
Abstract
We compared the genetic differentiation in the green sea urchin Strongylocentrotus droebachiensis from discrete populations on the NE Atlantic coast. By using eight recently developed microsatellite markers, genetic structure was compared between populations from the Danish Strait in the south to the Barents Sea in the north (56–79°N). Urchins are spread by pelagic larvae and may be transported long distances by northwards-going ocean currents. Two main superimposed patterns were identified. The first showed a subtle but significant genetic differentiation from the southernmost to the northernmost of the studied populations and could be explained by an isolation by distance model. The second pattern included two coastal populations in mid-Norway (65°N), NH and NS, as well as the northernmost population of continental Norway (71°N) FV. They showed a high degree of differentiation from all other populations. The explanation to the second pattern is most likely chaotic genetic patchiness caused by introgression from another species, S. pallidus, into S. droebachiensis resulting from selective pressure. Ongoing sea urchin collapse and kelp forests recovery are observed in the area of NH, NS and FV populations. High gene flow between populations spanning more than 22° in latitude suggests a high risk of new grazing events to occur rapidly in the future if conditions for sea urchins are favourable. On the other hand, the possibility of hybridization in association with collapsing populations may be used as an early warning indicator for monitoring purposes.
Similar content being viewed by others

Introduction
The green sea urchin Strongylocentrotus droebachiensis (O.F. Müller 1776) (Echinoidea) is a key species with severe impact on coastal ecosystems. By large-scale overgrazing of kelp forests in the Pacific (Estes et al. 1998), NW Atlantic (Steneck et al. 2004) and NE Atlantic (Norderhaug and Christie 2009), it has profound ecological and economic importance. Kelp forests are highly productive (Pedersen et al. 2012) and diverse (Norderhaug et al. 2012) systems and deliver ecosystem services including habitats (Norderhaug et al. 2002), feeding grounds (Norderhaug et al. 2005) and nursery areas (Godø et al. 1989), while sea urchin-dominated barren grounds are low-productive marine deserts (Ling et al. 2015).
S. droebachiensis has a broad Arctic-boreal distribution in the Atlantic and Pacific (Scheibling and Hatcher 2013). In the NE Atlantic, it is distributed from Denmark in the south [56°N, (Dahl et al. 2005)] to Svalbard (79°N, Gulliksen and Sandnes 1980) and Novaja Semlja (Propp 1977) in the north. A large cohesive barren ground area (up to 2000 km2) has dominated the coast of mid-Norway and North Norway and Russian Kola coast for more than four decades as a result of a sudden growth in the sea urchin populations (Sivertsen 1997). Observations from fishermen from around 1970 suggest that this barren was created quickly when urchins in high densities formed fronts and grazed all kelp forests between 63° 30′N on the Norway coast and beyond 71°N into Russian water (Norderhaug and Christie 2009). Small discrete urchin populations have also dominated fjords as far south as the Gullmarsfjord at the west coast of Sweden (back to the nineteenth century (Norderhaug and Christie 2009). Also, in the Danish Strait (at approximately 56°N), boulder reefs deeper than 13–15 m are heavily grazed by S. droebachiensis (Dahl et al. 2005). These S. droebachiensis populations thus prevail more than nine degrees south of the population on the Norwegian coast.
The distribution of benthic species with pelagic propagules is typically driven by the physical environment and how it has changed historically (Hoarau et al. 2007). Circulation patterns may be important for gene flow between urchin populations, carrying larvae from source to sink populations. The main current direction is from the south North Sea and Denmark and changes direction into the west flowing coastal current along the Norwegian Skagerrak coast which follows the coast northwards before it divides into two main currents heading north to Svalbard and east along the Barents Sea coast (Fig. 1; Sætre 2007). Tidal currents moving water in and out from the fjord every day (Sætre and Aure 2007) have the potential of carrying larvae from fjord to coastal water. Ocean currents may provide transport corridors between discrete populations in open marine water. However, large distances may represent barriers to dispersal of pelagic larvae between populations and result in high degree of isolation between populations of benthic marine animals (Reisser et al. 2011). Gyres along the Norwegian coast increase the retention time and may isolate populations locally. Also, echinoderms are sensitive to low salinity; hence, outflowing brackish surface water may represent barriers against dispersal of larvae out of fjords (Scheibling and Hatcher 2013). It has been hypothesized that urchin populations in Norwegian sill fjords may have been trapped in cold, saline deep water inside fjord basins and isolated since the last ice age (Fredriksen 1999). Historical geographical barriers created during the ice ages are important for the current distribution of shallow water marine species with pelagic dispersal propagules (Hu et al. 2011; Reisser et al. 2011).

