
Abstract
Background
Pompe disease (glycogen storage disease type II or acid maltase deficiency) is an autosomal recessive disorder caused by deficiency of the lysosomal enzyme acid α-glucosidase (GAA). Classic infantile-onset disease, characterized by cardiomegaly and profound weakness, leads to death in the first year of life from cardiorespiratory failure. Reversal of cardiomyopathy and improved motor function have been shown in clinical trials of rhGAA enzyme replacement therapy (ERT) with alglucosidase alfa (Myozyme), recently approved for clinical use. Increased survival potentially unmasks long-term complications of this previously lethal disease, including risk of skeletal fracture, recently identified at our institution and not previously reported in children with Pompe disease.
Objective
To report the risk of fracture in children with Pompe disease with increased survival with ERT.
Materials and methods
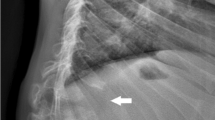
We present four cases of fracture in patients with classic infantile Pompe disease treated with ERT at our institution, and review a study database for additional reports of fracture in this population.
Results
We review 19 fractures in 14 children with Pompe disease on ERT.
Conclusion
Radiologists should be familiar with and vigilant for the association of fractures and increased survival on ERT in children with Pompe disease. We discuss potential mechanisms, implications for radiographic surveillance, potential intervention, and needs for further research.



Similar content being viewed by others


References
Hirschhorn R, Reuser AJ (2001) Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver AB, Sly W et al (eds) The metabolic and molecular bases of metabolic disease. McGraw Hill, New York, pp 3389–3420
Kishnani PS, Howell RR (2004) Pompe disease in infants and children. J Pediatr 144:S35–S43
van den Hout HM, Hop W, van Diggelen OP et al (2003) The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 112:332–340
Kishnani PS, Hwu WL, Mandel H et al (2006) A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 148:671–676
Kishnani PS, Steiner RD, Bali D et al (2006) Pompe disease diagnosis and management guideline. Genet Med 8:267–288
Amalfitano A, Bengur AR, Morse RP et al (2001) Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 3:132–138
Klinge L, Straub V, Neudorf U et al (2005) Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscul Disord 15:24–31
van den Hout H, Reuser AJ, Vulto AG et al (2000) Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet 356:397–398
Van den Hout JM, Kamphoven JH, Winkel LP et al (2004) Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 113:e448–e457
Van den Hout JM, Reuser AJ, de Klerk JB et al (2001) Enzyme therapy for Pompe disease with recombinant human alpha-glucosidase from rabbit milk. J Inherit Metab Dis 24:266–274
Kishnani PS, Nicolino M, Voit T et al (2006) Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr 149:89–97
Kishnani PS, Corzo D, Nicolino M et al (2007) Recombinant human acid alpha-glucosidase. Major clinical benefits in infantile-onset Pompe disease. Neurology 68:99–109
Lewiecki EM, Watts NB, McClung MR et al (2004) Official positions of the international society for clinical densitometry. J Clin Endocrinol Metab 89:3651–3655
Bianchi ML, Mazzanti A, Galbiati E et al (2003) Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos Int 14:761–767
Biggar WD, Bachrach LK, Henderson RC et al (2005) Bone health in Duchenne muscular dystrophy: a workshop report from the meeting in Cincinnati, Ohio, July 8, 2004. Neuromuscul Disord 15:80–85
Bothwell JE, Gordon KE, Dooley JM et al (2003) Vertebral fractures in boys with Duchenne muscular dystrophy. Clin Pediatr (Phila) 42:353–356
Burke SW, Jameson VP, Roberts JM et al (1986) Birth fractures in spinal muscular atrophy. J Pediatr Orthop 6:34–36
Granata C, Giannini S, Villa D et al (1991) Fractures in myopathies. Chir Organi Mov 76:39–45
Gray B, Hsu JD, Furumasu J (1992) Fractures caused by falling from a wheelchair in patients with neuromuscular disease. Dev Med Child Neurol 34:589–592
Hsu JD (1979) Extremity fractures in children with neuromuscular disease. Johns Hopkins Med J 145:89–93
Larson CM, Henderson RC (2000) Bone mineral density and fractures in boys with Duchenne muscular dystrophy. J Pediatr Orthop 20:71–74
McDonald DG, Kinali M, Gallagher AC et al (2002) Fracture prevalence in Duchenne muscular dystrophy. Dev Med Child Neurol 44:695–698
Vestergaard P, Glerup H, Steffensen BF et al (2001) Fracture risk in patients with muscular dystrophy and spinal muscular atrophy. J Rehabil Med 33:150–155
