvisan GmbH (Neu-Ulm, Germany), respectively. Concentrations of total platinum (from carboplatin) were determined by a validated inductively coupled plasma/mass spectrometry assay at Nuvisan GmbH (Neu-Ulm, Germany).
Tumor assessments were performed by the investigators at screening and every 6 weeks after the start of treatment until progressive disease (PD) according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.0).
Statistical analysis
AEs and antitumor activity were assessed in all patients who received at least one dose of afatinib. All analyses in this trial were descriptive and exploratory.
Results
Patients and treatment exposure
Thirty-eight patients were treated in the trial, with the most common tumor type (A/C, 58%; A/C/P, 65%) being non-small cell lung cancer (NSCLC). Additional baseline demographics and disease characteristics are shown in Table 1. Twelve patients received the combination of A/C and 26 received A/C/P. In the A/C arm, 10 (83%) patients completed at least one cycle and 6 (50%) patients completed at least six cycles of treatment; all patients discontinued study treatment due to PD. In the A/C/P arm, 24 (92%) patients completed at least one cycle and 11 (42%) patients completed at least six cycles of treatment. The primary reason for termination of study treatment in the A/C/P arm was PD in 21 patients (81%) and AEs, including DLTs, in 5 patients (19%). Median treatment duration was 106 days (range 13–390) for A/C and 85 days (range 6–401) for A/C/P.
MTD assessment
In the A/C arm, nine patients were evaluable for the determination of MTD (Fig. 1a; Table 2). In Cohort 1 (afatinib 20 mg plus carboplatin AUC6), three patients received combination treatment and no DLTs were reported. In Cohort 2 (afatinib 40 mg plus carboplatin AUC6), no DLTs were reported in the first three evaluable patients (two patients were not evaluable for MTD determination and were replaced), and the cohort was expanded to nine patients (one patient was not evaluable and was replaced). Of the six patients who were evaluable for MTD in Cohort 2, one experienced a DLT (grade 3 acneiform rash). The afatinib dose, here combined with a dose of carboplatin, was not further escalated from the standard monotherapy dose of 40 mg, consistent with standard of care. Afatinib 40 mg plus carboplatin AUC6 was defined as the recommended phase II dose (RP2D) in the A/C arm, without an MTD being reached.

Dose-escalation schema and incidence of DLTs in the A/C arm (a) and the A/C/P arm (b). A20 + C6, afatinib 20 mg/day + carboplatin AUC6; A40 + C6, afatinib 40 mg/day + carboplatin AUC6; A20 + P175 + C5, afatinib 20 mg/day + paclitaxel 175 mg/m2 + carboplatin AUC5; A20 + P175 + C6, afatinib 20 mg/day + paclitaxel 175 mg/m2 + carboplatin AUC6; A30 + P175 + C5, afatinib 30 mg/day + paclitaxel 175 mg/m2 + carboplatin AUC5; A40 + P175 + C5, afatinib 40 mg/day + paclitaxel 175 mg/m2 + carboplatin AUC5
In the A/C/P arm, 23 of 26 patients were evaluable for assessment of the MTD (Fig. 1b; Table 2). Of the seven patients who received treatment in Cohort 1 (afatinib 20 mg plus carboplatin AUC6 plus paclitaxel 175 mg/m2), six were evaluable for MTD and two had DLTs, which included grade 3 neutropenic sepsis with Clostridium difficile diarrhea in one patient and grade 3 mucositis in the other patient (Table 2). The carboplatin dose was decreased to AUC5 in Cohort 2 (afatinib 20 mg plus carboplatin AUC5 plus paclitaxel 175 mg/m2), in which three patients received treatment and no DLTs were reported. The afatinib dose was then increased to 40 mg in Cohort 3 (afatinib 40 mg plus carboplatin AUC5 plus paclitaxel 175 mg/m2), and five patients received treatment and were evaluable for MTD; two patients had DLTs (grade 3 fatigue and grade 3 mucositis in one patient; grade 3 febrile neutropenia in another). Owing to the DLTs observed, an intermediate afatinib dose of 30 mg was explored in Cohort 4 (afatinib 30 mg plus carboplatin AUC5 plus paclitaxel 175 mg/m2). Eight patients received this treatment and, of the six patients evaluable for MTD, two reported DLTs (grade 3 stomatitis; grade 3 mucositis). The next lower dose level (afatinib 20 mg plus carboplatin AUC5 plus paclitaxel 175 mg/m2) was then expanded with three patients, in addition to the three patients already treated with these doses in Cohort 2, and no DLTs were reported. Thus, afatinib 20 mg was identified as the MTD in combination with carboplatin AUC5 and paclitaxel 175 mg/m2. This dose was also identified as the RP2D for the A/C/P combination.
