
Abstract
DiGeorge syndrome (DGS) is commonly associated with both congenital heart disease (CHD) and immunologic abnormalities. While CHD may prompt consideration for heart transplantation (HTx), little is known about HTx management or outcomes in this group. The aim of this study was to describe the spectrum of patients with DGS who undergo HTx and report post-HTx outcomes. All pediatric HTx recipients (2002–2016) with DGS were identified using ICD codes from a linked billing and clinical registry database. Patient characteristics and outcomes were described and compared to non-DGS HTx recipients with CHD. Kaplan–Meier methods were used to assess overall survival, freedom from infection, and freedom from rejection. A total of 17 patients with DGS who underwent HTx at 12 different centers were included. Median age at HTx was 5 years (IQR 0–13 years). Steroids were used for induction in all patients in addition to thymoglobulin in 13/17 (76%) and IL2R antagonists in 3/17 (18%). Maintenance immunosuppression was a combination of tacrolimus or cyclosporine and mycophenolate or azathioprine in 16/17 (94%). Half received steroids at the time of discharge. There were six deaths (35%). The median post-HTx survival was 5.4 years with no difference in freedom from rejection, infection, or overall survival between patients with and without DGS. Patients with DGS undergoing HTx received standard immunosuppression. We found no difference in freedom from infection, rejection, or overall post-HTx survival compared to non-DGS patients, although the small size of our study resulted in limited statistical power. Given the potential for favorable outcomes, patients with DGS may be considered for HTx in the appropriate clinical setting.
Similar content being viewed by others


Introduction
DiGeorge syndrome (DGS), also known as 22q11.2 deletion syndrome, is a common genetic condition affecting approximately 1 in 4000 live births [1,2,3]. The chromosomal deletion in DGS impacts the development of the third and fourth pharyngeal pouches causing a variety of abnormalities, of which congenital heart disease (CHD) is prevalent. It is estimated that 77% of all patients with DGS have abnormalities of cardiac development [2], and it is one of the most common chromosomal causes of CHD, second only to trisomy 21 [4]. Patients with DGS may also manifest with hypocalcemia, palate deformities, developmental delay, and immune abnormalities [2, 5, 6]. The spectrum of immunologic deficiency in DGS is highly variable. Patients may have mild lymphopenia with minimal immunologic abnormality, but can also present with thymic aplasia and profound immunologic alterations [6].
Hemodynamically significant CHD may prompt consideration of heart transplantation (HTx) in patients with DGS. However, outcomes in this population are unknown. Additionally, management of immunosuppression in the post-HTx period may be complicated by the presence of immunodeficiency. The aim of this study was to describe the population of patients with DGS who have undergone HTx at pediatric hospitals in the United States, focusing on patient outcomes, management of immunosuppression, and the incidence of post-HTx infection and rejection.
Methods
This study utilized data from the Scientific Registry of Transplant Recipients (SRTR, Hennepin Healthcare Research Institute, Minneapolis, MN) and the Pediatric Health Information System (PHIS, Children’s Hospital Association, Lenexa, KS) administrative billing database. The SRTR data system includes data from all donors, wait-listed candidates, and transplant recipients in the U.S., submitted by the members of the Organ Procurement and Transplantation Network (OPTN). The Health Resources and Services Administration, U.S. Department of Health and Human Services provides oversight to the activities of the OPTN and SRTR contractors. The SRTR database includes data from every organ transplant and waitlist addition within the U.S. since October 1987. The PHIS database collects clinical and resource utilization data for hospital encounters from over 50 large children’s hospitals. Data captured by PHIS include inpatient hospitalizations, observation, ambulatory surgery, and emergency department encounters. The SRTR and PHIS databases were linked at the patient level using indirect identifiers, the results of which have been previously described [7].