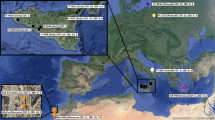
Map of the study area including sampling stations and a simplified illustration of the dominating currents along the coast from the Danish Belt Sea to the Barents Sea. Green arrows indicate the northbound coastal current, and red arrows indicate ocean currents from the NE Atlantic. Red circles and white boxes show the position and codes of the sample stations. See Table 1 for explanation of the codes. Oslofjorden (IO and D2), Lysefjorden (LY), Salangen (SI and SY) and Isfjorden (KW) are sill fjords, whereas the fjords represented by NH, NS VI and FV are open fjords/coastal areas. DV is located in the Danish Straits. The squares indicates groups (South and North) used in Migrate analysis
To what extent these small, southernmost populations supply larvae to populations further north is unknown. S. droebachiensis has high fecundity and is a free spawner with a planktonic larval stage lasting 4–21 weeks (Strathmann 1978; Hart and Scheibling 1988; Metaxas 2013); the dispersal potential is large exceeding 1000 km on the coasts of Nova Scotia despite relatively slow surface currents (Addison and Hart 2004). As the average planktonic larval stage duration for the species along the Norwegian coast is reported to be 16 weeks (Fagerli et al. 2013), high dispersal potential is also expected for NE Atlantic S. droebachiensis. Mature females are usually found February to April, spawning peaks in March, and the main settling period is during summer (Fagerli et al. 2013). In a study of population genetic structure of S. droebachiensis by the use of microsatellites, Addison and Hart (2004) found generally little differentiation between populations along the NW Atlantic coast, but considerable differentiation between one analysed population from the NE (North Norway) and populations in the NW Atlantic. They found no evidence of gene flow across the Atlantic, and the genetic difference between the NE and the NW Atlantic was even larger than the difference between populations in the NW Atlantic and populations from the Pacific coast. Marks et al. (2008) found indication of first stage speciation between S. droebachiensis from the NW and NE Atlantic. The NW Atlantic populations seem more closely related to populations from the N Pacific than NE Atlantic, but gene flow studies from the NE Atlantic are lacking.
The main aims of this study were to analyse genetic diversity and gene flow among S. droebachiensis populations in the NE Atlantic and assess possible links to physical features like ocean currents and fjord topography, as well as observed changes in this species distribution. Microsatellites recently developed for studying NE Atlantic populations were used in this study (Anglès d’Auriac et al. 2014). We compared differentiation across the species’ geographical distribution in the NE Atlantic (i.e. from Denmark in the south to Svalbard in the north) and from sill fjords, open fjords and coastal populations. Increased knowledge of gene flow between urchin populations and detection of genetic patterns disruption is important to assess risk for future grazing events and is thus highly relevant for the management of NE Atlantic coastal areas. For instance, a strong gene flow may show rapid recruitment ability and grazing events in the future. By contrast, detection of localized disruption of such a gene flow might be informative pertaining to ongoing ecosystem changes possibly affecting the equilibrium of the species and therefore associated grazing events along the Norwegian coast.
Materials and methods
Area of investigation and field sampling
The study area was chosen to cover the NE Atlantic distribution range of Strongylocentrotus droebachiensis. We included stations from the open coast as well as inside sill fjords and open fjords. Samples were primarily taken from 2 to 22 m depth, as abundant urchins were only found deep at the southern populations (Table 1). Sea urchins were collected by SCUBA divers at each of the 11 stations: 7 from open coastal areas or fjord mouths (DV, D2, NH, NS, VI, SY, FV, Table 1) and 4 from inside sills in fjords (IO, LY, SI, KW). Gonad material from 30 to 40 large urchins was sampled and fixed on site by ethanol. Sea urchins from Lysefjorden were collected and put in a styrofoam box and transported express to the laboratory in Oslo where samples were preserved within 8 h after they were collected.
Geographical distance calculations
Geographical distances were calculated as the distance from the southernmost population located close to the Vejrø Island in Denmark to all other stations further north. Since, to our knowledge, GIS maps (shape files or other) of ocean currents of the NE Atlantic do not exist, we created shape files in a GIS made from approximate, manually drawn paths following the main currents across Kattegat, along the Swedish and Norwegian coast, and crossing the Barents Sea to Svalbard (Fig. 1) based on a published map from Sætre and Aure (2007). Thus, the geographical distances calculated are not exact and the actual travelling distance may well be much longer, taking into account smaller currents and gyres and wind-driven deviations that may prolong the actual travel distance considerably. The geographical distance can thus be regarded as the least possible travel distance from Denmark and northwards following the main coastal currents and is assumed to be of sufficient precision for the purpose of this study.
Laboratory analysis
Rapid crude DNA extraction using 96 % ethanol-preserved gonad tissue was performed as described in Anglès d’Auriac et al. (2014). A total of 321 individuals were analysed using 10 microsatellites loci specifically developed for the Northeast Atlantic S. droebachiensis (Anglès d’Auriac et al. 2014). Among these microsatellites, Strdro-837 and Strdro-849 acted as polysomic and were not included in the analysis. Briefly, a 3-primer PCR approach was used using a M13 tail for the forward primers as well as labelled forward M13 primers in addition to the reverse primers. Simplex PCR amplifications were performed using iProof mastermix (Bio-Rad, Hercules, CA, USA) and a CFX96 thermocycler (Bio-Rad). The amplification products of up to four different microsatellite loci, each labelled with a different dye, were mixed for product size characterization using a 3730XL DNA analyser (Applied Biosystems, Foster City, CA, USA). Alleles were scored using GeneMapper software version 4.0 (Applied Biosystems).