Adverse events
The most frequent drug-related AEs of any grade in the A/C arm were rash (grouped term; n = 9, 75%), which was most commonly acneiform, fatigue (n = 8, 67%) and diarrhea (n = 7, 58%). Most AEs were of CTCAE grade 1 or 2 in intensity (Table 3), and the only non-hematologic treatment-related grade 3 AEs in this arm were rash, fatigue and diarrhea (one patient each; 8%). There were no treatment-related grade ≥ 4 AEs.
The most frequent drug-related AEs in the A/C/P arm were diarrhea (n = 23, 88%), rash (grouped term; n = 19, 73%) and fatigue (n = 18, 69%). Most AEs were of CTCAE grade 1 or 2 in intensity, and there were no grade ≥ 4 treatment-related AEs (Table 4). The most common non-hematologic treatment-related grade 3 AEs in this arm were mucositis (n = 4; 15%) and fatigue (n = 3; 12%). Seven (27%) patients, including one patient with Clostridium difficile infection and one patient with mucositis (discussed above as DLTs), had AEs leading to discontinuation of study treatment.
Pharmacokinetics
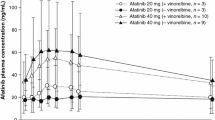
Table 5 summarizes key pharmacokinetic parameters for the RP2D cohorts of both treatment arms. Total platinum and paclitaxel showed a comparable exposure, based on Cmax and AUC, in the absence and presence of afatinib, suggesting no clinically relevant effect of afatinib on the pharmacokinetic parameters of these drugs. Afatinib reached peak plasma concentration 1–4 h after administration in the A/C arm and 3–8 h after administration in the A/C/P arm.
Antitumor activity
In the A/C arm, three patients (25%) had a confirmed partial response (PR), five (42%) had stable disease (SD) and one (8%) had PD; three patients (25%) were not evaluable for tumor response. Five patients (42%) achieved disease control (PR or SD) for at least 6 months. No ORs were seen in the seven patients with NSCLC receiving A/C treatment, but four patients (57%) had a best response of SD, one (14%) for at least 6 months. A response duration of 10.2 months was seen in one patient with a gastrointestinal neuroendocrine tumor.
In the A/C/P arm, five patients (19%) had a confirmed PR, eleven (42%) had SD and six (23%) had PD; four patients (15%) were not evaluable for tumor response. Of the 17 patients with NSCLC treated with A/C/P, ORs were reported for three patients (18%), and eight (47%) had SD. Four patients (15%), all of whom had NSCLC, achieved disease control (PR or SD) for at least 6 months. A response duration of 10.5 months was seen in one patient with NSCLC.
Discussion
In this phase I study, we assessed the safety and preliminary antitumor activity of afatinib in combination with carboplatin, and with carboplatin plus paclitaxel. In the A/C arm, dose escalation was not continued beyond afatinib 40 mg/day plus carboplatin AUC6 and so an MTD was not reached. The standard dose for afatinib monotherapy is 40 mg/day [10, 11]. Since manageable safety and antitumor activity were observed at this dose level with carboplatin AUC6, and no drug–drug interactions were observed, the RP2D was determined as afatinib 40 mg plus carboplatin AUC6. In the A/C/P arm, the MTD and RP2D were determined to be afatinib 20 mg plus carboplatin AUC5 plus paclitaxel 175 mg/m2. For this triplet combination, the afatinib single-agent dose needed to be de-escalated (from 40 to 20 mg) for the regimen to be tolerated. In both treatment combinations, afatinib at the RP2D was associated with a manageable safety profile. The most common drug-related AEs were rash, fatigue and diarrhea, and the majority of these were grade 1 or 2 in intensity. The AE profiles of the combinations were in line with the known individual safety profiles of afatinib, carboplatin and paclitaxel [5, 10, 12‒12].
Afatinib pharmacokinetic parameters were consistent with earlier studies of afatinib as a single agent [13, 14] and in combination [7], suggesting carboplatin and paclitaxel did not influence absorption and plasma concentrations of afatinib. Similarly, total platinum (from carboplatin) and paclitaxel plasma concentrations were similar in the absence and presence of afatinib, suggesting there were no relevant interactions between afatinib and paclitaxel and/or carboplatin in this study.
Evidence of antitumor activity was observed with both the A/C and A/C/P combinations. A/C was associated with disease control (PR or SD) in eight patients (67%), while disease control was observed in 16 patients (62%) receiving A/C/P. In this study, 58% and 65% of patients in the A/C and A/C/P arms, respectively, had NSCLC. Of these pretreated NSCLC patients, more than half in each treatment combination had disease control (57% in the A/C arm and 65% in the A/C/P arm), with 18% achieving an OR in the A/C/P arm. Of note, a response in one patient with NSCLC in the A/C/P combination arm was maintained for 10.5 months.