All pediatric (age < 18) HTx recipients with a diagnosis of CHD were identified from the linked database for inclusion. The presence of DGS was determined by the presence of ICD-9 or ICD-10 codes for DGS (279.11 or D82.1) or velo-cardio-facial syndrome (758.32 or Q93.81) at any encounter. The characteristics of HTx recipients with DGS were assessed using standard descriptive statistics. A comparison group was constructed and consisted of all pediatric patients with a diagnosis of CHD, excluding patients known to have other common genetic syndromes including Turner syndrome (758.6) and Down syndrome (758.0). Demographics were compared between patients with DGS and non-DGS patients with CHD. The Fisher’s exact test was used for categorical variables and the Wilcoxon rank sum test was used for continuous variables. The Kaplan–Meier method was used to assess post-HTx survival, censoring at death or last known follow-up, and compared using the log-rank test. Given the potential for age to significantly impact the analysis, a secondary analysis was performed using an age and gender-matched control group (five controls for each subject with DGS) for the non-DGS comparison population.
Immunosuppression in the immediate post-HTx period (induction immunosuppression) and at the time of hospital discharge (maintenance immunosuppression) were extracted from the PHIS database and assessed for the population with DGS. Hospitalization for infection in the post-HTx period was assessed using SRTR patient follow-up data, with details supplemented by PHIS ICD coding. Similarly, treatment for rejection was identified using SRTR follow-up data. The Kaplan–Meier method was used to assess freedom from hospitalization for infection and freedom from rejection. Comparisons between patients with and without DGS were done using the log-rank test.
All statistical analyses were performed in SAS version 9.4 (SAS Institute; Cary, NC) or STATA version 15 (StataCorp LLC; College Station, TX) with two-sided p < 0.05 considered statistically significant. This project was approved by the Vanderbilt University Institutional Review Board, SRTR, and PHIS.
Results
A total of 17 unique HTx recipients with DGS were identified from 12 different centers (Table 1). Patients with DGS undergoing HTx were most commonly < 1 year of age (47%) and were predominantly Caucasian (65%). The underlying cardiac abnormalities in patients with DGS varied, but conotruncal defects were common including truncus arteriosus in 3/17 (18%), left-sided obstructive lesions (i.e., interrupted aortic arch, coarctation) in 4/17 (24%), and double outlet right ventricle/tetralogy of fallot in 3/17 (18%). In addition to this, 4/17 (24%) patients had hypoplastic left heart/common ventricle as their underlying cardiac diagnosis.
A total of 1306 HTx recipients with CHD without DGS were identified as a comparison group. Demographics of patients with and without DGS are shown in Table 2. Mechanical support pre-HTx was uncommon in patients with DGS, with two patients (12%) supported with a ventricular assist device and none supported with ECMO at the time of HTx. There were no significant differences in pre-HTx characteristics between patients with and without DGS undergoing HTx. Similarly, there were no significant differences in post-HTx complications between groups including the need for post-HTx ECMO, dialysis, re-operation (cardiac or extra-cardiac), and the incidence of stroke, rejection, or chest tube drainage > 2 weeks. Total, pre-, and post-HTx length of stay were not significantly different between groups.
During the immediate post-HTx period, all patients received intravenous steroids. Thymoglobulin was the most common induction agent, used in 13/17 (77%) patients. Three patients received interleukin-2 receptor antagonists (18%). Two received basiliximab alone and one received daclizumab in conjunction with thymoglobulin. Two patients received no additional induction therapies aside from IV steroids (12%). Maintenance immunosuppression consisted of a calcineurin inhibitor (tacrolimus or cyclosporine) paired with a purine synthesis inhibitor (mycophenolate or azathioprine) in 16/17 (94.1%) patients (Table 1). One patient received tacrolimus monotherapy. Nearly half of all patients with DGS (8/17) received maintenance steroids.