Two populations, NH and NS, were found to be highly differentiated from the others. These were found in the middle of the sampling area. To exclude the possibility of this deviation resulting from sampling the wrong species, in particular S. pallidus (Gagnon and Gilkinson 1994), we sequenced part of the mitochondrial cytochrome oxydase I (COI) on three individuals from each NH and NS stations as well as from the Northernmost and Southernmost Norwegian stations, respectively, KW and D2. A 1056-bp COI fragment was amplified using the following primers: 5-ACACTTTATTTGATTTTTGG-3 (forward) and 5-CCCATTGAAAGAACGTAGTGAAAGTG-3 (reverse) described by Lee (2003); Balakirev et al. (2008). The phylogenetic analysis included 12 additional sequences from GenBank. PCR amplifications were performed using a CFX96 Bio-Rad thermocycler (Bio-Rad, Hercules, CA, USA) in 15 μl reaction volume containing 7.5 μl SsoFast or iProof mastermix (Bio-Rad), 0.1 μM of each primers and 2.5 μl sample (8 ng/μl DNA). Reaction volume was completed with sterile deionized water. PCR amplifications were carried out under the following conditions: a denaturing step for 2 min (SsoFast) or 30 s (iProof) at 98 °C, followed by 40 cycles of 98 °C for 10 s, 50 °C for 30 s and 72 °C for 40 s followed with a final extension at 72 °C for 2 min. Cycle sequencing was performed in both directions using amplification primers and BigDye Terminator version 3.1 kit (Life technologies, Applied Biosystems). One microlitre PCR template was used with 0.5 µl Terminator mix, 0.32 µl 10 µM forward or reverse primer, 1.75 µl Terminator 5X buffer in a final volume of 10 µl. Cycle sequencing was performed using an ABI 7500 qPCR machine (Life technologies, Applied Biosystems) as following: 96 °C for 1 min followed by 28 cycles of 96 °C for 10 s, 50 °C for 5 s and 60 °C for 4 min. Sequence purification was performed using BigDye XTerminator Purification kit (Life technologies, Applied Biosystems) adding to each PCR sample well 10 µl XTermination solution and 45 µl Sam solution, final volume of 65 µl. The PCR plate was then sealed and vortexed for 30 min prior to being processed by an ABI3730XL DNA analyser (Life technologies, Applied Biosystems). Trace files analyses and sequence alignments were performed using CodonCode Aligner version 5.1.5 (CodonCode Corporation), and the evolutionary history was inferred using the UPGMA method (unweighted pair group means analysis). The evolutionary distances were computed using the Kimura 2-parameter method [3] and are in the units of the number of base substitutions per site. The analysis involved 24 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 771 positions in the final dataset. The analysis was conducted using MEGA version 6.06 (Tamura et al. 2013).
Statistical analysis
Data files from GeneMapper were imported into Microsoft Excel (version 14) and formatted for analysis in GenAlEx (version 6.5.01 (Peakall and Smouse 2012). GenAlEx was used to calculate allele frequencies as well as for analysis of molecular variance (AMOVA; Excoffier et al. 1992) and principal coordinate analysis. This add-in was also used to export data for other programs, like MicroChecker [version 2.2.3 (Van Oosterhout et al. 2004)] which was applied for estimating null allele frequencies. We used Arlequin [version 3.5.1.2 (Excoffier and Lischer 2010)] to estimate deviations from Hardy–Weinberg equilibrium and linkage disequilibrium evaluation.
A search for substructures in genetic variation was conducted in Structure version 2.3.3 (Pritchard et al. 2000) implemented at https://lifeportal.uio.no/. The analysis was run with 106 burn-in iterations followed by 106 Markov chain Monte Carlo steps, applying an admixture model with correlated allele frequencies utilizing location information (LOCPRIOR and LOCISPOP set to 1, but USEPOPINFO set to 0). Results of 10 independent runs with K = 1–10 clusters were input to Structure Harvester (Earl and vonHoldt 2012) to evaluate probabilities of K with the Evanno et al. (2005) method and then summarized in CLUMPP version 1.1.2 (Jakobsson and Rosenberg 2007) and finally plotted with Distruct (Rosenberg 2004).
Most loci showed considerable heterozygote deficiencies, which resulted in significant departures from HW expectations. This involved all loci except Strdro-97 and 7209 and indicated the possible presence of null alleles although using simplex PCR reduces the possible occurrence of false-negative loci amplification results. We applied corrections as suggested by the Brookfield1 algorithm (Brookfield 1996) implemented in MicroChecker (Van Oosterhout et al. 2004), which entails replacing one of two alleles in a homozygous state with a missing value. We chose the Brookfield1 approach as being the most conservative, i.e. requiring the least manipulation of data. This correction removed most of the HW deviations, although some remained. Later analyses were performed on the corrected as well as the original data. We observed linkage disequilibria between certain loci in some populations, but no pairs of loci showed signs of linkage across all populations. Thus, all loci were retained in further analyses.
Genetic differentiation between populations was estimated as Jost’s D (obtained in GenAlEx) as well as F ST, based on null-allele-corrected allele frequencies. For Jost’s D, we used null allele corrections as decribed above, while F ST values were computed in the FreeNA software (Chapuis and Estoup 2007). This software estimates null allele frequencies using the EM algorithm (Dempster et al. 1977) and then calculates an F ST matrix applying the ENA correction (Chapuis and Estoup 2007) based on adjusted frequencies of visible alleles (thus disregarding the null alleles).
Effects of null alleles on inbreeding (fixation index, F IS) were analysed with INEST version 2.0 (Chybicki and Burczyk 2009), applying the default Bayesian approach using 300,000 steps, sampling every 100 steps and discarding the first 30,000 steps as burn-in.
Testing for selection on the eight loci was conducted in BayeScan version 2.1 (Foll and Gaggiotti 2008), applying default settings. Data were corrected for null alleles prior to these analyses, and only Group 1 populations and individuals as identified by Structure (see ‘Results’ section) were included.