Talim B, Malaguti C, Gnudi S et al (2002) Vertebral compression in Duchenne muscular dystrophy following deflazacort. Neuromuscul Disord 12:294–295
Germain DP, Benistan K, Boutouyrie P et al (2005) Osteopenia and osteoporosis: previously unrecognized manifestations of Fabry disease. Clin Genet 68:93–95
Pastores GM, Meere PA (2005) Musculoskeletal complications associated with lysosomal storage disorders: Gaucher disease and Hurler-Scheie syndrome (mucopolysaccharidosis type I). Curr Opin Rheumatol 17:70–78
Apkon SD (2002) Osteoporosis in children who have disabilities. Phys Med Rehabil Clin N Am 13:839–855
Brunner R, Doderlein L (1996) Pathological fractures in patients with cerebral palsy. J Pediatr Orthop B 5:232–238
Henderson RC, Lark RK, Gurka MJ et al (2002) Bone density and metabolism in children and adolescents with moderate to severe cerebral palsy. Pediatrics 110(1 Part 1):e5
Jiang SD, Dai LY, Jiang LS (2006) Osteoporosis after spinal cord injury. Osteoporos Int 17:180–192
Quinlivan R, Roper H, Davie M et al (2005) Report of a Muscular Dystrophy Campaign funded workshop Birmingham, UK, January 16th 2004. Osteoporosis in Duchenne muscular dystrophy; its prevalence, treatment and prevention. Neuromuscul Disord 15:72–79
Kinali M, Banks LM, Mercuri E et al (2004) Bone mineral density in a paediatric spinal muscular atrophy population. Neuropediatrics 35:325–328
Douvillez B, Braillon P, Hodgkinson I et al (2005) Pain, osteopenia and body composition of 22 patients with Duchenne muscular dystrophy: a descriptive study. Ann Readapt Med Phys 48:616–622
Krishnamurthy V, Hanna R, Mackey JM et al (2005) Osteopenia in Pompe disease: a case series presentation. Paper presented at the meeting of the Society of Inherited Metabolic Disorders, Monterey, CA
Oktenli C (2000) Renal magnesium wasting, hypomagnesemic hypocalcemia, hypocalciuria and osteopenia in a patient with glycogenosis type II. Am J Nephrol 20:412–417
Huang MH, Barrett-Connor E, Greendale GA et al (2006) Hyperkyphotic posture and risk of future osteoporotic fractures: the Rancho Bernardo study. J Bone Miner Res 21:419–423
Orchowski J, Polly DW Jr, Klemme WR et al (2000) The effect of kyphosis on the mechanical strength of a long-segment posterior construct using a synthetic model. Spine 25:1644–1648
Chad KE, Bailey DA, McKay HA et al (1999) The effect of a weight-bearing physical activity program on bone mineral content and estimated volumetric density in children with spastic cerebral palsy. J Pediatr 135:115–117
Goemaere S, Van Laere M, De Neve P et al (1994) Bone mineral status in paraplegic patients who do or do not perform standing. Osteoporos Int 4:138–143
Gudjonsdottir B, Mercer VS (2002) Effects of a dynamic versus a static prone stander on bone mineral density and behavior in four children with severe cerebral palsy. Pediatr Phys Ther 14:38–46
Ward K, Alsop C, Caulton J et al (2004) Low magnitude mechanical loading is osteogenic in children with disabling conditions. J Bone Miner Res 19:360–369
Bianchi ML (2005) How to manage osteoporosis in children. Best Pract Res Clin Rheumatol 19:991–1005
Hawker GA, Ridout R, Harris VA et al (2005) Alendronate in the treatment of low bone mass in steroid-treated boys with Duchennes muscular dystrophy. Arch Phys Med Rehabil 86:284–288
Wagner KR, Lechtzin N, Judge DP (2007) Current treatment of adult Duchenne muscular dystrophy. Biochim Biophys Acta 1772:229–237
Financial disclosures
The clinical trials with rhGAA were supported by grants from Genzyme Corporation at the various sites that patients were treated. P.S.K. has received research/grant support and honoraria from Genzyme Corporation. P.S.K. is a member of the Pompe Disease Advisory Board for Genzyme Corporation. L.E.C. has received honoraria from Genzyme Corporation and research support from the Leal Foundation. S.D. and J.M. have received honoraria from Genzyme Corporation. Y.T.C. has served as a consultant for Genzyme Corporation. rhGAA, in the form of Genzyme’s product, Myozyme, has now been approved by the US FDA and the European Union as therapy for Pompe disease. Duke University and inventors of the method of treatment and predecessors of the cell lines used to generate the enzyme (rhGAA) used in the clinical trials could benefit financially pursuant to the University’s Policy on Inventions Patents and Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Laura E. Case and Rabi Hanna contributed equally to this study.
Rights and permissions
About this article
Cite this article
Case, L.E., Hanna, R., Frush, D.P. et al. Fractures in children with Pompe disease: a potentiallong-term complication. Pediatr Radiol 37, 437–445 (2007). https://doi.org/10.1007/s00247-007-0428-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-007-0428-y