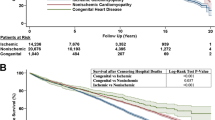
All but one patient with DGS (94%) survived to hospital discharge following HTx. There were five additional deaths in the DGS cohort following hospital discharge. Causes of death varied including one each with respiratory failure, cerebral hemorrhage, and coronary vasculopathy, and two deaths related to infection. The lone patient who did not survive the transplant hospitalization died from sepsis and multi-organ failure 154 days post-HTx. The other patient whose death was related to infection died 3.8 years post-HTx from a viral infection and acute respiratory distress syndrome. Overall post-transplant survival is shown in Fig. 1. The median survival for patients with DGS was 5.4 years, compared to a median survival for the non-DGS CHD cohort of > 15 years (log rank p = 0.473).

Post-transplant survival in patients with and without DiGeorge syndrome
The secondary analysis included 85 controls matched 5:1 by age and gender to the cohort with DGS. Post-transplant survival for this group compared to patients with DGS is shown in Fig. 2. There was no difference in overall post-HTx survival between groups (log rank p = 0.869).

Post-transplant survival in patients with and without DiGeorge syndrome using age and gender-matched controls
Eight (47%) patients with DGS were hospitalized for infection post-HTx, compared to 557 (42%) in the non-DGS cohort (p = 0.806). Most hospitalizations for infection occurred remote from HTx with the median time to infection of 1.1 years (IQR 1–2 years) (excluding the patient who died 154 days post-HTx). Freedom from hospitalization for infection is shown in Fig. 3. There was no significant difference between patients with and without DGS (log rank p = 0.834).

Freedom from hospitalization for infection in patients with and without DiGeorge syndrome
At least one episode of treated rejection occurred in 6 DGS (35.3%) and 428 (32.4%) non-DGS patients during follow-up (p = 0.797). The median time to the first rejection episode in the DGS cohort was 381 days (IQR 154–395 days). The earliest episode of rejection occurred 39 days post-HTx (N = 1). Freedom from rejection is shown in Fig. 4. There was no significant difference between patients with and without DGS (log rank p = 0.892).

Freedom from rejection in patients with and without DiGeorge syndrome
Discussion
This analysis provides novel data about HTx in children with DGS, in whom immunodeficiency is a common part of the underlying syndrome. We found that overall survival after HTx and freedom from rejection or infection were not different between HTx recipients with and without DGS. Though the small cohort size limits our power to detect differences, no prior report has specifically examined HTx outcomes for children with DGS. While median survival in the DGS population of 5.4 years appears suboptimal, this may be due to small patient numbers with more limited follow-up. Importantly, we demonstrate the potential for favorable outcomes with follow-up of > 10 years in select patients. Therefore, the presence of DGS alone should not serve as an absolute contraindication to HTx.
Immunologic abnormalities vary considerably in DGS, ranging from minimal to profound immunodeficiency. Fortunately, severe immunodeficiency is rare, occurring in approximately 0.5–1% of patients with DGS [1, 6]. It is important to recognize that there is likely a significant selection bias in our cohort, with more significantly immunocompromised patients being excluded from transplant consideration. Therefore, the results of our analysis may not be generalizable to all patients with DGS and careful evaluation of each patient on an individual basis is warranted. Despite the clear advantages of utilizing a linked database, the available data do not provide enough data granularity to further characterize the immune status or the immunologic evaluation performed prior to listing for included patients. Additionally, it is impossible to know if deaths related to infections in patients with DGS were associated with an inherent immunodeficiency.
Our analysis demonstrates that patients with DGS, despite possible baseline immunodeficiency, receive routine immunosuppression with trends in induction and maintenance immunosuppression mirroring current practice variation [8]. However, the long-term impact of immunosuppressive therapy in patients with potential immunodeficiencies is unknown. Both patients who died from infectious complications received induction therapy with thymoglobulin. However, the small number of included patients with few infectious events in our study preclude assessment of the associations between immunosuppression and post-transplant complications.
