
Abstract
Cr3+ can substitute for Al3+ in the crystal structure of kyanite, Al2O(SiO4). Cr3+-rich solid solutions along the binary Al2O(SiO4)–Cr2O(SiO4) joint were synthesized under distinct high-pressure conditions. Sample crystals of Cr1+xAl1−xO(SiO4) with x = 0.025(12) and 0.188(8) have been synthesized at pressures of 7 GPa and temperatures up to 1200 °C. Unit-cell dimensions and single-crystal X-ray diffraction of the new Cr3+-rich kyanite-type phases show a linear increase of lattice parameters a, b and c with increasing Cr3+ content. Cr3+ replaces Al3+ on the octahedrally coordinated M-sites with Cr3+ preferentially occupying large [MO6]-octahedra. Cr3+ substitution for Al3+ is highest on the M3 site in a sequence of Cr3+(M3) > Cr3+(M2) > Cr3+(M1) > Cr3+(M4) following the polyhedral volumes V(M3O6) > V(M2O6) > V(M1O6) > V(M4O6). The compressibility of Cr1.19Al0.81O(SiO4) has been determined via precise lattice parameters up to 6.00(4) GPa. The pressure–volume data fitted to the third-order Birch–Murnaghan equation of state yielded an isothermal bulk modulus (KT0) of 196(16) GPa and a pressure derivative K′T0 of 2(4) at V0 = 310.3(1) Å3. The value of KT0 is in accordance to the Anderson–Anderson relation but it is slightly smaller than the respective value for natural Cr3+-free kyanite.
Similar content being viewed by others

Introduction
The Al2O(SiO4) polymorphs are amongst the most important rock-forming minerals of the Earth’s crust where they typically appear as minor constituents of metapelites and Al-rich metabasites. They serve as important indicators for regional and contact metamorphic P–T conditions from greenschist to granulite-facies P–T conditions. In appropriate bulk compositions, the high-P polymorph kyanite may be stable at upper mantle P–T conditions as evidenced by kyanite inclusions in diamond (e.g., Prinz et al. 1975; Smith et al. 2009) and the frequent presence of kyanite in mantle eclogites (e.g., Carswell et al. 1981; Smyth et al. 1989). High P–T experiments (Schmidt et al. 1997; Konzett et al. 2008) indicate that the upper P-stability limit of kyanite defined by the reaction kyanite = corundum + stishovite is located at P ≥ 12–15 GPa in the T range 800–1500 °C, thus extending the stability of kyanite to transition zone depths of ∼ 360–450 km.
The only crystal chemical substituent in natural kyanite that has been found so far in significant concentrations is Cr3+ with all known occurrences of kyanites with > 1 wt% Cr2O3 was reported from high-P environments (e.g., Cooper 1980; Delor and Leyreloup 1986; Gil Ibarguchi et al. 1991). The highest naturally occuring Cr2O3 contents known so far are in the range 15.6–17.0 wt% and were reported from chromite-bearing meta-gabbroic eclogites of the Eastern Alps and the Karpathians (Negulescu and Sabau 2012; Janák et al. 2015; Hauzenberger et al. 2016). P–T conditions estimated for the formation of these eclogitic kyanites are in the range 2.1–2.8 GPa and 700–900 °C. Cr3+-rich kyanites that are also known to be from upper mantle settings. Sobolev et al. (1968) and Pivin et al. (2011) reported the presence of kyanites with up to 12.7 and 11.8 wt% Cr2O3 from kimberlite-hosted grosspydite and clinopyroxenite xenoliths. Fe3+, too, is a common substituent in natural kyanite, however, with concentrations not exceeding approximately 1 wt% Fe2O3 and usually on the order of a few hundred to a few thousand µg/g. Other elements such as Ti, V, Mg, Ca or Na show concentrations ≤ 150–200 µg/g (e.g., Pivin et al. 2011; Müller et al. 2005, 2016; Pyka et al. 2014).