We estimated gene flow between geographical regions using Migrate version 3.36.11 (Beerli 2006, 2009). This approach is based on a coalescence model and provides mutation-scaled estimates of effective population sizes (N e) and migration parameters between populations under specified dispersal scenarios (models) using genetic data. Posterior distributions of parameters (effective population sizes and migration rates) were generated by Bayesian inference using Markov Chain Monte Carlo runs. Basically, we ran three migration models: migration from A to B only, from B to A only or both ways. Posterior model probabilities were compared using Bayes factors. Parameters were free to vary over intervals specified as priors, set for each run. We applied uniform prior distributions of effective population size Θ from 0 to 200 and for migration rate M from 0 to 100. Mutation rate was constant over all loci. We applied a static heating scheme of 4 chains with temperatures proposed by the program (1, 1.5, 3 and 106 degrees), with swapping intervals set at 10. Only Group 1 individuals (as identified by the Structure analysis) were included in the datasets, since Group 2 individuals only occurred in a few areas, whereas Group 1 was present at all but one sampling stations. Since there was evidence for selection on five out of eight loci (see ‘Results’ section from BayeScan runs), only three loci were included in the dataset (Strdro-97-R, Strdro-7209 and Strdro-5563). We pooled the DV, IO, D2 and LY samples into one ‘South’ group and individuals from NH, VI, SI and SY into one ‘North’ group (Fig. 1).
Results
Genetic diversity and differentiation
All loci were polymorphic, with between 11 (Strdro-97 and 7209) and 26 (Strdro-5563) alleles detected per locus (Table 2). Populations did not differ markedly in allelic richness, with average number of alleles ranging between 7.12 and 10.0 per locus (Table 3). The number of private alleles ranged from none at stations DV and IO to 6 at station KW (=average 0.75 per locus, Table 3). Genetic diversity (expected heterozygosity) varied only slightly, from the highest estimate in population NH (0.69) to the lowest in SI (0.56). Observed heterozygosity was highest in LY (0.43) and lowest in KW (0.3) (Table 3).
Genetic differentiation between populations is here presented as estimates of F ST and Jost’s D EST estimates (Table 3). Although several populations differed significantly from each other, the genetic differentiation between populations was generally low (D EST < 0.06) over a wide geographical range (56–79°N). However, two coastal stations (NH Helløya and NS Skogsholmen at 65°N in the middle of the distribution area) stood apart, showing a much higher level of divergence (D EST 0.11–0.32, Table 4). Estimates of F ST were generally lower than D EST, ranging from 0.01 to 0.10. Nonetheless, F ST estimates also differed significantly from zero in exactly the same pairwise comparisons as D EST.
Only five loci (Strdro-97, 1356, 4147, 5563 and 7209) differentiated significantly (p < 0.05) between populations in an overall calculation, resulting in an overall D EST of 0.085 (SE 0.050). Among these five loci, Strdro-97 and 7209 contributed little to differentiation (D EST 0.009 and 0.012), while the remaining three loci showed stronger differentiation (D EST 0.116–0.471). This was due to marked differences in allele frequencies at these loci, particularly at Strdro-5563 and 1356. Figure 2 shows allele frequencies on the latter locus, while similar figures for other loci are given in Supplementary Information.

Allele frequencies by populations shown for locus Strdro-1356. See Supplement 1 for the other 7 loci
F IS estimates in INEST became lower in all populations when null alleles were allowed in the calculations, and this model was preferred over the alternative by the DIC criterion in all cases. Estimates of F IS varied from 0.022 (population LY) to 0.22 (population IO, Table 5). However, the posterior 95 % probability intervals included zero for all population estimates. Thus, all F IS estimates were not significantly different from zero, indicating no significant inbreeding in these populations. Increasing the number of MCMC steps from 300,000 to 600,000 had negligible effects on the estimates of F IS as well as the posterior distributions.
An analysis of molecular variance (AMOVA) with Group 1 animals partitioned most of the genetic variance (96.4 %) to within populations and the remaining 3.6 % among populations (Table 6). Variation within individuals was suppressed in this analysis. AMOVA results did not change with null-allele-corrected data.
Analysis of population structure clearly indicated two subgroups (Fig. 3). With K = 2, the highest rate of change (delta K = 479.3; 32 times higher than any other step) and mean log probability of K levelled off at higher K. Concordant with the genetic differentiation estimates, one cluster dominated stations from south to north, except the NH and NS station in mid-Norway, which were mainly allocated to the second cluster, and the FV station where individuals were evenly divided among clusters (Fig. 4).

Clustering of 8-locus genotypes with Structure in two groups (K = 2 preferred by both log probability of K and by the Evanno method). Group 1, coloured in orange, gathers most individuals and populations, whereas group 2, coloured in blue, is found in NS, NH and about half of FV individuals

Genetic versus geographical distance. Scaled differentiation (F ST/(1 − F ST)) for each population compared with the southernmost Belt Sea (DV) population. Based on the Structure results shown in Fig. 3, NH and FV are both split in two, NH1, NH2, FV1 and FV2. Statistics are based on the linear regression between the two variables, with the intercept forced to 0 (Danish population, DV)
Isolation by distance (IBD)
Based on the expectation that larval transport occurs primarily from south to north following the Norwegian coastal current, we plotted an estimate of genetic differentiation between the southernmost population from the Belt Sea (DV) and all other populations versus estimated distance between stations (Fig. 4). Linearized F ST values (i.e. F ST/(1 − F ST)) were then plotted against estimated distances from the southernmost site DV, to test a hypothesis of IBD given an assumed dominant northwards transport of pelagic larvae.