It is also important to recognize that the immune abnormalities in DGS may vary over time [9, 10]. While impaired T cell function is prominent in infancy, older patients with DGS also demonstrate impaired B cell function and resulting deficiency in humoral immunity [11]. Additionally, T cell counts decline over time in healthy individuals, a phenomenon also found in patients with DGS [10]. Therefore, continued immunologic surveillance for this population is important, and likely becomes even more critical in the setting of systemic immunosuppression following HTx.
Immune dysregulation is a prominent feature of DGS, as evidenced by the increasing recognition of autoimmune diseases in this population [6]. The etiology of autoimmune diseases in this group is unclear, but evidence suggests that imbalance of T cell subsets with resulting B cell dysregulation may play a role [6, 12]. The impact of altered T cell responses in the presence of a transplanted organ is unclear. Our study demonstrated no difference in freedom from rejection between patients with and without DGS, suggesting that the risk of rejection is not higher in this group. Autoimmune disease can also occur post-HTx in the absence of DGS [13]. It remains unclear if routine immunosuppression may exacerbate or increase the frequency which autoimmune disease occurs in patients with DGS.
There are other significant comorbidities that occur in patients with DGS, which may have implications in the post-HTx period. Cognitive dysfunction and learning disabilities are commonly encountered in patients with DGS [14, 15], occurring in > 90% of patients [1]. Recent data suggest that the presence of cognitive delay should not serve as a contraindication to solid organ transplantation, as long-term patient outcomes are similar to those without evidence of cognitive delay [16,17,18,19,20]. However, these studies also highlight the importance of an adequate social support structure following transplantation in patients with cognitive impairment. Psychiatric disorders are also frequently encountered (60% of adults with DGS [1]) and may complicate the post-HTx course. Other potentially important factors which may complicate post-HTx management include hypocalcemia, hypothyroidism, urinary tract abnormalities, renal abnormalities, thrombocytopenia, failure to thrive, and a greater risk of malignancy [1, 21]. All of these factors have the potential to impact post-HTx outcomes in patients with DGS. Therefore, involvement of a multidisciplinary care team is likely a critical step both pre- and post-HTx in patients with DGS.
There are limited data describing the outcomes of patients with DGS following HTx. To our knowledge, there is only one prior report which also utilized the PHIS database. Broda and colleagues reported the outcomes of 64 patients with chromosomal anomalies (12 with DGS) following HTx [22]. They demonstrated similar 1-year post-HTx survival between patients with and without a chromosomal anomaly. In contrast to our study, the study by Broda et al. documented that patients with chromosomal anomalies have significantly longer post-HTx length of stay. This suggests that the post-HTx course in patients with DGS may be less complicated compared to other chromosomal abnormalities. Therefore, it is important to assess the risk of each genetic comorbidity in isolation when considering patients for HTx, as grouping chromosomal anomalies may be misleading.
Our study has inherent limitations. The small number of included patients with DGS limits our ability to perform a multivariable analysis. Therefore, there may be confounding factors not adjusted for in the analysis that may impact the outcomes observed. The PHIS database does not include all U.S. centers performing HTx in children. Therefore, some patients with DGS who have undergone HTX may have been missed. However, the PHIS database includes many of the highest volume pediatric HTx centers and therefore we suspect the population included is representative. As with all databases, erroneous data are possible, and thus some patients identified as having DGS may have been misclassified. However, the prominence of conotruncal defects in our cohort suggests that our analysis accurately identified the population of interest. The higher than expected prevalence of hypoplastic left heart may be explained by successful surgical repair of CHD more commonly associated with DGS, precluding the need for HTx. As previously discussed, there is almost certainly a selection bias in our cohort with only patients with DGS and few comorbidities being listed for HTx. There is insufficient data granularity in the linked PHIS-SRTR database to assess the degree of immunodeficiency in patients with DGS. Because of this, it is also impossible to assess if underlying immunodeficiency contributed to post-HTx deaths related to infection in patients with DGS. Lastly, while the linkage between the PHIS and SRTR databases allowed assessment of immunosuppression, dosages were unable to be assessed. Therefore, we are unable to account for any dosage adjustments that may have been made to account for any potential immunodeficiencies in this population.