High P–T experiments in the system SiO2–Al2O3–Cr2O3 have shown that at 3 GPa and 1300 °C, up to 38 wt% Cr2O3 can be substituted into the synthetic analogue of kyanite which is equivalent to 25 mol% Cr2O(SiO4) exchange component (Langer and Seifert 1971). Based on these experiments, the present study was conducted to further explore the limits of Cr3+ substitution in kyanite-structure type Cr1 + xAl1−xO(SiO4), assuming that P > 3 GPa will stabilize Cr1 + xAl1−xO(SiO4) with > 25 mol% Cr2O(SiO4) component. Moreover, the experimental study was performed to shed light on the site occupancies, the distribution of Cr3+ within the kyanite crystal’s structure, and the impact of Cr3+ substitution on the elastic properties of kyanite. In this study, we investigated the crystal chemistry of two selected synthetic Cr3+-rich single-crystal samples, corresponding to compositions with 51.3 and 59.3 mol% Cr2O(SiO4), using single-crystal X-ray diffraction.
Synthesis and chemical composition
The starting materials (Table 1) were made from synthetic high-purity (≥ 99.99%) SiO2, Al2O3 and Cr2O3 powders that were mixed in appropriate stoichiometric proportions, homogenized in ethanol and dried at 150 °C for at least 48 h. The oxide mixtures were then filled into Pt tubes with 2.0/1.8 mm outer/inner diameter and a length of ≤ 3.5 mm and welded shut. The high-P–T synthesis experiments (Table 2) were conducted with a 500 t Walker-type multi-anvil device at the University of Innsbruck using 18/11 assemblies with pre-fabricated MgO-based ceramic octahedra and a graphite furnace combined with pyrophyllite gaskets. After the experiment, the sample capsules were removed from the assemblies, embedded longitudinally in epoxy resin and ground to expose the run products within the sample capsule (Fig. 1) as suitable for both electron microprobe analysis (EMPA) and extraction of crystal fragments for single-crystal X-ray diffraction studies. The composition of phases (Table 3) was determined with a JEOL 8100 electron microprobe using 15 kV acceleration voltage and 20 nA beam current. The duration of measurement was 20 s on peaks and 10 s on backgrounds of the Si-Kα, Al-Kα and Cr-Kα lines. Pure synthetic SiO2 and Al2O3, and natural chromite were used as standard materials. Raw counts were corrected with the PRZ correction procedure. MA53 resulted in crystals with a size of a few tens of micrometres and a bulk composition of Cr1.05Al0.95SiO5, very close to CrAlSiO5. In run MA69, crystals grew in a fine-grained matrix to form up to several 100 μm in size. The average composition of these crystals determined by electron microprobe was Cr1.33Al0.66SiO5.

Back-scattered electron images of run products from runs a MA53 and b MA69 (cf. Table 2). Abbreviation “ky” stands for Cr1+xAl1−xO(SiO4). c Stereo-photomicrograph of run product MA69 using a fibre-optical light source. Small matrix crystal show the intense emerald-green colour of Cr1.19Al0.81O(SiO4), whereas large crystals appears black
Single-crystal diffraction
Several single crystal fragments of intense emerald-green colour (Fig. 1) were selected under polarised light for clarity and optically sharp extinction under crossed polarizers, and finally checked for sharp XRD Bragg peak profiles thus giving small uncertainties for the refined lattice parameters. Subsequently a crystal from run MA69 with the size of 80 × 140 × 140 µm3 was mounted for single X-ray diffraction intensity data collection of a full sphere on an Oxford Diffraction Gemini four-circle diffractometer, equipped with a Ruby CCD detector and using graphite-monochromatized Mo-Kα radiation (50 kV, 40 mA) at the University of Innsbruck. Data collection was performed at room temperature. Data reduction included intensity integration, background and Lorentz-polarisation correction performed with the CrysAlisPRO Version 1.171.36.20 software package (Rigaku 2015). The morphologies of the crystal fragments were approximated by faces and an analytical absorption correction based on these indexed faces was