
Abstract
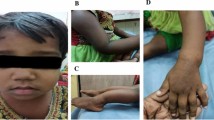
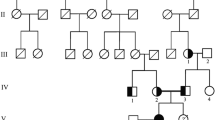
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is an autosomal recessive disorder caused by mutations in the CLDN16 or CLDN19 genes, encoding claudin-16 and claudin-19 in the thick ascending limb of Henle’s loop. In patients with claudin-19 mutations, severe ocular involvement (macular coloboma, pigmentary retinitis, nystagmus, or visual loss) has been described. In this report, we presented a 12-year-old girl with rickets, polyuria, and polydipsia. She was the daughter of consanguineous parents, and she had a history of recurred hypocalcemic and hypomagnesemic tetany. On physical examination, bilateral horizontal nystagmus and severe myopia were detected. Laboratory examination revealed hypomagnesemia, hypocalcemia, hypercalciuria, nephrocalcinosis, and renal stone. A clinical diagnosis of FHHNC caused possibly by claudin-19 mutation was decided with the ocular findings. DNA analysis revealed a novel homozygous missense mutation c.241C>T in the CLDN19 gene. In conclusion, in a patient with hypomagnesemia, hypercalciuria, nephrocalcinosis, and ocular findings, a diagnosis of FHHNC caused by claudin-19 mutation should be considered. This is the first study of FHHNC in Chinese population. Our findings of the novel mutation c.241C>T in exon 2 add to the list of more than 16 mutations of CLDN19 gene reported.




Similar content being viewed by others

References
Michelis MF et al (1972) Decreased bicarbonate threshold and renal magnesium wasting in a sibship with distal renal tubular acidosis. (Evaluation of the pathophysiological role of parathyroid hormone). Metabolism 21(10):905–920
Weber S et al (2000) Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis maps to chromosome 3q27 and is associated with mutations in the PCLN-1 gene. Eur J Hum Genet 8(6):414–422
Muller D, Kausalya PJ, Bockenhauer D, Thumfart J, Meij IC, Dillon MJ, Van’T HW, Hunziker W (2006) Unusual clinical presentation and possible rescue of a novel claudin-16 mutation. J Clin Endocrinol Metab 91:3076–3079
Simon DB et al (1999) Paracellin-1, a renal tight junction protein required for paracellular Mg2 + resorption. Science 285(5424):103–106
Konrad M et al (2006) Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79(5):949–957
Haisch L et al (2011) The role of tight junctions in paracellular ion transport in the renal tubule: lessons learned from a rare inherited tubular disorder. Am J Kidney Dis 57(2):320–330
Schlingmann KP, Konrad M, Seyberth HW (2004) Genetics of hereditary disorders of magnesium homeostasis. Pediatr Nephrol 19(1):13–25
Angelow S, Ahlstrom R, Yu AS (2008) Biology of claudins. Am J Physiol Renal Physiol 295(4):F867–F876
Godron A et al (2012) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol 7(5):801–809
Faguer S, Chauveau D, Cintas P, Tack I, Cointault O, Rostaing L, Vargas-Poussou R, Ribes D (2011) Renal, ocular, and neuromuscular involvements in patients with CLDN19 mutations. Clin J Am Soc Nephrol 6:355–360
Claverie-Martin F, Garcia-Nieto V, Loris C, Ariceta G, Nadal I, Espinosa L, Fernandez-Maseda A, Anton-Gamero M, Avila A, Madrid A, Gonzalez-Acosta H, Cordoba-Lanus E, Santos F, Gil-Calvo M, Espino M, Garcia-Martinez E, Sanchez A, Muley R (2013) Claudin-19 mutations and clinical phenotype in spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS One 8:e53151
Al-Shibli A, Konrad M, Altay W, Al MO, Al-Gazali L, Al AI (2013) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC): report of three cases with a novel mutation in CLDN19 gene. Saudi J Kidney Dis Transpl 24:338–344
Hou J, Renigunta A, Konrad M, Gomes AS, Schneeberger EE, Paul DL, Waldegger S, Goodenough DA (2008) Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest 118:619–628
Naeem M, Hussain S, Akhtar N (2011) Mutation in the tight-junction gene claudin 19 (CLDN19) and familial hypomagnesemia, hypercalciuria, nephrocalcinosis (FHHNC) and severe ocular disease. Am J Nephrol 34:241–248
Terry S, Nie M, Matter K, Balda MS (2010) Rho signaling and tight junction functions. Physiology (Bethesda) 25:16–26
Heiskala M, Peterson PA, Yang Y (2001) The roles of claudin superfamily proteins in paracellular transport. Traffic 2:93–98
Vargas-Poussou R et al (2008) Report of a family with two different hereditary diseases leading to early nephrocalcinosis. Pediatr Nephrol 23(1):149–153
Kasapkara CS et al (2011) A novel mutation of the claudin 16 gene in familial hypomagnesemia with hypercalciuria and nephrocalcinosis mimicking rickets. Genet Couns 22(2):187–192
Kari JA, Farouq M, Alshaya HO (2003) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Pediatr Nephrol 18(6):506–510
Rodriguez-Soriano J, Vallo A (1994) Pathophysiology of the renal acidification defect present in the syndrome of familial hypomagnesaemia-hypercalciuria. Pediatr Nephrol 8:431–435
Knoers NV (2009) Inherited forms of renal hypomag nesemia: an update. Pediatr Nephrol 24(4):697–705
Konrad M et al (2008) CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 19(1):171–181
Zimmermann B et al (2006) Hydrochlorothiazide in CLDN16 mutation. Nephrol Dial Transpl 21(8):2127–2132
Nijenhuis T et al (2005) Enhanced passive Ca2 + reabsorption and reduced Mg2 + channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115(6):1651–1658
Acknowledgments
This study was supported by a Grant from The Ministry of Science and Technology of the People’s Republic of China (National Science and Technology Major Projects for “Major New Drugs Innovation and Development” 2008ZX09312-016). We also thank National Natural Science Foundation of China (No.81070687 and 81170805), Beijing Natural Science Foundation (No. 7121012), Scientific Research Foundation of Beijing Medical Development (No. 2007-3029), and National Key Program of Clinical Science (WBYZ2011-873) for their support.
Conflict of interest
Tao Yuan, Qianqian Pang, Xiaoping Xing, Xi Wang, Yuhui Li, Jingjun Li, Xueyan Wu, Mei Li, Ou Wang, Yan Jiang, Jin Dong, and Weibo Xia declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This study was approved by the Department fo Scientific Research of PUMCH. All participants signed informed consent at the time of their participation in the study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Tao Yuan and Qianqian Pang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yuan, T., Pang, Q., Xing, X. et al. First Report of a Novel Missense CLDN19 Mutations Causing Familial Hypomagnesemia with Hypercalciuria and Nephrocalcinosis in a Chinese Family. Calcif Tissue Int 96, 265–273 (2015). https://doi.org/10.1007/s00223-014-9951-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-014-9951-7