Conclusion
Children with DGS demonstrate overall post-HTx survival that is not statistically different compared to patients without DGS. Despite receiving standard induction and maintenance immunosuppression, patients with DGS did not demonstrate differences in freedom from infection or rejection. While our study may be underpowered to detect a difference, these results suggest that patients with DGS may be considered for HTx in the appropriate clinical setting. However, careful evaluation of each patient on an individual basis is warranted. Further research is needed to delineate the optimal immunosuppression strategy and long-term post-HTx risks in this group.
References
Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J International 22q11.2 Deletion Syndrome C (2011) Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr 159(2):332–339 e331. https://doi.org/10.1016/j.jpeds.2011.02.039
McDonald-McGinn DM, Sullivan KE (2011) Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore) 90(1):1–18. https://doi.org/10.1097/MD.0b013e3182060469
McDonald-McGinn DM, Zackai EH (2008) Genetic counseling for the 22q11.2 deletion. Dev Disabil Res Rev 14(1):69–74. https://doi.org/10.1002/ddrr.10
Goodship J, Cross I, LiLing J, Wren C (1998) A population study of chromosome 22q11 deletions in infancy. Arch Dis Child 79(4):348–351
Alsoufi B, McCracken C, Shashidharan S, Deshpande S, Kanter K, Kogon B (2017) The impact of 22q11.2 deletion syndrome on surgical repair outcomes of conotruncal cardiac anomalies. Ann Thorac Surg 104(5):1597–1604. https://doi.org/10.1016/j.athoracsur.2017.04.019
Gennery AR (2012) Immunological aspects of 22q11.2 deletion syndrome. Cell Mol Life Sci 69(1):17–27. https://doi.org/10.1007/s00018-011-0842-z
Godown J, Thurm C, Dodd DA, Soslow JH, Feingold B, Smith AH, Mettler BA, Thompson B, Hall M (2017) A unique linkage of administrative and clinical registry databases to expand analytic possibilities in pediatric heart transplantation research. Am Heart J 194:9–15. https://doi.org/10.1016/j.ahj.2017.08.014
Rossano JW, Dipchand AI, Edwards LB, Goldfarb S, Kucheryavaya AY, Levvey Rn BJ, Lund LH, Meiser B, Yusen RD, Stehlik J, International Society for H, Lung T (2016) The Registry of the International Society for Heart and Lung Transplantation: nineteenth pediatric heart transplantation report-2016; focus theme: primary diagnostic indications for transplant. J Heart Lung Transplant 35(10):1185–1195. https://doi.org/10.1016/j.healun.2016.08.018
Jawad AF, Prak EL, Boyer J, McDonald-McGinn DM, Zackai E, McDonald K, Sullivan KE (2011) A prospective study of influenza vaccination and a comparison of immunologic parameters in children and adults with chromosome 22q11.2 deletion syndrome (digeorge syndrome/velocardiofacial syndrome). J Clin Immunol 31(6):927–935. https://doi.org/10.1007/s10875-011-9569-8
Jawad AF, McDonald-Mcginn DM, Zackai E, Sullivan KE (2001) Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr 139(5):715–723. https://doi.org/10.1067/mpd.2001.118534
Derfalvi B, Maurer K, McDonald McGinn DM, Zackai E, Meng W, Luning Prak ET, Sullivan KE (2016) B cell development in chromosome 22q11.2 deletion syndrome. Clin Immunol 163:1–9. https://doi.org/10.1016/j.clim.2015.12.004
Zemble R, Luning Prak E, McDonald K, McDonald-McGinn D, Zackai E, Sullivan K (2010) Secondary immunologic consequences in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Immunol 136(3):409–418. https://doi.org/10.1016/j.clim.2010.04.011
