
Abstract
Depression is a major health problem with a high prevalence and a heavy socioeconomic burden in western societies. It is associated with atrophy and impaired functioning of cortico-limbic regions involved in mood and emotion regulation. It has been suggested that alterations in neurotrophins underlie impaired neuroplasticity, which may be causally related to the development and course of depression. Accordingly, mounting evidence suggests that antidepressant treatment may exert its beneficial effects by enhancing trophic signaling on neuronal and synaptic plasticity. However, current antidepressants still show a delayed onset of action, as well as lack of efficacy. Hence, a deeper understanding of the molecular and cellular mechanisms involved in the pathophysiology of depression, as well as in the action of antidepressants, might provide further insight to drive the development of novel fast-acting and more effective therapies. Here, we summarize the current literature on the involvement of neurotrophic factors in the pathophysiology and treatment of depression. Further, we advocate that future development of antidepressants should be based on the neurotrophin theory.
Similar content being viewed by others


Introduction
Depression has emerged over the past decades as a major debilitating disease with a high prevalence in occidental populations, resulting in profound social and economic burden (Lopez and Murray 1998; Nestler et al. 2002; Pincus and Pettit 2001; Wittchen et al. 2011). Despite recent advances in neuroscience research, the neurobiological mechanisms underlying the pathophysiology of depression remain poorly understood. The development and course of major depressive disorder (MDD) are likely to be mediated by a complex interaction between genetic and environmental factors, and the associated heterogeneity of the disease makes it difficult to develop effective therapeutic treatments (Keers and Uher 2012). So far, many classes of antidepressants have been discovered and marketed for the treatment of depression. However, currently available antidepressants display significant limitations, including a delayed onset of action, low response rates, and relapse after treatment cessation, which remain major drawbacks for a disease with relatively high suicide rates (Angst et al. 2002). To date, clinical and preclinical studies have linked depression to structural and cellular alterations, such as neuronal loss and synaptic dysfunction, in cortico-limbic brain regions controlling mood and emotions (Duman and Aghajanian 2012). Among many candidates, neurotrophic growth factors and related signaling pathways constitute major players in neuroplasticity, and current evidence indicates that impairment in growth factor signaling is associated with depressed mood (Pittenger and Duman 2008). Interestingly, currently prescribed antidepressants have been shown to increase neuroplasticity when exerting their therapeutic effects (Tardito et al. 2006). Hence, a deeper understanding of the exact molecular, cellular, and structural plasticity mechanisms involved in antidepressant action might lead to the identification of key effectors and provide further insight into the development of novel fast-acting and more effective therapies. Here, we summarize the current literature on the implication of neurotrophic factors and associated signaling pathways in depression and antidepressant treatments. First, a brief overview of the brain regions and circuits implicated in the pathophysiology of depression and in the response to antidepressants is highlighted. Further, evidence for the involvement of neurotrophic factors and associated signaling pathways in depression and its current treatment is described. Finally, a perspective towards the development of novel antidepressant drugs is given.
Brain regions and neurocircuits involved in depression: neuroanatomical evidence
Amygdala
The amygdala is an integrant part of the limbic system implicated in cognitive and emotional processing, in particular that involved in fear and anxiety (Aggleton 1993; LeDoux 2000). This infers to this structure a central role in the regulation of emotion and in consequence, in mood-related pathology. Although volumetric magnetic resonance imaging (MRI) studies so far revealed contrasting results, with studies showing either an increase (Frodl et al. 2003; Lange and Irle 2004; Vassilopoulou et al. 2013) or a decrease in amygdalar volume in depressed patients (Bellani et al. 2011; Kronenberg et al. 2009; Lorenzetti et al. 2009), most of the functional MRI (fMRI) studies showed an increased activity of the amygdala in depressed patients during encoding of negative but not neutral or positive stimuli. Indeed, studies in depressed patients have shown exaggerated left and right amygdala activity when confronted with emotional facial expressions (Canli et al. 2005; Peluso et al. 2009; Sheline et al. 2001). Similarly, Drevets et al. using positron emission tomography (PET) imaging reported an increase in amygdala activation and metabolism in MDD patients (Drevets 2003). The fact that higher amygdala activity was often observed after negative stimuli would explain the higher ability for depressed individuals to encode and remember negative rather than positive information, therefore contributing to the negative bias observed in depressed patients (Groenewold et al. 2013; Hamilton and Gotlib 2008).
Interestingly, in studies using diffusion tensor imaging or MRI, abnormal microstructure and connectivity of the amygdala and the medial prefrontal cortex (PFC) patients (Arnold et al. 2012) as well as reduced functional coupling between the amygdala and the supragenual PFC (Matthews et al. 2008) were reported in remitted MDD. These findings suggest that MDD might result, at least in part, from a failed ability to co-activate a top-down cognitive control network during emotion processing (Matthews et al. 2008). This is supported by data from Pezawas et al. (2005) who reported that, in individuals carrying the short (s) allele of a variable number of tandem repeats in the 5′ promoter region of the serotonin transporter gene (5-HTTLPR), increased anxiety—and consequently an increased risk for depression—was associated with a reduced amygdala–anterior cingulate cortex connectivity. These data further support the hypothesis of the alteration of the negative feedback from the PFC to the amygdala in depressed patients (Pezawas et al. 2005).
In rodent, while a decrease in basolateral amygdalar volume has been associated with an increase in both fear and stress reactivity in mice (Yang et al. 2008), an enhanced dendritic arborization, elongation, and spine density, providing evidence for increased synaptic connectivity within the amygdala, were reported after chronic stress exposure in rats (Vyas et al. 2006; Vyas et al. 2002). These data were in line with those showing an enhanced activity of the basolateral amygdala and long-lasting anxiety-like responses in rats that received repeated injections of urocortin, an agonist of corticotropin releasing factor (CRF) receptors (Rainnie et al. 2004). This further suggests that depression-like behavior is associated with increased amygdala activity.