applied. Because of the small size of crystals from MA53 X-ray intensity data of a crystal from MA53, measuring approximately 10 × 10 × 20 µm3, were collected using a Stoe StadiVari diffractometer equipped with a Dectris Pilatus 300 K pixel detector (with a 450 µm silicon layer) and using a 100 W air-cooled Incoatec IµS microfocus source (50 kV, 1 mA) with a Mo target and high-brilliance 2D-focussing Quazar multilayer mirrors at the University of Vienna. Recorded images were integrated using X-Area 1.76 (STOE & Cie GmbH, Germany 2015) and integrated intensities were corrected for absorption. The crystal structures were solved by charge flipping using the program SUPERFLIP (Palatinus and Chapuis 2007) and refined with the program JANA2006 (Petříček et al. 2014). Experimental details of data collection and crystal structure refinement are summarised in Table 4, atomic coordinates, interatomic distances and polyhedral volumes (Balić-Žunić and Vicković 1996) of the refined crystal structures are given in Tables 5 and 6. The crystal structure refinement of the data measured from a crystal of MA53 was performed with scattering factors for neutral atoms with anisotropic displacement parameters for all atoms. Due to an unfavourable ratio of measured reflections to refined parameter the refinement of the dataset measured from a crystal of run MA69 was performed with anisotropic displacement parameters for the cation positions and isotropic displacement parameters for the oxygen atoms. For the list of anisotropic displacement parameters refer to the deposited cif-file. In the initial stages of the crystal structure refinement Al and Cr atoms were placed on all M sites with an Al:Cr ratio of 1:1 for the crystal of MA53 and 1:2 for the crystal of run MA69. The substitution of Cr3+ for Al3+ was subsequently refined and found to be in excellent agreement for crystals of MA53 (Tables 3, 4): according to microprobe analysis the chemical composition is Cr1.05Al0.95O(SiO4) and the chemical formula from single-crystal structure refinement is Cr1.03Al0.98O(SiO4). However, for crystals of run MA69 a refined composition of Cr1.19Al0.81O(SiO4) was reproducibly obtained from the single-crystal data. This formula exhibits 6 mol% Cr2O(SiO4) less than the composition Cr1.33Al0.67O(SiO4) expected from synthesis and from the electron-microprobe analysis. Considering the given texture of inclusion-containing macrocrystals in a fine-grained matrix (cf. Fig. 1b), it cannot be ruled out that sub-microscopic Cr2O3 inclusions are responsible for apparently higher Cr3+-contents in the EMPA analysis. Microprobe data might thus reflect compositions adding up Cr1+xAl1−xO(SiO4) and Cr2O3-inclusions whereas the site-occupations derived from measured XRD intensities exclusively correspond to the kyanite lattice. Moreover, the crystal composition derived from site-occupation factors is confirmed by the fact that lattice-related data points follow the Vegard-type linear trend line of individual lattice parameters and the unit-cell volume versus Cr2O(SiO4)-content (Fig. 2a, b). Plotting the lattice parameters (Fig. 2a) of Cr1+xAl1−xO(SiO4) together with data reported by Langer and Seifert (1971) against the Cr2O(SiO4)-content, the values for all lattice parameters increase with increasing Cr2O(SiO4)-content due to the larger ionic radius of Cr3+ in comparison to that of Al3+ (ionic radii of Cr3+ and Al3+ in octahedral coordination: 0.615 Å and 0.535 Å, respectively; Shannon 1976). Assuming a content of 59.4 mol% Cr2O(SiO4) [i.e., Cr1.19Al0.81O(SiO4)] the dependence of the unit-cell parameters can be represented by the linear regression equation as follows: a = 7.121(1) + 0.00184(2)X, b = 7.851(3) + 0.00259(5)X, c = 5.569(1) + 0.00195(2)X Å, and V = 293.0(2) + 0.283(5)X Å3, [X = mol% Cr2O(SiO4)]. The values of reliability parameter χ2 are 0.99911, 0.99732, 0.99915 and 0.99815, whereas χ2-values for a linear regression assuming a Cr2O(SiO4) content of 66.7% [Cr1.33Al0.67O(SiO4) composition] range from 0.90069 to 0.98841.