Species identity
The S. droebachiensis mitochondrial partial COI sequences from this study are available at the European Nucleotide Archive: http://www.ebi.ac.uk/ena/data/view/LN828950-LN828961. Our sequences clustered with another sequence from Norway (positions 5894-7003 in GenBank accession number AM900391, Fig. 5). Three sequences from the NW Atlantic grouped in a separate clade within S. droebachiensis, while S. pallidus sequences formed a distinct sister clade to S. droebachiensis. Thus, there is no doubt about the maternal species affiliation of our study populations, whereas possible S. pallidus paternal hybridization cannot be concluded upon with this analysis alone.

UPGMA (unweighted pair group means analysis) tree for the COI sequences using Kimura 2-parameter substitution model. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (10,000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. In addition to the 12 S. strongylocentrotus individuals sequenced for this study shown by full blue circles, 12 sequences were obtained from GenBank: 4 S. strongylocentrotus, 4 S. pallidus and 4 S. purpuratus sequences. All access numbers are indicated in the figure
Gene flow in Migrate
Our main purpose in estimating gene flow was to test the hypothesis that S. droebachiensis larvae primarily disperse from south to north following the coastal current. Hence, the primary objective was to compare migration models, rather than obtain absolute estimates of number of migrants. For the composite North and South populations, we found that the model of northwards migration had a much higher probability than the alternatives (Table 7). We were unable to obtain satisfactory estimates of effective population size (Θ) the ‘North’ group, which showed considerable variation of the estimate across the prior space as reflected in the differences between modes, median and mean estimates (Table 8). By contrast, the estimates for the ‘South’ group and the migration rate converged well, with the corresponding estimates closely tied.
The effective population size of the South’ group was clearly smaller than that of the ‘North’ group. Even so, the estimated mutation-scaled migration rate M from ‘South’ to ‘North’ was high (Table 8).
Discussion
Two patterns superimposed could be distinguished of genetic differentiation of the NE Atlantic S. droebachiensis populations. The first pattern showed a consistent weak differentiation across its latitudinal distribution range which could be explained by an isolation by distance model (Fig. 4). This pattern is consistent with larvae being spread with ocean currents coming from the area of the southernmost population in the Danish Belt Sea area, going to the Norwegian Skagerrak coast and onwards by the coastal current turning west and north before dividing to the west coast of Svalbard and East Finnmark, respectively. The Danish (and southern Norwegian populations) may be fed with larvae from reef populations in the southeast North Sea and Danish Skagerrak. Parts of the current along the Danish Skagerrak coast turns south and penetrates into the Belt Sea area on deep water, while parts mix with Baltic water and later becomes the Norwegian coastal current.
This study thus supports the hypothesis that urchin populations in fjords and on the coast are generally not isolated, rather there seems to be relatively high gene flow from southern to northern populations and further into east Finnmark and west Svalbard, respectively. Northwards-flowing currents transport larvae from population to population, at least occasionally. With a larval phase lasting on average 16 weeks (Fagerli et al. 2013), larvae may be carried long distances from the source population before settling, providing an effective northwards-directional gene flow in the studied geographical area. The degree of isolation was relatively small across a large geographical area (over 3300 km from 56 to 79°N). Approximately 96 and 4 % of the observed genetic variation was found within and between the populations included in Group 1, respectively (according to AMOVA, Table 6). This is in line with what was found by Addison and Hart (2004) who found no differentiation among populations from Atlantic Canada consistent with high level of genetic flow or a low rate of genetic drift. There were thus few signs of isolation of populations even inside sill fjords, suggesting that sea urchins are in general not isolated by fjord circulation in sill fjords as earlier hypothesized by Fredriksen (1999), but there is likely considerable supply of larvae from upstream areas. The reason why sea urchin populations are only being found inside fjords in south Norway therefore seems not to be a lack of supply of larvae, but rather unfavourable conditions outside the fjords. This is discussed further below. LY Lysefjorden population showed some indications of higher isolation. While it did not deviate much in the isolation by distance model (Fig. 4), it had a relatively high numbers of private alleles (Table 4). The Lysefjorden is characterized by a shallow sill (13 m) and very little water exchange with the outside fjord water (Aure et al. 2001). Urchins are only found on rather deep water (from 22 m and below), and this population show signs of being trapped in deep water for long periods by the fjord circulation, and the results may show signs of higher isolation compared to the coastal and other fjord populations. This may indicate that physical fjord features may isolate some populations more than others.