Interestingly, antidepressants have been shown to normalize most of these defects. Indeed, a meta-analysis from MRI studies showed that amygdalar volume was increased in medicated patients (Hamilton et al. 2008). Given that amygdala hyperactivity could lead to the amygdalar volume reduction observed in depression (Siegle et al. 2003), it could therefore be expected that antidepressants would decrease amygdala activity as well. Indeed, several meta-analyses concluded that antidepressants facilitate positive emotional stimuli processing in MDD patients and reduce the activity of negative emotions (Delaveau et al. 2011), concomitant with a normalization of amygdala activity (Chen et al. 2014; Victor et al. 2010).
Hippocampus
The hippocampus is also a major structure within the limbic system known to be highly vulnerable to stress and other environmental factors. This region is critical in diverse cognitive processes and in the regulation of emotions (Bartsch and Wulff 2015). MRI analyses revealed reduced hippocampal volume in patients suffering from both first episode and recurrent depression (Bremner et al. 2000; Cole et al. 2011; Frodl et al. 2007). Besides, a correlation between volume reductions and total duration of major depression has been reported more than 20 years ago (Sheline et al. 1996). These results have been further confirmed by several meta-analyses that show, e.g., a decreased hippocampal volume only in MDD patients having suffered from depression for 2 years or who had more than one episode (McKinnon et al. 2009) or evidencing deficit in hippocampal volume deficits in recurrent but not in first episode MDD patients (Schmaal et al. 2016), although this last study gave rise to some debate (Fried and Kievit 2016). It has also been proposed that reductions in hippocampal volume may not antedate illness onset but that hippocampal volume may decrease most in the early years after illness onset (MacQueen et al. 2003). Post-mortem analysis in MDD patients suggested an increase in the density of pyramidal, granule, and glial cells combined with a decrease of soma size of pyramidal cells (Stockmeier et al. 2004). This could indicate that the decrease in cellular neuropil might account for the reduced hippocampal volume found in depressed subjects.
Furthermore, disrupted functional hippocampal connectivity within the prefrontal and parietal cortex has been revealed by fMRI in MDD patients (Cao et al. 2012; Delaveau et al. 2011; Jaworska et al. 2015; Milne et al. 2012; Toki et al. 2014).
Animal studies revealed that chronic stress causes atrophy of apical dendrites of pyramidal neurons in the CA3 region of the hippocampus (Magarinos et al. 1996; Vyas et al. 2002; Watanabe et al. 1992). Exposure to excess glucocorticoids in rats showed decreased apical branching numbers and apical dendrite length (Woolley et al. 1990), suggesting a role for the activation of the hypothalamo-pituitary-adrenal (HPA) axis in remodeling hippocampal morphology. A wealth of data have clearly evidenced a correlation between hyperactivity of the HPA axis and the development of depression (Frodl and O’Keane 2013). Indeed, HPA axis activation is mainly triggered by stress, which has been shown to be strongly involved in inducing depression (Kendler et al. 1995). In addition, various models of chronic stress exposure in rodents have indicated a decrease in neurogenesis in the dentate gyrus (DG) of the hippocampus, involving a reduced proliferation, survival, and differentiation of neural stem cells (Eisch and Petrik 2012).
When treated with antidepressants (Boldrini et al. 2013; Fu et al. 2013; Sheline et al. 2003) or electroconvulsive therapy (ECT) (Nordanskog et al. 2014), depressed patients showed increased hippocampal volume. Interestingly, a meta-analysis reported that antidepressants could decrease the hypersensitivity to negative stimuli by decreasing hippocampus hyperactivation (Delaveau et al. 2011). Similarly, antidepressant such as imipramine could restore the total number of cells in the hippocampus impaired by social defeat stress in mice (Van Bokhoven et al. 2011). In addition, the same antidepressant could increase the number of hippocampal neurons in Flinders Sensitive Line rats, a genetic rat model of depression that shows impaired cell proliferation (Chen et al. 2010).
Altogether, these findings provide further evidence of the crucial role of the hippocampus in depression.
Prefrontal cortex
The PFC is functionally connected with several brain structures, for processing sensory input and mediating executive motor functions. The ventromedial PFC and the orbitofrontal cortex are involved in the cognitive processing of emotional stimuli originating from the limbic system (e.g., amygdala, ventral striatum, hippocampus, and hypothalamus) and are especially engaged in memory consolidation and retrieval (Ongur and Price 2000; Price 1999). As such, the PFC plays a major role in regulating the appropriate emotional response such as fear or anxiety. Moreover, the PFC has been associated with decision-making, personality expression, social behavior, and hedonic responses (Mitterschiffthaler et al. 2003).
Neuroimaging studies showed a reduction in size of multiple areas of the PFC in subjects diagnosed with MDD (Bremner et al. 2002; Drevets 2000). In line with those studies, post-mortem brain analysis of depressed patients revealed reduced neural cell size and neural and glial cell densities as well as synapse number in the dorsolateral and subgenual PFC (Cotter et al. 2002; Kang et al. 2012; Öngür et al. 1998; Rajkowska et al. 1999). The PFC is strongly connected with the amygdala and the hippocampus and the activity of its different subdivisions has been widely studied in depressed patients. Hence, although studies seemed consistent in showing a lower activity of dorsolateral PFC in resting state analyses of MDD patients (Fitzgerald et al. 2008; Hamilton et al. 2012; Limon et al. 2016; Zhang et al. 2015a; Zhong et al. 2016), meta-analyses of this region during task processing and especially in response to emotional stimuli with a negative valence have shown either a higher activity (Miller et al. 2015; Wang et al. 2015b; Zhang et al. 2013) or a lower activity (Groenewold et al. 2013; Hamilton et al. 2012; Zhang et al. 2015a). This might be due to the various parameters of the studies such as age of individuals, severity of depression, or whether there were medicated or not. The last point corroborates the hypothesis of impaired executive functions leading to emotional biases and dysregulation in MDD. Furthermore, the study of other PFC subregions tended to show a hyperactivity of the ventrolateral, the orbitofrontal, and ventromedial PFC in MDD patients (Groenewold et al. 2013; Limon et al. 2016; Miller et al. 2015).