a Lattice parameter and b volume of Cr1+xAl1−xO(SiO4) with increasing Cr3+ content plotted from this study (red diamonds) in comparison to the data reported by Langer and Seifert (1971) (blue squares). Notice that the error bars are smaller than the symbols
Equation-of-state measurement
Equation-of-state measurements of a single crystal of Cr1.19Al0.81O(SiO4) were performed through static compression at room temperature in an ETH-type diamond anvil cell (Miletich et al. 1999), using anvils with culets of 0.6 mm diameter and a gasket prepared from 0.25 mm thick stainless steel was pre-indented to a thickness of approximately 100 µm. The sample chamber of 250 µm diameter was drilled with an electronic spark eroder. A ruby chip and a quartz single crystal were loaded together with the specimen crystal and a 16:3:1 mixture of methanol–ethanol–water was used as hydrostatic pressure-transmitting medium. Ruby was used as internal pressure standard (Jacobsen et al. 2008) for a quick estimation of the pressure upon pressure change. After each change in pressure, the sample was left to settle for 36–48 h and subsequently high-precision measurements of the unit-cell parameters of Cr1.19Al0.8O(SiO4) and of quartz were performed at pressures up to 6.00(4) GPa on a four-circle Siemens P4 diffractometer, equipped with an Eulerian cradle and a point detector with slits parallel and perpendicular to the diffraction plane. Non-monochromatized MoKα radiation from a standard sealed-tube source, operated at 40 kV and 35 mA power setting, was used for the measurement of the Bragg intensities of the peak positions obtained for MoKα1/2 radiation. The setting angles of diffracted Bragg peaks were recorded with a point detector (crystal-to-detector distance 170 mm), the slits of which were set to 2 and 9 mm, respectively, for scan directions within and perpendicular to the diffraction plane. Each reflection was centred in eight positions according to King and Finger (1979), so as to eliminate systematic aberrations for diffractometer settings and sample positioning. During the fitting of reflection profiles, the α1–α2 peak splitting was treated by setting constraints on both the position and intensity (α2/α1 = 0.53) of the α2-peak components as implemented in the SINGLE software (Angel and Finger 2011). The pressure of the respective data point was determined from the lattice parameters of quartz (Scheidl et al. 2016). Equation-of-state parameters were fitted using the program EoSFit7-GUI (Gonzalez-Platas et al. 2016), while the strain analyses were performed using Win_Strain Vers. 4.11 (Angel 2011).
The crystal of Cr1.03Al0.98O(SiO4) used for the single-crystal intensity data collection was covered with insoluble epoxy glue and could therefore not be used for EoS measurements. Unfortunately, it was not possible to extract another single crystal of sufficient size and quality to collect volume–pressure data from MA53.
Results and discussion
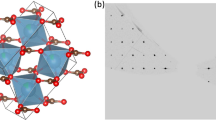
The crystallographically unconventional cell setting with lattice parameter c < a < b has been retained since the first crystal structure description of Taylor and Jackson (1928) and eases the comparison to the crystal structures of the two Al2O(SiO4)5 polymorphs, sillimanite and andalusite. The crystal structure of kyanite (Nárayo-Szabe et al. 1929; Burnham 1963; Winter and Ghose 1979) is a distorted cubic closed packing of oxygen atoms in an arrangement of the densely packed layers parallel to (110), in which Si atoms occupy 10% of the tetrahedral sites and Al atoms 40% of the octahedral sites M1–M4. The cation distribution follows a fashion that densely packed layers with only octahedrally coordinated cations alternate with relatively loose packed layer with octahedrally and tetrahedrally coordinated cations. Within the octahedra-only layer edge sharing, [M1O6] and [M2O6] octahedra form chains parallel to the c-axis and share edges with two additional [M3O6] and [M4O6] octahedra in the adjacent mixed tetrahedral–octahedral layer on alternating sides of the chain in a step-like fashion (Fig. 3a). Viewed along the c-axis, a broad layer-like package of octahedra parallel to (100) are connected by [SiO4] tetrahedra (Fig. 3b). Gatta et al. (2006, 2009) have described this octahedral arrangement as building block units with a zig-zag evolution along the b-axis.