Mouledoux JH, Albers EL, Lu Z, Saville BR, Moore DJ, Dodd DA (2014) Clinical predictors of autoimmune and severe atopic disease in pediatric heart transplant recipients. Pediatr Transplant 18(2):197–203. https://doi.org/10.1111/petr.12205
Gerdes M, Solot C, Wang PP, Moss E, LaRossa D, Randall P, Goldmuntz E, Clark BJ III, Driscoll DA, Jawad A, Emanuel BS, McDonald-McGinn DM, Batshaw ML, Zackai EH (1999) Cognitive and behavior profile of preschool children with chromosome 22q11.2 deletion. Am J Med Genet 85(2):127–133
Simon TJ, Bish JP, Bearden CE, Ding L, Ferrante S, Nguyen V, Gee JC, McDonald-McGinn DM, Zackai EH, Emanuel BS (2005) A multilevel analysis of cognitive dysfunction and psychopathology associated with chromosome 22q11.2 deletion syndrome in children. Dev Psychopathol 17(3):753–784. https://doi.org/10.1017/S0954579405050364
Kamin DS, Freiberger D, Daly KP, Oliva M, Helfand L, Haynes K, Harrison CH, Kim HB (2016) What is the role of developmental disability in patient selection for pediatric solid organ transplantation? Am J Transplant 16(3):767–772. https://doi.org/10.1111/ajt.13519
Prendergast C, McKane M, Dodd DA, Godown J (2017) The impact of cognitive delay on pediatric heart transplant outcomes. Pediatr Transplant. https://doi.org/10.1111/petr.12896
Wightman A, Bartlett HL, Zhao Q, Smith JM (2017) Prevalence and outcomes of heart transplantation in children with intellectual disability. Pediatr Transplant. https://doi.org/10.1111/petr.12839
Wightman A, Hsu E, Zhao Q, Smith J (2016) Prevalence and outcomes of liver transplantation in children with intellectual disability. J Pediatr Gastroenterol Nutr 62(6):808–812. https://doi.org/10.1097/MPG.0000000000001071
Wightman A, Young B, Bradford M, Dick A, Healey P, McDonald R, Smith J (2014) Prevalence and outcomes of renal transplantation in children with intellectual disability. Pediatr Transplant 18(7):714–719. https://doi.org/10.1111/petr.12339
Stevens T, van der Werff Ten Bosch J, De Rademaeker M, Van Den Bogaert A, van den Akker M (2017) Risk of malignancy in 22q11.2 deletion syndrome. Clin Case Rep 5(4):486–490. https://doi.org/10.1002/ccr3.880
Broda CR, Cabrera AG, Rossano JW, Jefferies JL, Towbin JA, Chin C, Shamszad P (2018) Cardiac transplantation in children with Down syndrome, Turner syndrome, and other chromosomal anomalies: a multi-institutional outcomes analysis. J Heart Lung Transplant 37(6):749–754. https://doi.org/10.1016/j.healun.2018.01.1296
Disclosures
The data reported here have been supplied by the Hennepin Healthcare Research Institute (HHRI) as the contractor for the Scientific Registry of Transplant Recipients (SRTR). The interpretation and reporting of these data are the responsibility of the author(s) and in no way should be seen as an official policy of or interpretation by the SRTR or the U.S. Government.
Funding
This project was supported through internal funding from the Katherine Dodd Faculty Scholar Program at Vanderbilt University (JG). Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K23HL123938 (Bethesda, MD) (JS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to disclose related to the content of this manuscript.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was waived by Vanderbilt IRB.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Woolman, P., Bearl, D.W., Soslow, J.H. et al. Characteristics and Outcomes of Heart Transplantation in DiGeorge Syndrome. Pediatr Cardiol 40, 768–775 (2019). https://doi.org/10.1007/s00246-019-02063-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-019-02063-w