Regarding preclinical studies, chronic restraint stress caused a significant reduction in the number and length of apical dendritic branches of pyramidal neurons in PFC areas (Cook and Wellman 2004). Similar results were observed in rats that received chronic administration of corticosterone. In this rodent model for stress-related disorders, a drastic dendritic reorganization of pyramidal neurons was also reported in the medial PFC (Wellman 2001). Furthermore, in rats expressing a depressive-like behavior, cell activity measured by c-Fos immunoreactivity was decreased in the ventromedial PFC (Lim et al. 2015). These data were supported by a study showing a decrease in both the excitatory and inhibitory neuronal functions in the PFC and a disturbed neurotransmitter homeostasis in the social defeat mouse model of depression (Veeraiah et al. 2014).
PFC abnormalities observed either in MDD patients or in rodent models are partly corrected by antidepressants. An 8-week escitalopram treatment was shown to reduce irregular high functional connectivity in the bilateral dorsal medial PFC in MDD patients (Lyttle et al. 2015; Wang et al. 2015a) and, in rats, a 2-week administration of a selective serotonin reuptake inhibitor (SSRI), fluvoxamine, restored dendritic length and spine densities but not cortical thickness after early-life stress exposure (Lyttle et al. 2015). Furthermore, it appeared that both high- and low-frequency electrical ventromedial PFC stimulations in rat models of depression attenuated depressive-like behavior (Bruchim-Samuel et al. 2016; Lim et al. 2015).
Ventral striatum
Finally, a preponderant role for the ventral striatum has also been reported in major depression. The fundamentals of the natural reward system are attributed to the dopaminergic connections between the ventral tegmental area (VTA) and the nucleus accumbens (NAc). In this respect, the NAc and the VTA may play a role in mediating the anhedonic symptoms of depression (Nestler and Carlezon Jr. 2006; Russo and Nestler 2013; Yadid and Friedman 2008). Depressed patients show an attenuated activation of the VTA-NAc pathway or the NAc itself when compared to normal patients in fMRI analyses (Epstein et al. 2006; Furman et al. 2011; Pizzagalli et al. 2009; Smoski et al. 2009). In rodent models of depression, a reduced dopaminergic activity in the NAc (Shirayama and Chaki 2006) with a disturbed burst firing of VTA neurons was also observed (Friedman et al. 2008).
Deep brain stimulation (DBS) of the NAc has been shown to influence the functionality of efferent projections of the NAc to the hippocampus (Settell et al. 2017). DBS targeting the NAc was shown to exert antidepressant, anxiolytic, and hedonic effects, notably in treatment-resistant depression (Bewernick et al. 2010; Giacobbe et al. 2009; Schlaepfer et al. 2008). These effects have been proven to be long-lasting and stable for up to 4 years (Bewernick et al. 2012; Malone Jr. et al. 2009). While MDD patients demonstrated reduced ventral striatal activation during anticipation of gain and loss, a treatment with the SSRI escitalopram was able to normalize this hyporesponsiveness (Stoy et al. 2012). Preclinical studies also showed that, in response to NAc DBS, more neuronal precursors were found in the dentate gyrus of the hippocampus, hinting at enhanced adult neurogenesis (Schmuckermair et al. 2013). NAc DBS may also alter the morphology of the PFC, with increases in apical and basilar dendrite length (Falowski et al. 2011).
Overall, despite few discrepant findings, studies so far suggest that MDD is associated with structural and functional changes in brain regions, especially those described above, and alterations in their mutual connectivity (for summary, see Fig. 1). These changes are the likely result of alterations in neuroplastic processes that regulate synaptic connectivity and maintain neuronal integrity. Understanding the nature and molecular underpinnings—including neurotrophic signaling—of these processes may provide new clues for succesful pharmacological interventions. The next sections explore alterations of neuroplasticity in the context of MDD.

Summary of the neuroanatomical changes observed in MDD patients. In the MDD brain, the dotted lines correspond to the brain volume in a healthy patient. In the HIP and the dlPFC, the alternative blue and red colors show the discrepancies reported in the different studies regarding their activities in MDD patients. The thinner arrows and lines show reduced connectivity between the regions. dlPFC, dorsolateral prefrontal cortex; vm/vlPFC, ventromedial/ventrolateral prefrontal cortex; HIP, hippocampus; AMY, amygdala; VTA, ventral tegmental area. See text for more details. This illustration was taken from “Servier medical art” (http://www.servier.fr/servier-medical-art)
Neuroplasticity changes in MDD and the effects of antidepressant therapies
Neuroplasticity: definition
Neuroplasticity can be defined as the ability of the nervous system to respond to intrinsic or extrinsic stimuli by reorganizing its structure, function, and connections (Cramer et al. 2011). It includes different mechanisms as described in an excellent review by Castren and Hen (2013). One of them is neurogenesis, the formation of newborn neurons in proliferative areas. The regions identified so far in the rodent adult brain are the subventricular zone (SVZ) and the subgranular zone (SGZ) of the DG in the hippocampus. Another mechanism of plasticity is the modification of mature neuronal morphology, involving axonal and dendritic arborization and pruning, an increase in spine density, and synaptogenesis. At a functional level, long-term potentiation (LTP) is the main mechanism mediating plasticity. The transcriptional regulation of genes involved in neuroplasticity by epigenetic mechanisms also contributes to synaptic plasticity. Altogether, these processes mediated the dynamic and adaptive changes in synaptic strength (Castren and Hen 2013).