Crystal structure of Cr1+xAl1−xO(SiO4) a in a view approximately along the a-axis highlighting the edge-sharing chain of [M1O6]–[M2O6] octahedra parallel to the c-axis with [M3O6] and [M4O4] octahedra attached on either side. Large [M2O6] and [M3O6] octahedra with high Cr3+ content are plotted in green, small [M1O6] and [M4O6] octahedra with lower Cr3+ occupation in blue, [SiO4] tetrahedra in red. b Larger block of the crystal structure of Cr1+xAl1−xO(SiO4) showing broad layer-like packages of octahedra parallel to (100) interconnected by [SiO4] tetrahedra
Cr3+ substitution occurs in the crystal structure of Cr1+xAl1−xO(SiO4) through replacement of Al3+ on all of the M-positions. Langer and Seifert (1971) have raised the question if one of the octahedra may be preferentially occupied by Cr3+. Their powder diffraction data on Cr1+xAl1−xO(SiO4) with up to 25% of the Al3+ replaced by Cr3+ did not allow a reliable refinement of the distribution of Al3+/Cr3+ on the M-sites. According to their findings on the intensity ratio of the (300) and (200) reflections, they concluded that the M1- and M2-site might be highly enriched with Cr3+ over Al3+. However, in contrast to this expected result, the highest Cr3+ content in the refined crystal structures of Cr1+xAl1−xO(SiO4) is found for M3 followed by M2 and M1, whereas the lowest Cr3+ concentration can be found on the M4 site (Table 6). The finding can be satisfyingly explained by considering the polyhedral volumes (Tables 5, 6; Fig. 4), which follow the same sequence V(M3O6) > V(M2O6) > V(M1O6) > V(M4O6). This leads to the suggestion, that the larger octahedra can substitute more of the relatively larger Cr3+ atoms than the smaller octahedra. Interestingly, a comparison with the high-pressure data measured on natural Cr3+-free kyanite (Yang et al. 1997a) shows an almost linear increase of the octahedral volumes as a function of V/V0, which confirms the above assumption (Fig. 4).

Polyhedral volumes of individual [MO6] octahedra in Cr1+xAl1−xO(SiO4) as a function of relative unit-cell volume. Data for V/V0 < 1 from Yang et al. (1997a). Inset: Cr3+ content of individual [MO6] octahedra as a function of composition
Unit-cell parameters were collected at room temperature to pressures of up to 6.00(4) GPa, (Table 7). The EoS parameters V0, KT0, K′T0, a, b, c and their axial compressibilities M′T0 were computed by fitting the P–V, P–a, P–b, and P–c data by a third-order Birch–Murnaghan EoS formulation. On a quick glance, the compressibility of the lattice parameters a, b, and c (Fig. 5a) look isotropic. However, detailed examination reveals a slight anisotropy: the axial compressibilities given in Table 8 show that Cr1+xAl1−xO(SiO4) is least compressible along the c-axis. To facilitate comparison with existing data of Cr3+-free kyanite (Yang et al. 1997a, b; Liu et al. 2009), we performed the analyses of lattice strain on compression between 1 bar and 4.6 and 5.9 GPa, respectively. The eigenvalues of the strain ellipsoid (e1 = 185(8) × 10− 5, e2 = 168(8) × 10− 5, e3 = 148(10) × 10− 5) are very similar to those in Cr3+-free kyanite (e1 = 188(3) × 10− 5, e2 = 169(7) × 10− 5, e3 = 140(3) × 10− 5) thus confirming approximately isotropic elasticity as expected for a close packing of the oxygens, with the direction of lowest compressibility approaching the c-axis. This is a general feature of the Al2O(SiO4) polymorphs with edge-sharing octahedral chains along the c-axis (Ralph et al. 1984; Yang et al. 1997b; Comodi et al. 1997; Friedrich et al. 2004; Burt et al. 2006). However, in the close-packed crystal structure of kyanite, the octahedra share several edges in a very complex manner. This results in a more uniformly distributed compression, as discussed by Yang et al. (1997a). Figure 5b–d show the effect of pressure on the unit-cell parameters a, b, and c in Cr1.19Al0.81O(SiO4) in comparison to the respective data measured in natural Cr3+-free kyanite. It is obvious that due to being isotypic Cr3+-free kyanite and Cr1+xAl1−xO(SiO4) show the same pressure response. The pressure response of the unit-cell angles α, β, and γ is shown in Fig. 5e–g: α increases, whereas β and γ decrease, if only slightly, with increasing pressure. This trend has also been observed for natural Cr3+-free kyanite with high-precision single-crystal data from Yang et al. (1997a) being exceptionally similar, whereas observations from Friedrich et al. (2004) and Liu et al. (2009) show a more distributed scatter.