The second main differentiation pattern, to our surprise, showed that two coastal populations in mid-Norway (65°N), NH and NS, as well as the northernmost population of continental Norway (70°N) FV, were very different genetically from the other populations and showed high degree of isolation from all other populations. This was indicated both by population differentiation estimates (D EST, R ST) and by the clear indication of two distinct clusters in Structure. Punctual presence of high diversity patches in the midst of genetically homogenous populations of a species covering large geographical areas is not uncommon, especially among marine invertebrates (Larson and Julian 1999). This phenomenon, chaotic genetic patchiness, has also been described for sea urchin populations which have experienced collapse caused by disease outbreaks (Addison and Hart 2004). The apparent paradox of such a pattern showing high genetic diversity among collapsing populations may be associated with introgression (Harper et al. 2007). The COI sequences we obtained showed unequivocally that the NS and NH individuals had a S. droebachiensis maternal origin, whereas hybridization with males from the closely related species S. pallidus could not be eliminated. Indeed, it is known that ova of S. droebachiensis can be fertilized by sperm of S. pallidus giving viable hybrids with pigmentation resembling S. droebachiensis in the adult stage (Strathmann 1981), and asymmetric introgression from S. pallidus into S. droebachiensis was further demonstrated (Addison and Pogson 2009). Hybridization between S. droebachiensis and S. pallidus was also suggested as early as 1952 in three specimens from the Trondheims fjord by (Vasseur 1952). Hence, the most likely explanation for this high diversity observed in areas where S. droebachiensis populations are collapsing is that a high selection pressure has paved the way for asymmetric introgression from S. pallidus into S. droebachiensis. These sea urchin populations in mid-Norway are currently collapsing and retreating northwards. Fagerli et al. (2013) found low urchin settlement around NS Skogsholmen compared to Hammerfest (near the North Cape at 71°N). The retreat is probably caused by ocean warming (Fagerli et al. 2013) and increasing predation on sea urchin recruits from northwards expanding Cancer pagurus and Carcinus maenas crabs as a result from warming (Fagerli et al. 2014). From the opposite side, invasive crabs from Russian waters, Paralithodes camtschaticus, invade the East Finnmark coast. This species was introduced to Kola (Russia) from the Pacific for marine cultivation purposes during the 1960s and has since the 1990s extended its range westwards into East Finnmark waters (Oug et al. 2011). Dense king crab populations in shallow water, collapse in sea urchin populations and recovery of kelp have been observed locally on the Russian coast during the last decade (Gudimov et al. 2003) and recently on the Norwegian coast (Christie and Gundersen 2014). While it is not known what have caused urchin collapse in this area, S. droebachiensis is a major prey for king crabs during spring when sea urchin recruits are settling (Pavlova 2009). If introgression is an early warning sign of urchin populations progressing towards collapse, larger areas shifting from sea urchin to kelp domination may be expected in the future.
The observed genetic differentiation in the NH and NS populations could also possibly be explained by hydrographical features: gyres south of Lofoten (i.e. the archipelago west of station VI in Fig. 1) may increase the retention time and trap pelagic larvae. South of Lofoten the current turns away from the coast and around Lofoten. South of Lofoten and inside the coastal current large gyres are formed (Aas 1994; Sætre 2007) that may isolate or delay larvae to such a degree that settlement only occurs locally. Isolation may be further increased because this is a very shallow area (locally referred to as ‘boot sea’ to illustrate that it is almost possible to take on boots and wade offshore). Larvae could also be brought to this part of the coast by ocean currents from Scotland, regularly or occasionally (Fig. 1). The Lofoten gyre or transport from Scottish water, however, cannot explain why the FV populations shared a similar differentiation and we find them thus less likely. Isolation would also be expected to lower diversity (as indicated for LY Lysefjorden) and not the observed increase in diversity.
The HWE disequilibrium we observe on the 321 samples with all 8 loci (Table 2) is very similar to that observed with the 96 samples used to establish the microsatellite method (Anglès d’Auriac et al. 2014), showing consistency in the observed disequilibrium. Such HWE disequilibrium has been previously observed in S. droebachiensis as for example with 3 of the 4 microsatellites used in a prior study (Addison and Hart 2004), hence suggesting that S. droebachiensis may naturally deviate from HWE as it has been reported to be the case for many other marine invertebrates (Brownlow et al. 2008).Our findings have important implications for the risk of future grazing events. Widely spread larvae from Danish Skagerrak and fjords in southern Norway imply a high risk of new grazing events which can be expected to occur rapidly in the future under favourable conditions for the green sea urchin. While climate variation cannot be managed, kelp forest resilience can. Therefore, emphasis should be focused on strengthening kelp forest state resilience to withstand future sea urchin blooms. Our findings can also be used for developing monitoring indicators. Regime shifts between sea urchin-dominated and kelp forest states which are being observed along the Norway coast occur typically suddenly and come as a surprise. Therefore, early warning signals for these types of events are difficult to identify (Möllmann et al. 2015). Our findings may provide an opportunity to develop tools for predicting and monitor sea urchin population collapse before and as they occur.