Several randomized trials have evaluated the potential clinical activity of the combination of EGFR-targeted agents with chemotherapy in advanced cancers, particularly NSCLC. Phase III studies in the first-line NSCLC setting in populations unselected for EGFR mutation have shown varying degrees of clinical efficacy for EGFR-targeted agent–chemotherapy combinations versus chemotherapy alone [15‒17]. In the phase III TRIBUTE trial, 1059 treatment-naïve patients with NSCLC were randomized to first-line carboplatin and paclitaxel combined with either erlotinib or placebo. No survival benefit was observed with erlotinib plus chemotherapy versus placebo plus chemotherapy in the overall trial population; however, a substantial prolongation of survival was observed in the ‘never smoked’ subgroup (22.5 vs 10.1 months; hazard ratio [HR] 0.49, 95% confidence interval [CI] 0.28–0.85; p = 0.01) [15]. Similarly, in the phase III TALENT trial, 1172 treatment-naïve patients with NSCLC were randomized to first-line gemcitabine and cisplatin combined with either erlotinib or placebo, with no survival benefit observed in the erlotinib group. In a small subgroup of patients who ‘never smoked’, overall survival (OS) and PFS were increased with chemotherapy and erlotinib, compared to chemotherapy alone [16]. In contrast to the TALENT and TRIBUTE studies, the phase III FAST-ACT2 trial, conducted primarily in Asia, demonstrated significantly improved PFS (HR 0.57, 95% CI 0.47–0.69; p < 0.0001) and OS (HR 0.79, 95% CI 0.64–0.99; p = 0.0420) with first-line chemotherapy (gemcitabine and platinum; Days 1 and 8 of a 4-week cycle) plus erlotinib (Days 15–28 of each cycle) over chemotherapy alone in 451 treatment-naïve patients with NSCLC. Benefit was primarily shown in those with EGFR mutation-positive disease; however, tumor samples were available in only 53% of the intent-to-treat population [17].
The combination of EGFR-targeted agents with chemotherapy has also been explored in the relapsed/refractory NSCLC setting. A randomized, phase II study in 165 patients with non-squamous NSCLC previously treated with one prior platinum-based chemotherapy regimen showed that pemetrexed plus erlotinib significantly improved PFS, OS and time to treatment failure versus pemetrexed alone; however, the combination was associated with an increase in grade 3/4 AEs [18]. We have previously reported on the combination of afatinib and weekly paclitaxel in patients with advanced solid tumors [5]. This regimen has since been assessed in a phase III trial of patients with relapsed/refractory NSCLC following ≥ 1 line of chemotherapy, and whose tumors had progressed after disease control of ≥ 12 weeks with erlotinib or gefitinib, and thereafter, afatinib. The combination of afatinib with paclitaxel significantly improved tumor response and PFS compared with paclitaxel alone in patients who had EGFR tyrosine kinase inhibitor (TKI)-resistant (including afatinib) disease [19]. Conversely, in the phase III IMPRESS study, gefitinib plus cisplatin and pemetrexed in patients with EGFR mutation-positive advanced NSCLC and acquired resistance to first-line gefitinib did not prolong PFS in the overall population versus cisplatin plus pemetrexed [20]. Indeed, OS was inferior in the experimental arm of this trial, although there was a suggestion of improved outcomes in patients lacking the T790M resistance mutation [21].
In the current study, the safety and clinical activity of two new afatinib combinations in patients with advanced solid tumors are presented. The RP2Ds of oral afatinib in these new combinations were defined as 40 mg/day with carboplatin AUC6, and 20 mg/day with carboplatin AUC5 and paclitaxel 175 mg/m2. These regimens may be of potential interest for further study, for example, in patients with squamous NSCLC; in selected populations with EGFR mutations; and, particularly for the combination of afatinib and carboplatin, in elderly populations and patients with an ECOG PS of ≥ 2.