A third-order Birch–Murnagham equation of state (EoS) was fit into fourteen pressure–volume data to yield an isothermal bulk modulus K0T of 196 ± 16 GPa, a pressure derivative K′0T of 2 ± 4 GPa and a refined V0 of 310.3(1). For a truncation to second order, i.e., when K′0T is fixed to 4.0, K0T refines to a value of 188 ± 4 GPa. Values for K0 of natural kyanite have been reported in the range of 156(10)–202(15) (see Table 2 in Liu et al. 2009 and Table 2 in Friedrich et al. 2004). Considering only the latest and most accurate EoS data measured on single crystals (Yang et al. 1997a; Friedrich et al. 2004) and powdered samples (Liu et al. 2009) computed for a Birch–Murnaghman of second order, the reported K0T values of 193(1), 190(3) and 201(2) GPa, respectively, are slightly larger than K0T of 188(4) of Cr1.19Al0.81O(SiO4). This is in accordance with observations made by Anderson and Anderson (1970) for isostructural materials, i.e., that the product of volume and bulk modulus follows a constant value. Comparing the V0K0T-products for the above-mentioned natural kyanite samples, i.e., 56,611 ± 210, 55,803 ± 650 and 58,692 ± 712 Å3 GPa, respectively, with the one of Cr1.19Al0.81O(SiO4) of 58,327 ± 877 Å3 GPa, it is determined that the Anderson–Anderson statement is fulfilled within the obtained standard deviations.
Summary
It has been demonstrated that substitution of Cr3+ in the kyanite crystal structure type is possible up to at least 59.3 mol% Cr2O(SiO4) component. Cr3+ substitutes for Al3+ and gives rise to a linear increase of the unit-cell parameters a, b, c and V. In contrast to previous assumptions (Langer and Seifert 1971), Cr3+ preferentially occupies the larger [M3O6] and [M2O6] octahedra, whereas Cr3+ replacement on the smaller octahedra around the M1- and M4-site occurs to a smaller degree. The pressure response of Cr1.19Al0.81O(SiO4) in regards to the behaviour of the lattice parameters is comparable to the one of Cr3+-free kyanite with a, b, c, β, and γ decreasing with increasing pressure, whereas α increases. As expected from the Anderson–Anderson relationship (Anderson and Anderson 1970), Cr1.19Al0.81O(SiO4) has a lower compressibility in comparison to Cr3+-free kyanite.