Neuroplasticity changes in MDD
While neuroplasticity in rodents has been well documented during the last decades, the study of neuroplasticity in the human brain largely remains indirect, mostly because of methodological limitations as well as ethical constraints.
However, some alternative methods such as the assessment of the hippocampal DG volume by MRI, magnetic resonance spectroscopy (Bergmann et al. 2015; Ho et al. 2013), and more recently the use of 14C in genomic DNA labeling (Spalding et al. 2013) have provided some valuable information on human neuroplasticity. Most of the post-mortem studies have now evidenced that in adult humans, new neurons continued to be generated with a modest decline during aging (Eriksson et al. 1998; Reif et al. 2006; Spalding et al. 2013). Although recently questioned by Sorrells et al. (2018) who suggested that human neuroplasticity could differ from other species, it has been nevertheless confirmed that in human DG, about 700 new neurons were generated per day whatever the age (Boldrini et al. 2018).
Beside neurogenesis, it is also rather well-established that LTP can be induced in the human CNS with similar molecular mechanisms than those observed in rodent models (Bliss and Cooke 2011). Human LTP has first been demonstrated in isolated cortical tissue obtained from patients undergoing surgery, and recent studies have shown that LTP-like phenomena can be obtained in the human cortex as well by using repetitive presentations of sensory stimuli while recording event-related potentials from the scalp (Clapp et al. 2005a, b).
In MDD, neuroimaging and post-mortem studies in humans indicate that structural changes are often observed in the course of this pathology. Structural MRI studies have revealed reduced hippocampal volume in individuals during a depressive episode in comparison to patients in remission (Kempton et al. 2011), while increased hippocampal dendritic atrophy and cell death as well as reduced LTP and BDNF expression have also been reported (Miller and Hen 2015; Pittenger 2013). While MRI studies were rather consistent with the observation of a reduced DG size in patients with depression or anxiety disorders (Boldrini et al. 2013; Bremner et al. 2000; Cole et al. 2011; Frodl et al. 2007; Huang et al. 2013), post-mortem studies in depressed patients showed important disparities regarding neurogenesis, showing either no difference (Reif et al. 2006) or a decrease in the number of DG progenitor cells (Lucassen et al. 2010). Although these findings make it tempting to speculate on reduced levels of neurogenesis in MDD, further investigations making use of more specific techniques are needed to better understand the dynamics of adult neurogenesis in MDD. However, other attempts to measure human brain plasticity in the course of MDD have demonstrated functional alterations such as those observed at LTP level. For example, visual-evoked potential amplitudes in the visual pathway were, compared to matched control subjects, decreased in patients with depression (Normann et al. 2007; Bubl et al. 2015) and also in bipolar disorder patients (Elvsashagen et al. 2012).
In preclinical depression-like models as well, consistent data have reported a decrease in the proliferation and survival of hippocampal neurons when the HPA axis was dysregulated. Hence, using proliferation and survival cell markers such as BrdU, Ki-67, or DCX, impaired neurogenesis was observed in rodent models of depressive-like behavior. These models included transgenic mice (Paizanis et al. 2010), corticosterone-induced mouse model of depression/anxiety (Zhang et al. 2014b), or rats subjected to chronic mild stress (Morais et al. 2017 and see Anacker and Hen 2017 for a review). Beside neurogenesis, numerous studies have also reported that synaptic plasticity was also greatly impaired in stress models of depression (Pittenger 2013). Severe stress has also been shown to inhibit LTP (Kim and Diamond 2002) and enhance LTD (Xu et al. 1997) in the hippocampus and in prefrontal pyramidal cells (Goldwater et al. 2009).
Altogether, these data strongly suggest synaptic plasticity is strongly affected in MDD, at both structural and functional levels, and that these alterations are similar to those evidenced in rodent studies.
Effects of antidepressant therapies on plasticity
Electroconvulsive therapy
Recent clinical studies showed that electroconvulsive therapy (ECT) promoted structural plasticity including increased volume and morphometric changes in the hippocampus and the amygdala along with improved clinical responses, especially in patients with a smaller hippocampal volume (Joshi et al. 2016; Nordanskog et al. 2010; Tendulkar et al. 2013). ECT may also reduce cortical excitability and, thereby reverse increases in the excitability of cerebral cortex, during treatment-resistant depression (Sackeim et al. 1983). In addition, low-frequency trains of transcranial magnetic stimulation (rTMS) applied on several regions of the brain to induce LTP- and LTD-like changes in neuronal activity produced identical effects to those achieved with ECT in depressive patients (Fitzgerald and Daskalakis 2011).
Similar results have been demonstrated in rodent models of depression (Nakamura et al. 2013; Schloesser et al. 2015) and non-human primates (Perera et al. 2007).
Exercise
Exercise has also shown beneficial effects on plasticity. Although it has been proven that exercise in MDD patients reduced depressive symptoms (Herring et al. 2012; Ota and Duman 2013; Schuch et al. 2016), neuroplasticity per se has not yet been monitored in patients in these conditions, due to the limitations mentioned in section “Neuroplasticity changes in MDD.” However, a meta-analysis has suggested that acute aerobic, but not strength exercise, increases basal peripheral BDNF concentrations, although this effect was only transient (Knaepen et al. 2010). A recent study showed an increase in synchronous neuronal responses during a task that requires an upregulation of cognitive control when exercise was combined with meditation (Alderman et al. 2016). In preclinical studies, swimming showed antidepressant-like effects on anhedonia in stressed rodents along with a normalization of a stress-induced BDNF mRNA expression decrease (Jiang et al. 2014). A 21-day exercise regimen in rats transiently increased LTP in the DG of the hippocampus (Radahmadi et al. 2016), although recent data suggested that, if motor activity could exert positive effects on cognitive processes, it was under very controlled conditions (D’Arcangelo et al. 2017) and that acute swim stress could also led to LTD (Tabassum and Frey 2013).