References
Aas E (1994) De norske farvann. Institute of Geophysics, University of Oslo, Oslo
Addison JA, Hart MW (2004) Analysis of population genetic structure of the green sea urchin (Strongylocentrotus droebachiensis) using microsatellites. Mar Biol 144:243–251. doi:10.1007/s00227-003-1193-6
Addison JA, Pogson GH (2009) Multiple gene genealogies reveal asymmetrical hybridization and introgression among strongylocentrotid sea urchins. Mol Ecol 18:1239–1251. doi:10.1111/j.1365-294X.2009.04094.x
Anglès d’Auriac M, Hobæk A, Christie H, Gundersen H, Fagerli C, Haugstetter J, Norderhaug K (2014) New microsatellite loci for the green sea urchin Strongylocentrotus droebachiensis using universal M13 labelled markers. BMC Res Notes 7:699
Aure J, Strand Ø, Skaar A (2001) Framtidige muligheter for havbruk i Lysefjorden. Fisken og havet, nr 9
Balakirev ES, Pavlyuchkov VA, Ayala FJ (2008) DNA variation and symbiotic associations in phenotypically diverse sea urchin Strongylocentrotus intermedius. Proc Natl Acad Sci 105:16218–16223. doi:10.1073/pnas.0807860105
Beerli P (2006) Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22:341–345. doi:10.1093/bioinformatics/bti803
Beerli P (2009) How to use migrate or why are markov chain monte carlo programs difficult to use? In: Bertorelle G, Bruford MW, Haue HC, Rizzoli A, Vernesi C (eds) Population genetics for animal conservation. Cambridge University Press, Cambridge UK, pp 42–79
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455. doi:10.1046/j.1365-294X.1996.00098.x
Brownlow R, Dawson D, Horsburgh G, Bell J, Fish J (2008) A method for genotype validation and primer assessment in heterozygote-deficient species, as demonstrated in the prosobranch mollusc Hydrobia ulvae. BMC Genet 9:55
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. doi:10.1093/molbev/msl191
Christie H, Gundersen H (2014) From sea urchin deserts to rich kelp forests: crabs and climate as drivers of ecosystem shifts in southern Nordland and eastern Finnmark. FRAM, Print version: ISSN 1893-5532, Online version: ISSN 8193-5540
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113. doi:10.1093/jhered/esn088
Dahl K, Lundsteen S, Tendal OS (2005) Mejlgrund og Lillegrund. En undersøgelse af biologisk diversitet på et lavvandet område med stenrev i Samsø Bælt. DMU Report 529
Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc: Ser B (Methodol) 39:1–38
Earl D, vonHoldt B (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. doi:10.1007/s12686-011-9548-7
Estes JA, Tinker MT, Williams TM, Doak DF (1998) Killer whale predation on sea otters linking oceanic and nearshore ecosystems. Science 282:473–476. doi:10.1126/science.282.5388.473
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: applications to human mitochondrial DNA restriction data. Genetics 131:479–791
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Fagerli CW, Norderhaug KM, Christie HC (2013) Lack of sea urchin settlement may explain kelp forest recovery in overgrazed areas in Norway. Mar Ecol Prog Ser 488:119–132. doi:10.3354/Meps10413
Fagerli CW, Norderhaug KM, Christie H, Pedersen MF, Fredriksen S (2014) Predators of the destructive sea urchin Strongylocentrotus droebachiensis on the Norwegian coast. Mar Ecol Prog Ser 502:207–218. doi:10.3354/meps10701
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993. doi:10.1534/genetics.108.092221
Fredriksen K (1999) Vertikalfordeling og livshistorie hos kråkebollen Strongylocentrotus droebachiensis i Drøbaksundet. MSc thesis, University of Oslo, Oslo
Gagnon J-M, Gilkinson KD (1994) Discrimination and distribution of the sea urchins Strongylocentrotus droebachiensis (O.F. Müller) and S. pallidus (G.O. Sars) in the Northwest Atlantic. Sarsia 79:1–11. doi:10.1080/00364827.1994.10413542
Godø OR, Gjøsæter J, Sunnanå K, Dragesund O (1989) Spatial distribution of 0-group gadoids off mid-Norway. Rapports et Procès-verbaux des Réunions du Conseil International pour l’Exploration de la Mer, Copenhague, pp 273–280
Gudimov AV, Gudimova EN, Pavlova LV (2003) Effect of the red king crab Paralithodes camtschaticus on the Murmansk coastal macrobenthos: the first estimates using sea urchins of the genus Strongylocentrotus as an example. Dokl Biol Sci 393:539–541. doi:10.1023/B:DOBS.0000010317.69725.47
Gulliksen B, Sandnes O (1980) Marine bunndyrsamfunn, nøkkelarter og felteksperimenter på hardbunn. Fauna 33:1–9
Harper FM, Addison JA, Hart MW (2007) Introgression versus immigration in hybridizing high-dispersal echinoderms. Evolution 61:2410–2418. doi:10.1111/j.1558-5646.2007.00200.x
Hart MW, Scheibling RE (1988) Heat waves, baby booms, and the destruction of kelp beds by sea urchins. Mar Biol 99:167–176. doi:10.1007/BF00391978
Hoarau G, Coyer JA, Veldsink JH, Stam WT, Olsen JL (2007) Glacial refugia and recolonization pathways in the brown seaweed Fucus serratus. Mol Ecol 16:3606–3616. doi:10.1111/j.1365-294X.2007.03408.x
Hu ZM, Uwai S, Yu SH, Komatsu T, Ajisaka T, Duan DL (2011) Phylogeographic heterogeneity of the brown macroalga Sargassum horneri (Fucaceae) in the northwestern Pacific in relation to late Pleistocene glaciation and tectonic configurations. Mol Ecol 20:3894–3909. doi:10.1111/j.1365-294X.2011.05220.x
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. doi:10.1093/bioinformatics/btm233
Larson RJ, Julian RM (1999) Spatial and temporal genetic patchiness in marine populations and their implications for fisheries management. Cal Coop Ocean Fish 40:94–99