References
Anderson DL, Anderson OL (1970) The bulk modulus–volume relationship for oxides. J Geophys Res 75:3494–3500
Angel RJ (2011) Win_Strain: a program to calculate strain tensors from unit-cell parameters. http://www.rossangel.com/text_strain.htm. Accessed 3 Jan 2019
Angel RJ, Finger LW (2011) SINGLE: a program to control single-crystal diffractometers. J Appl Crystallogr 44:247–251
Balić-Žunić T, Vicković I (1996) IVTON—a program for the calculation of geometrical aspects of crystal structures and some crystal chemical applications. J Appl Crystallogr 29:305–306
Burnham CW (1963) Refinement of the crystal structure of kyanite. Z Kristallogr 118:337–360
Burt JB, Ross NL, Angel RJ, Koch M (2006) Am Mineral 91:319–326. https://doi.org/10.2138/am.2006.1875
Carswell DA, Dawson JB, Gibb FGF (1981) Equilibration conditions of upper-mantle eclogites: implications for kyanite-bearing and diamondiferous varieties. Mineral Mag 44:79–89
Comodi P, Zanazzi PF, Poli S, Schmidt MW (1997) High-pressure behaviour of kyanite: compressibility and structural deformations. Am Mineral 82:452–459
Cooper AF (1980) Retrograde alteration of chromian kyanite in metachert and amphibolite whiteschist from the Southern Alps, New Zealand, with implications for uplift on the Alpine Fault. Contrib Mineral Petrol 75:153–164
Delor CP, Leyreloup AF (1986) Chromium-rich kyanite in an eclogite from the Rouergue area, French Massif Central. Mineral Mag 50:537–537
Friedrich A, Kunz M, Winkler B, Le Bihan T (2004) High-pressure behavior of sillimanite and kyanite: compressibility, decomposition and indications of a new high-pressure phase. Z Kristallogr 219:324–329
Gatta GD, Nestola F, Walter JM (2006) On the thermo-elastic behaviour of kyanite: a neutron powder diffraction study up to 1200 °C. Mineral Mag 70:309–317
Gatta GD, Rotiroti N, Zucali M (2009) Plastic deformations in kyanites by tectonometamorphic processes: a single-crystal X-ray diffraction study. Mineral Mag 73:359–371
Gil Ibarguchi JI, Mendia M, Girardeau J (1991) Mg- and Cr-rich staurolite and Cr-rich kyanite in high-pressure ultrabasic rocks (Cabo Ortegal, northwestern Spain. Am Mineral 76:501–511
Gonzalez-Platas J, Alvaro M, Nestola F, Angel RJ (2016) EosFit7-GUI: a new GUI tool for equation of state calculations, analyses and teaching. J Appl Crystallogr 49:1377–1382
Hauzenberger C, Taferner H, Konzett J (2016) Genesis of chromium-rich kyanite in eclogite-facies Cr-spinel-bearing gabbroic cumulates, Pohorje Masif, Eastern Alps. Am Mineral 101:448–460
Jacobsen SD, Holl CM, Adams KA, Fischer RA, Martin ES, Bina CR, Lin JF, Prakapenka VB, Kubo A, Dera P (2008) Compression of single-crystal magnesium oxide to 118 GPa and a ruby pressure gauge for helium pressure media. Am Mineral 93:1823–1828
Janák M, Uher P, Ravna EK, Kullerud K, Vrabec M (2015) Chromium-rich kyanite, magnesiostaurolite and corundum in ultrahigh-pressure eclogites (examples from Pohorje Mountains, Slovenia and Tromso Nappe, Norway. Eur J Mineral 27:377–392
King H, Finger LW (1979) Diffracted beam crystal centering and its application to high-pressure crystallography. J Appl Crystallogr 12:374–378
Konzett J, Frost DJ, Proyer A, Ulmer P (2008) The Ca-Eskola component in eclogitic clinopyroxenen as a function of pressure, temperature and bulk composition: an experimental study to 15 GPa with possible implications for the formation of oriented SiO2-inclusions in omphacite. Contrib Mineral Petrol 155:215–228
Langer K, Seifert F (1971) High pressure–high temperature-synthesis and properties of chromium kyanite, (Al, Cr)2SiO5. Z Anorg Allg Chem 383:29–39
Liu X, Shieh SR, Fleet ME, Zhang L (2009) Compressibility of natural kyanite to 17.5 GPa. Prog Nat Sci 19:1281–1286
Miletich R, Reifler H, Kunz M (1999) The “ETH” diamond-anvil cell design for single-crystal diffraction at non-ambient conditions. Acta Crystallogr A55:Abstr P08.CC. 001