Antidepressant treatments
Few studies have directly addressed the effects of antidepressant therapy on neuroplasticity in the human brain. Imaging studies have provided data showing that, e.g., in MDD patients taking medication for 3 years, the left hippocampus increased in volume compared to the beginning of the study (Frodl et al. 2008). Interestingly, the volume of the left hippocampus has also been shown to enlarge under lithium treatment in elderly bipolar patients, probably through a neuroprotective effect (Zung et al. 2016).
Regarding neurogenesis per se, the analysis of post-mortem brains of MDD patients treated with nortriptyline and clomipramine showed an increase of neural progenitor cells and dividing cells in the DG, as compared to healthy controls (Boldrini et al. 2009). In addition, the same group reported that both the fewer mature granule neurons and the smaller DG and granule cell layer volume found in post-mortem brain tissue of depressive patients were reversed by antidepressant treatment (Boldrini et al. 2013). Interestingly, a meta-analysis concluded that depressed patients with a decreased hippocampal volume showed lower response/remission rates after antidepressant treatment (Colle et al. 2016).
In a rodent model of depression, both reduced hippocampal proliferation and increased cell death were reversed by chronic administration of antidepressants (Pilar-Cuellar et al. 2013). Adult hippocampal neurogenesis was proposed to be required for the therapeutic action of antidepressants (David et al. 2009; Djavadian 2004; Klempin et al. 2013; Sahay and Hen 2007; Santarelli et al. 2003). Accordingly, chronic treatment with a SSRI such as fluoxetine increased hippocampal neurogenesis through the generation of newborn cells in the DG (Boldrini et al. 2009; Encinas et al. 2006; Malberg et al. 2000; Santarelli et al. 2003; Surget et al. 2011). In addition, serotonin and noradrenaline reuptake inhibitor (SNRI) antidepressants, like SSRIs, also modulate neurogenesis and plasticity. Neurogenesis in the DG of the hippocampus was increased following chronic venlafaxine administration to rats (Mostany et al. 2008). Similarly, chronic venlafaxine treatment proved to be efficient in preventing the deleterious effects of restraint stress on hippocampal neurogenesis and BDNF protein expression (Xu et al. 2006). Likewise, tricyclic antidepressants (TCAs) have also been shown to modulate hippocampal neurogenesis. Clomipramine was able to counteract the stress-induced inhibition of proliferation in the hippocampus (Liu et al. 2008). Chronic imipramine and desipramine treatment increased cell proliferation in the SGZ (Pechnick et al. 2011; Santarelli et al. 2003; Schiavon et al. 2010).
In addition to their effects on neurogenesis, evidence has also been generated that antidepressants can regulate other types of plasticity. Treatments with classical antidepressants are indeed able to modulate LTP within the hippocampus (Wang et al. 2008) and chronic fluoxetine administration has also been demonstrated to increase synaptic plasticity in naive rats (Stewart and Reid 2000).
Overall, these data showed that antidepressants have a rather beneficial effect on neuroplasticity. However, they still show lack of efficacy in some depressed patients. Indeed, MDD is a complex disorder of which the etiology remains unclear and that cannot be simplified by neuroplasticity dysfunctions as the only targetable factor.
Neurotrophins and other growth factors
Growth factors have been proven to be strongly involved in regulating plasticity by impacting upon almost all of the neuroplasticity processes mentioned in section “Neuroplasticity changes in MDD and the effects of antidepressant therapies,” including neuronal survival, differentiation, and proliferation (Crutcher 1986; Lu et al. 2014). Accordingly, this section focuses on the main studied growth factors that have been described thus far, from their general functions to their role in MDD. This is summarized in Table 1 and Fig. 2.

Neurotrophin level changes observed in MDD patients with or without antidepressant treatment. Observed changes in neurotrophin levels, in MDD patients whether or not under antidepressant treatment, represented in the different brain areas involved in depression, and in blood (IGF-1 only). The blue color corresponds to a low level of neurotrophin while the red color shows a high level. Arrows with the single strand represent RNA expression changes and arrows with circles protein level changes. PFC, prefrontal cortex; Acc, anterior cingulate cortex; c.brainstem, caudal brainstem; CSF, cerebrospinal fluid; HIP, hippocampus. See text for more details. This illustration was taken from “Servier medical art” (http://www.servier.fr/servier-medical-art)
Brain-derived neurotrophic factor or BDNF
General function
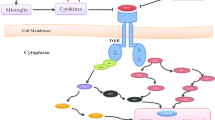
BDNF is a neurotrophin involved in the growth, differentiation, and survival of neurons and has also been shown to represent an important factor in the regulation of neurogenesis and synaptic plasticity (Lu et al. 2014). It exerts its neurotrophic effects by activating the tropomyosin-related kinase receptor B (TrkB) (Chao and Hempstead 1995). It also binds, albeit with a lower affinity, to the p75NTR receptor, which is generally known to promote proteolysis and apoptosis (Boulle et al. 2012). BDNF is abundantly expressed in the mammalian brain, with the highest concentrations found in the hippocampus and cortex (Ernfors et al. 1990).
BDNF and neuroplasticity
Several in vitro studies have been conducted in order to unravel the effects of BDNF on plasticity. Indeed, when PC12 cells transfected with TrkB were stimulated with BDNF for 48 h, neurite outgrowth was increased compared to the non-treated cells (Cazorla et al. 2011). Interestingly, in growth medium B27-deprived primary hippocampal cells, BDNF stimulation was able to promote dendritic outgrowth and spine formation (Park et al. 2016) and this neuroplastic effect is probably achieved through intracellular signaling cascades (Cavanaugh et al. 2001; Obrietan et al. 2002). As such, BNDF has been shown to increase the activity of the mitogen-activated protein kinase (MAPK) cascade promoting survival in neural cell cultures (Hetman et al. 2002).