Lee Y-H (2003) Molecular phylogenies and divergence times of sea urchin species of Strongylocentrotidae, Echinoida. Mol Biol Evol 20:1211–1221. doi:10.1093/molbev/msg125
Ling SD, Scheibling RE, Rassweiler A, Johnson CR, Shears N, Connell SD, Salomon AK, Norderhaug KM, Pérez-Matus A, Hernández JC, Clemente S, Blamey LK, Hereu B, Ballesteros E, Sala E, Garrabou J, Cebrian E, Zabala M, Fujita D, Johnson LE (2015) Global regime shift dynamics of catastrophic sea urchin overgrazing. Philos Trans R Soc B. doi:10.1098/rstb.2013.0269
Marks JA, Biermann CH, Eanes WF, Kryvi H (2008) Sperm polymorphism within the sea urchin Strongylocentrotus droebachiensis: divergence between Pacific and Atlantic oceans. Biol Bull 215:115–125
Metaxas A (2013) Larval ecology of echinoids. In: Lawrence JM (ed) Sea urchins: biology and ecology. Elsevier, New York, pp 381–412
Möllmann C, Folke C, Edwards M, Conversi A (2015) Marine regime shifts around the globe: theory, drivers and impacts. Philos Trans R Soc Lond B Biol Sci. doi:10.1098/rstb.2013.0260
Norderhaug KM, Christie HC (2009) Sea urchin grazing and kelp re-vegetation in the NE Atlantic. Mar Biol Res 5:515–528. doi:10.1080/17451000902932985
Norderhaug KM, Christie H, Rinde E (2002) Colonisation of kelp imitations by epiphyte and holdfast fauna: a study of mobility patterns. Mar Biol 141:965–973. doi:10.1007/s00227-002-0893-7
Norderhaug KM, Christie H, Fosså JH, Fredriksen S (2005) Fish–macrofauna interactions in a kelp (Laminaria hyperborea) forest. J Mar Biol Assoc Uk 85:1279–1286. doi:10.1017/S0025315405012439
Norderhaug KM, Christie H, Andersen GS, Bekkby T (2012) Does the diversity of kelp forest macrofauna increase with wave exposure? J Sea Res 69:36–42. doi:10.1016/j.seares.2012.01.004
Oug E, Cochrane SJ, Sundet J, Norling K, Nilsson H (2011) Effects of the invasive red king crab (Paralithodes camtschaticus) on soft-bottom fauna in Varangerfjorden, northern Norway. Mar Biodivers 41:467–479. doi:10.1007/s12526-010-0068-6
Pavlova LV (2009) Estimation of foraging on the sea urchin Strongylocentrotus droebachiensis (Echinoidea: Echinoida) by the red king crab Paralithodes camtschaticus (Malacostraca: Decapoda) in coastal waters of the Barents Sea. Russ J Mar Biol 35:288–295. doi:10.1134/S1063074009040038
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. doi:10.1093/bioinformatics/bts460
Pedersen MF, Nejrup LB, Fredriksen S, Christie H, Norderhaug KM (2012) Effects of wave exposure on population structure, demography, biomass and productivity of the kelp Laminaria hyperborea. Mar Ecol Prog Ser 451:45–60. doi:10.3354/meps09594
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Propp MV (1977) Ecology of the sea urchin Strongylocentrotus droebachiensis of the Barents Sea: metabolism and regulation of abundance. Sov J Mar Biol 3:27–37
Reisser CMO, Wood AR, Bell JJ, Gardner JPA (2011) Connectivity, small islands and large distances: the Cellana strigilis limpet complex in the Southern Ocean. Mol Ecol 20:3399–3413. doi:10.1111/j.1365-294X.2011.05185.x
Rosenberg NA (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. doi:10.1046/j.1471-8286.2003.00566.x
Sætre R (2007) The Norwegian coastal current. Tapir Academic Press, Trondheim
Sætre R, Aure J (2007) Characteristic circulation features. In: Sætre R (ed) The Norwegian coastal current. Tapir Academic Press, Trondheim, pp 99–114
Scheibling RE, Hatcher BG (2013) Strongylocentrotus droebachiensis. In: Lawrence JM (ed) Sea urchins: biology and ecology developments in aquaculture and fisheries science. Elsevier, New York, pp 381–412
Sivertsen K (1997) Geographic and environmental factors affecting the distribution of kelp beds and barren grounds and changes in biota associated with kelp reduction at sites along the Norwegian coast. Can J Fish Aquat Sci 54:2872–2887. doi:10.1139/f97-186
Steneck RS, Vavrinec J, Leland AV (2004) Accelerating trophic-level dysfunction in kelp forest ecosystems of the western North Atlantic. Ecosystems 7:323–332. doi:10.1007/s10021-004-0240-6
Strathmann R (1978) Length of pelagic period in echinoderms with feeding larvae from the Northeast Pacific. J Exp Mar Biol Ecol 34:23–27. doi:10.1016/0022-0981(78)90054-0
Strathmann RR (1981) On barriers to hybridization between Strongylocentrotus droebachiensis (O.F. Müller) and S. Pallidus (G.O. Sars). J Exp Mar Biol Ecol 55:39–47. doi:10.1016/0022-0981(81)90091-5
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol. doi:10.1093/molbev/mst197
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. doi:10.1111/j.1471-8286.2004.00684.x
Vasseur E (1952) Geographic variation in the Norwegian sea-urchins, Strongylocentrotus droebachiensis and S. pallidus. Evolution 6:87–100. doi:10.2307/2405506
Acknowledgments
We would like to thank Stein Fredriksen, University of Oslo, for sampling from Svalbard and Erling Svensen, Marinereparatørene and Nomi Maria Holter-Norderhaug for help during field work and shipping of samples from Oslo- and Lysefjorden, Norway. We would also like to thank our colleague Jens Thaulow for advice on BayeScan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: S. Uthicke.
Reviewed by: M. Gonzalez-Wanguemert and an undisclosed expert.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Norderhaug, K.M., Anglès d’Auriac, M.B., Fagerli, C.W. et al. Genetic diversity of the NE Atlantic sea urchin Strongylocentrotus droebachiensis unveils chaotic genetic patchiness possibly linked to local selective pressure. Mar Biol 163, 36 (2016). https://doi.org/10.1007/s00227-015-2801-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-015-2801-y