Müller A, Wanvik JE, Kronz A (2005) Norwegian kyanite quartzites—potential resources of high purity quartz? Geol Survey Norway Report No. 2005.039
Müller A, van den Kerkhof AM, Selbekk RS, Broekmans MATM (2016) Trace element composition and cathodoluminescence of kyanite and its petrological implications. Contrib Mineral Petrol 171:70. https://doi.org/10.1007/s00410-016-1280-6 (1–17)
Nárayo-Szabo S, Taylor WH, Jackson W (1929) The structure of kyanite. Z Kristallogr 71:117–130
Negulescu E, Sabau G (2012) Chromium-rich lawsonite in high-Cr eclogites from the Fagaras Massif (South Carpathians) American Geophysical Union, Fall Meeting 2012, abstract #V23D-2862
Palatinus L, Chapuis G (2007) Superflip—a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J Appl Crystallogr 40:786–790
Petříček V, Dusek M, Palatinus L (2014) Crystallographic computing system JANA2006: general features. Z Kristallogr 229(5):345–352. https://doi.org/10.1515/zkri-2014-1737
Pivin M, Berger J, Demaiffe D (2011) Nature and origin of an exceptional Cr-rich kyanite-bearing clinopyroxenite from Mbuji-Mayi kimberlite (DCR). Eur J Mineral 23:257–268
Prinz M, Manson VD, Hlava PF, Keil K (1975) Inclusions in diamonds: garnet lherzolite and eclogite assemblages. Phys Chem Miner 9:797–815
Pyka P, Gaweda A, Szopa K, Müller A, Sikorska M (2014) Petrogenesis of kyanite-quartz segregations in the mica schists of the Western Tatra Mountains (Slovakia). Mineralogia 45:99–120
Ralph RL, Finger LW, Hazen RM, Ghose S (1984) Compressibility and crystal-structure of andalusite at high pressure. Am Mineral 69:513–519
Rigaku (2015) CrysAlisPro, Yarnton, Oxfordshire, England
Scheidl KS, Kurnosov A, Trots DM, Boffa-Ballaran T, Angel RJ, Miletich R (2016) Extending the single-crystal quartz pressure gauge up to hydrostatic pressure of 19 GPa. J Appl Crystallogr 49:2129–2137
Schmidt MW, Poli S, Comodi P, Zanazzi PF (1997) High-pressure behavior of kyanite: decomposition of kyanite into stishovite and corundum. Am Mineral 82:460–466
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst A32:751–767
Smith CB, Bulanova GP, Kohn SC, Milledge HJ, Hall AE, Griffin BJ, Pearson DG (2009) Nature and genesis of Kalimantan diamonds. Lithos 112S:822–832
Smyth JR, Caporuscio FA, McCormick TC (1989) Mantle eclogites: evidence of igneous fractionation in the mantle. Earth Planet Sci Lett 93:133–141
Sobolev NV, Kuznetsova IK, Zyuzin NI (1968) The petrology of grospydite xenoliths from the Zagadochnaya kimberlite pipe in Yakutia. J Petrol 9:253–280
STOE & Cie GmbH (2015) X-Area, Germany
Taylor W, Jackson W (1928) The structure of cyanite, Al2SiO5. Proc R Soc London 119:132–146
Winter JK, Ghose S (1979) Thermal expansion and high-temperature crystal-chemistry of the Al2SiO5 polymorphs. Am Mineral 64:573–586
Yang H, Downs RT, Finger LW, Hazen RM, Prewitt CT (1997a) Compressibility and crystal structure of kyanite, Al2SiO5, at high pressure. Am Mineral 82:467–474
Yang H, Hazen RM, Finger LW, Prewitt CT, Downs RT (1997b) Compressibility and crystal structure of sillimanite, Al2SiO5, at high pressure. Phys Chem Miner 25:39–47. https://doi.org/10.1007/s002690050084
Acknowledgements
Open access funding provided by University of Innsbruck and Medical University of Innsbruck. C.H. wishes to express her gratitude to Biljana Krüger for helpful discussion. R.M. and T.P. are grateful to the Grant BE532003 of the University of Vienna. Constructive reviews from Hexiong Yang and an anonymous reviewer contributed to improving the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hejny, C., Konzett, J., Pippinger, T. et al. Pressure-enforced Cr substitution in Cr1+xAl1−xO(SiO4), synthetic analogues of kyanite. Phys Chem Minerals 46, 583–594 (2019). https://doi.org/10.1007/s00269-019-01024-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-019-01024-2