In vivo evidences also support the critical role of BDNF in plasticity. In particular, mutation studies have demonstrated the role of this neurotrophin in structural and synaptic plasticity. Total BDNF deficiency is lethal and most of the mice lacking BDNF die during the second postnatal week (Ernfors et al. 1994). However, heterozygous BDNF knockout mice survive into adulthood and the use of these mice evidenced that BDNF was required for several forms of LTP (Aarse et al. 2016). This was in agreement with data that showed that BDNF infusion in the rat hippocampus induced LTP and triggered synaptic strengthening (Bramham 2007; Ying et al. 2002). At morphological level, these mice display a specific hippocampal volume reduction (Lee et al. 2002; Magarinos et al. 2011) similarly to what was found in heterozygous TrkB mice (von Bohlen und Halbach et al. 2003) but in contrast to p75NTR-deficient mice (Dokter et al. 2015), suggesting a link between hippocampal volume and BDNF-mediated TrkB signaling (von Bohlen und Halbach et al. 2003; von Bohlen Und Halbach and von Bohlen Und Halbach 2018). In addition, in the hippocampus, BDNF increases the total length, but not the branching, of apical dendrites within the CA1 stratum radiatum, without affecting basal dendrites in the stratum oriens (Alonso et al. 2004). However, in mutant mice in which bdnf excision mediated by Cre recombinase led to an almost total disappearance of BDNF in the brain, the volume of the hippocampus was mostly unchanged except for small changes in dendritic branches in restricted segments and signs of a modest delay in spine maturation (Rauskolb et al. 2010). Although these data and other (Baquet et al. 2004) indicate a temperate effect of BDNF in the maintenance of the cellular architecture of the adult brain, most of the studies evidenced its central role in neuronal plasticity.
BDNF in MDD
BDNF in MDD has been largely documented. However, many of the studies addressed peripheral (blood) BDNF concentration, with the assumption that blood BDNF could be a biomarker reflecting that of brain tissue (see, e.g., Klein et al. 2011). However, there is no evidence that serum BDNF is related to brain BDNF and neuroplasticity. The origin of serum BDNF has been now clearly documented and was demonstrated to come from the progenitors of platelets (Chacón-Fernández et al. 2016). Nevertheless, lowered serum concentrations of BDNF have often been associated with MDD (Karege et al. 2002, 2005; Sen et al. 2008, Molendijk et al. 2011). A thorough meta-analysis by Molendijk et al. showed that, despite demonstrable study heterogeneity, serum BDNF levels are overall lower in depressed patients (Molendijk et al. 2014). These findings were recently confirmed by two other meta-analytic efforts (Polyakova et al. 2015; Kishi et al. 2017). However, the significant variation between available assays (Polacchini et al. 2015) prevents the use of serum BDNF (and other neurotrophins) levels as a reliable biomarker for mood disorders.
Nevertheless, within the CNS, a reduction in BDNF and TrkB expression in the hippocampus and PFC has been reported in post-mortem brain tissue of suicide victims (Dwivedi et al. 2003; Pandey et al. 2008). One of the most common functional single nucleotide polymorphism (SNP) in the Bdnf gene is rs6265, causing a valine to methione substitution at codon 66 (val66met). This polymorphism affects the activity-dependent secretion of BDNF (Egan et al. 2003). Although at first, a negative association between the val66met polymorphism and hippocampal volume has been reported (Frodl et al. 2007), this observation was not supported by a meta-analysis that did not evidence any association between hippocampal volume and the val66met genotype in neuropsychiatric patients, perhaps because of the too many different disorders analyzed in this study (Harrisberger et al. 2015). Yet again, several other meta-analyses did confirm the association between the val66met polymorphism and an increased susceptibility to mood disorders (Hosang et al. 2014; Li et al. 2016; Yan et al. 2014; Zou et al. 2010). Finally, a recent study showed that subjects with the Met allele of the Bdnf gene had an increased risk for depression (Youssef et al. 2018). In this study, post-mortem analyses showed that depressed patients also have lower BDNF levels in the anterior cingulate cortex (ACC) and the caudal brainstem compared to non-depressed subjects, providing further evidence implicating low brain BDNF and the BDNF Met allele in major depression risk (Youssef et al. 2018).
BDNF and antidepressant treatments
The effect of antidepressants on BDNF expression and the role of BDNF in the treatment of MDD have been widely studied (see Castren and Kojima 2017 for a review). Interestingly, it has been shown that BDNF expression was increased in post-mortem brains of depressed patients treated with antidepressant drugs as compared to non-treated patients (Chen et al. 2001; Dunham et al. 2009).
The involvement of BDNF in the efficacy of antidepressant treatments has been demonstrated mainly in rodent models. Lee et al. developed a val66met mouse analogue, i.e., BDNFmet/met mice (Chen et al. 2006), and subsequent experiments showed that this genetic variant decreased fluoxetine efficacy through impaired synaptic plasticity defined by a reduction of theta-burst stimulation-induced LTP as well as impairments in the survival of newborn cells in the hippocampus (Bath et al. 2012; Ninan et al. 2010) and a deficit in synaptic transmission in the PFC (Pattwell et al. 2012). In addition, BDNF expression in the brain of rats was upregulated after chronic antidepressant drug exposure and ECT (Altar et al. 2003; Balu et al. 2008; Jacobsen and Mork 2004; Nibuya et al. 1995). Interestingly, some studies have shown that antidepressant-like effects observed in mouse models of depression after chronic administration of antidepressants were reversed by TrkB antagonist injection (Boulle et al. 2016; Ma et al. 2016; Yasuda et al. 2014) and that inhibition of TrkB signaling blocked the effects of antidepressants (Saarelainen et al. 2003). Interestingly, in addition to its effects on BDNF expression, autophosphorylation of TrkB by antidepressants has also been reported by the group of Castren (Rantamaki et al. 2007; Saarelainen et al. 2003) suggesting that TrkB can be transactivated independently from neurotrophins, as demonstrated in the case of glucocorticoid receptors (Jeanneteau et al. 2008). Indeed, a study by Rantamaki et al. (2011) showed a rapid action of imipramine on TrkB phosphorylation in either presence or absence of BDNF, suggesting that antidepressants do not require BDNF to activate TrkB, an effect that was independent of 5-HTT. These results highlight the potential direct effect of the BDNF-TrkB signaling pathway in the mechanism of action of antidepressants (Autry and Monteggia 2012) and evidenced that TrkB is a valuable target to treat depression.