References
Solca F, Dahl G, Zoephel A et al (2012) Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther 343:342–350
Sequist LV, Yang JC, Yamamoto N et al (2013) Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31:3327–3334
Wu YL, Zhou C, Hu CP et al (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15:213–222
Soria JC, Felip E, Cobo M et al (2015) Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol 16:897–907
Suder A, Ang JE, Kyle F et al (2015) A phase I study of daily afatinib, an irreversible ErbB family blocker, in combination with weekly paclitaxel in patients with advanced solid tumours. Eur J Cancer 51:2275–2284
Solca F, Baum A, Himmelsbach F et al (2006) Efficacy of BIBW 2992, an irreversible dual EGFR/HER2 receptor tyrosine kinase inhibitor, in combination with cytotoxic agents. Eur J Cancer Suppl 4:172
Spicer J, Irshad S, Ang JE et al (2017) A phase I study of afatinib combined with paclitaxel and bevacizumab in patients with advanced solid tumors. Cancer Chemother Pharmacol 79:17–27
European Medicines Agency (2015) Abraxane—European Public Assessment Report (summary). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000778/WC500020431.pdf
Griggs JJ, Mangu PB, Anderson H et al (2012) Appropriate chemotherapy dosing for obese adult patients with cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 30:1553–1561
European Medicines Agency (2013) Giotrif 20 mg film-coated tablets: Summary of Product Characteristics, Annex 1. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/00228.0/WC500152392pdf
Food and Drug Administration (2016) Gilotrif prescribing information. http://www.accessdata.fdagov/drugsatfda_docs/label/2016/201292s009lblpdf
EMC (2014) Carboplatin 10 mg/ml concentrate for solution for infusion. https://www.medicines.org.uk/emc/medicine/25716
Wind S, Schmid M, Erhardt J et al (2013) Pharmacokinetics of afatinib, a selective irreversible ErbB family blocker, in patients with advanced solid tumours. Clin Pharmacokinet 52:1101–1109
Murakami H, Tamura T, Takahashi T et al (2012) Phase I study of continuous afatinib (BIBW 2992) in patients with advanced non-small cell lung cancer after prior chemotherapy/erlotinib/gefitinib (LUX-Lung 4). Cancer Chemother Pharmacol 69:891–899
Herbst RS, Prager D, Hermann R et al (2005) TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 23:5892–5899
Gatzemeier U, Pluzanska A, Szczesna A et al (2007) Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol 25:1545–1552
Wu YL, Lee JS, Thongprasert S et al (2013) Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): a randomised, double-blind trial. Lancet Oncol 14:777–786
Dittrich C, Papai-Szekely Z, Vinolas N et al (2014) A randomised phase II study of pemetrexed versus pemetrexed + erlotinib as second-line treatment for locally advanced or metastatic non-squamous non-small cell lung cancer. Eur J Cancer 50:1571–1580
Schuler M, Yang JC, Park K et al (2016) Afatinib beyond progression in patients with non-small-cell lung cancer following chemotherapy, erlotinib/gefitinib and afatinib: phase III randomized LUX-Lung 5 trial. Ann Oncol 27:417–423
Soria JC, Wu YL, Nakagawa K et al (2015) Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol 16:990–998
Soria J-C, Kim S-W, Wu Y-L et al (Gefitinib/Chemotherapy vs Chemotherapy in EGFR Mutation-Positive NSCLC Resistant to First-Line Gefitinib: IMPRESS T790M Subgroup Analysis. In: 16th World Conference on Lung Cancer, Denver, CO, September 6–9 2015) Oral presentation 17.08
Acknowledgements
This study was supported financially by Boehringer Ingelheim, as well as by the UK Department of Health via the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) award to Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust (and NIHR Clinical Research Facility), and to The Institute of Cancer Research and Royal Marsden Hospital NHS Foundation Trust. King’s College London and the Royal Marsden hold Experimental Cancer Medicine Centre grants. Katy Pelling, an employee of Boehringer Ingelheim International GmbH during the time of manuscript development, provided substantial contributions towards data assembly, analysis and interpretation, and manuscript writing. Medical writing assistance, funded by Boehringer Ingelheim, was provided by Laura Winton of GeoMed, an Ashfield Company, part of UDG healthcare plc.
Author information
Authors and Affiliations
Contributions
Conception and design: MU-F, JB, JS. Collection and assembly of data: MO, DS, JB, KT, MU-F, KP, JB, JS. Data analysis and interpretation: TAY, MU-F, XJ, SW, JB, JS. Provision of study material or patients: JB. Manuscript writing: MO, DS, JB, KT, TAY, MU-F, KP, XJ, SW, JB, JS. Final approval of manuscript: MO, DS, JB, KT, TAY, MU-F, KP, XJ, SW, JB, JS.
Corresponding author
Ethics declarations
Conflict of interest
J. Bhosle has received non-financial support from Boehringer Ingehleim. J. de Bono has received grants, personal fees and non-financial support from AstraZeneca, Genentech and Merck Sharp & Dohme; personal fees from Pfizer Oncology, Janssen Oncology and Merck Serono; and grants and personal fees from Sanofi Aventis and GlaxoSmithKline. J. Spicer reports institutional receipt of honoraria from Boehringer Ingelheim. M. Uttenreuther-Fischer, K. Pemberton, X. Jin and S. Wiebe are employees of Boehringer Ingelheim International GmbH. All other authors report no potential conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
O’Brien, M.E.R., Sarker, D., Bhosle, J. et al. A phase I study to assess afatinib in combination with carboplatin or with carboplatin plus paclitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 82, 757–766 (2018). https://doi.org/10.1007/s00280-018-3661-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-018-3661-1