Brain region-specific BDNF effects
In rodents, direct infusion of BDNF in the hippocampus (Deltheil et al. 2008, 2009; Shirayama et al. 2002; Sirianni et al. 2010) and midbrain (Siuciak et al. 1997) showed antidepressant-like effects. In contrast, an opposite, pro-depressive effect was reported after infusion of BDNF in the VTA or the NAc (Eisch et al. 2003). The same disparity was present using region-specific knockdown of BDNF expression. Impairment of BDNF signaling in the DG of the hippocampus (Taliaz et al. 2010) elicited pro-depressive behavior, whereas knockdown of BDNF in the NAc had an antidepressive effect (Berton et al. 2006). Interestingly, conditional knockout in the forebrain resulted in an increase in depressive-like behavior in female but not male mice (Monteggia et al. 2007) and, furthermore, decreased the efficacy of the antidepressant desipramine (Monteggia et al. 2004). This conditional knockout of BDNF in the forebrain displayed the same sex-specific incongruity in stress-induced depressive-like behavior (Autry et al. 2009). A different study using adeno-associated viral-mediated knockout of BDNF in the DG and CA1-region of the hippocampus showed that a loss of BDNF function in the hippocampus attenuated antidepressant drug treatment efficacy (Adachi et al. 2008).
Taken together, these data suggest that BDNF could be considered as a key therapeutic molecule against depression (Allen et al. 2015).
Fibroblast growth factor or FGF
General function
The fibroblast growth factor (FGF) family has been described as a major player in proliferation and maturation of neurons in the SVZ and SGZ of the hippocampal DG (Woodbury and Ikezu 2014). The FGF family is composed of 18 ligands and 4 subtypes of receptors. FGF1 is expressed mostly in neurons while FGF2 is expressed in both neurons and glial cells. FGF1 and FGF2 are the most studied ligands of this family and have been shown to be dysregulated in mood disorders (Turner et al. 2006; Turner et al. 2012). They can bind all four receptor subtypes in order to activate the phospholipase C-γ (PLC-γ1 MAPK and AKT pathways (Turner et al. 2006; Turner et al. 2012). In addition, FGF1 and FGF2 were evidenced to play a critical role in the regulation of synaptic plasticity (Di Liberto et al. 2014).
FGF in neuroplasticity
Intracerebroventricular (ICV) injection of FGF2 induced neurogenesis in both the SVZ and SGZ (Jin et al. 2003; Mudo et al. 2009; Rai et al. 2007). Mice with a complete loss-of-function FGF2 allele showed a significant decrease in newly generated neurons but no reduction in proliferating cells (Werner et al. 2011). The additional increase in cell death in the hippocampus indicated a faulty neurogenesis following FGF2 knockout (Werner et al. 2011). Conditional knockout experiments with FGF receptor 1 (FGFR1)-null mice show defective LTP and neurogenesis (Zhao et al. 2007), suggesting that the FGF2-FGFR1 interaction might represent an important mediator of neurogenesis.
FGF in MDD
Post-mortem brain analysis in humans revealed a lower expression of FGF1 and FGF2 in the dorsolateral PFC and the ACC of patients with MDD (Evans et al. 2004). In addition, FGF2 was decreased in the hippocampus of depressed patients, whereas FGFR1 was increased (Gaughran et al. 2006). In rodents, Turner et al. reported reduced mRNA expression of FGF2 and its main receptor FGFR1 in the CA1, CA2, CA3, and DG following social defeat stress, a well-established model of depression (Turner et al. 2008a). Injection of FGF2 was also shown to reduce depressive-like behavior in rats (Turner et al. 2008b). A more recent study also reported that increased cell proliferation in the PFC following FGF2 infusions might also be involved in the antidepressant actions of FGF2 (Elsayed et al. 2012).
Vascular endothelial growth factor or VEGF
General function
Vascular endothelial growth factor (VEGF) is primarily known for its induction of angiogenesis and modulation of vascular permeability during embryogenesis and growth, as well as pathological events such as in tumorigenesis. It can bind to different receptors: receptor tyrosine kinases (VEGFR) 1 and 2 with a higher affinity for VEGFR1. In addition, mounting evidence suggests that VEGF can be considered as a potent neurotrophic factor, inducing neurogenesis, neuronal survival and proliferation, glia survival, and glia migration (Carmeliet and Ruiz de Almodovar 2013).
VEGF in neuroplasticity
Interestingly, experimental studies showed that VEGF displays robust neuroprotective effects in cell models of ischemia and hypoxia (Jin et al. 2000) as well as a positive effect on neuronal growth, maturation, and proliferation under normoxic conditions (Khaibullina et al. 2004; Rosenstein et al. 2003; Silverman et al. 1999; Sondell et al. 1999; Zhu et al. 2003). A role in the development of dendrites and axons has also been described for VEGF (Khaibullina et al. 2004; Licht et al. 2010; Rosenstein et al. 2003; Sondell et al. 1999). Moreover, ICV administration of VEGF increased neuroprotection and neurogenesis in the adult rat brain after ischemia (Sun et al. 2003). More specifically, ICV administration of VEGF increased neurogenesis in both the SVZ and the SGZ of the DG with enhanced proliferation of neurons, astroglia, and endothelial cells (Jin et al. 2002), while VEGF-B knockout mice showed impaired neurogenesis (Sun et al. 2006). Hence, the overexpression of VEGF in the hippocampus using an adeno-associated viral vector in rats resulted in increased neurogenesis and was associated with improved learning and memory (Cao et al. 2004; During and Cao 2006). Interestingly, VEGF can promote neurogenesis by stimulating endothelial cells in order to release other neurotrophic factors (Yamada 2016). In addition, ependymal cells can synthesize VEGF leading to a stimulation of the VEGFR2 and inducing proliferation of neuronal precursors and enhanced formation of new neurons in the hippocampus (Nowacka and Obuchowicz 2012).
VEGF in MDD
A multitude of studies have investigated the plasma concentration of VEGF in MDD patients, but the data and interpretation remain conflicting possibly due to the differences in study designs (see review by Clark-Raymond and Halaris 2013) In addition, as mentioned above, there is currently no evidence that blood levels of neurotrophins reflect those of the brain, making causal inferences of the role of VEGF in MDD based on blood concentrations rather speculative. Nevertheless, angiogenesis seemed to be mediating the SSRI-induced upregulation of neurogenesis through VEGF (Yamada 2016). Indeed, higher hippocampal angiogenesis and neurogenesis have been found in SSRI-treated MDD patients when compared with untreated or healthy individuals (Boldrini et al. 2012). Chronic stress in rats decreased the expression of VEGF and its receptor in the hippocampus (Heine et al. 2005). Furthermore, VEGF is required for the proliferation of neural stem-like cells in the hippocampus following ECT treatment (Elfving and Wegener 2012; Segi-Nishida et al. 2008). In a similar manner, VEGF also seems to be required for the behavioral action of various antidepressant drugs in rodent models of depression (Greene et al. 2009; Sun et al. 2012; Warner-Schmidt and Duman 2007; Warner-Schmidt and Duman 2008).
Glial cell line-derived neurotrophic factor or GDNF
General function
Glial cell line-derived neurotrophic factor (GDNF) was first discovered in a glial cell line but is expressed in many brain regions. It is a member of the transforming growth factor β (TGF-β) superfamily and is important for neuronal survival especially for dopaminergic and serotonergic neurons. It binds to the GDNF-family receptors α1 (GFRα1) activating tyrosine kinase signaling (Sharma et al. 2016).
GDNF in neuroplasticity
Experimental studies in animal models evidenced a neuroprotective role of GDNF, and ICV infusion of GDNF increased progenitor cell proliferation in the DG (Dempsey et al. 2003) and SVZ (Kobayashi et al. 2006). Similarly, infusion of GDNF in the striatum of rats increased progenitor cell proliferation in the hippocampus and substantia nigra (Chen et al. 2005). Moreover, GDNF induced differentiation of DG-derived neural precursors into astrocytes in vitro (Boku et al. 2013). Further, the use of an adeno-associated viral vector that induced overexpression of GDNF in the rat cortex provided neuroprotection against ischemia-induced injury (Tsai et al. 2000).
GDNF in MDD
Only a few clinical studies examined the role of GDNF in MDD, and contrasting findings between brain and blood expression are reported (Sharma et al. 2016). A recent post-mortem brain analysis showed an increase of (GDNF) expression in the parietal cortex of MDD patients (Michel et al. 2008). To our knowledge, this is the only region that has been studied in humans with respect to GDNF, although several studies have investigated the serum level of GDNF in MDD (Diniz et al. 2012a, b; Pallavi et al. 2013; Zhang et al. 2008) and under antidepressant treatment (Zhang et al. 2008).
In addition, antidepressant exposure increased GDNF release in a rat C6 glioblastoma cell line (Hisaoka et al. 2001), whereas lithium treatment in rats resulted in increased GDNF concentrations in the PFC and occipital cortex but a decrease in the hippocampus (Angelucci et al. 2003b). In mice exposed to chronic ultra-mild stress, an increase in Gdnf mRNA expression was observed in the hippocampus. This modification was partly reversed by chronic administration of the antidepressant agomelatine (Boulle et al. 2014). Furthermore, work on the same mouse model showed that GDNF, with other neurotrophins, could be involved in mediating the behavioral response to antidepressants (Uchida et al. 2011). Thus far, the exact involvement of GDNF in the etiology of depression is not fully understood, but its neuroprotective capacity might make it an interesting future target for antidepressant treatment.
Insulin-like growth factor or IGF-1
General function
Insulin-like growth factor (IGF-1) and its receptor IGF-1R are found in many tissues including the brain (Aberg et al. 2006). It influences growth and differentiation processes (Frysak et al. 2015). IGF-1 has been designated as a potential therapeutic target for neurodegenerative diseases such as MDD (Szczesny et al. 2013).
IGF-1 in neuroplasticity
IGF-1 induced differentiation of neuronal precursors (Anderson et al. 2002; Arsenijevic and Weiss 1998) and proved to be neuroprotective in cerebellar granule neurons in vitro (D’Mello et al. 1993). IGF-1 knockout mice showed a decrease in total brain size and SGZ volume, further supporting the importance of IGF-1 in neurodevelopment (Beck et al. 1995). Developmental research in mice has revealed the importance of IGF-1 in hippocampal neurogenesis and synaptogenesis (O’Kusky et al. 2000). Furthermore, ICV infusion of IGF-1 ameliorated the age-related decline in hippocampal neurogenesis (Lichtenwalner et al. 2001), while peripheral administration of this growth factor could induce hippocampal neurogenesis in rats (Åberg et al. 2000).
IGF-1 in MDD
Although clinical studies showed somewhat inconsistent findings, they however mainly revealed higher IGF-1 levels in the serum of depressed patients, which declined during effective antidepressant treatment (Bot et al. 2016; Kopczak et al. 2015; Szczesny et al. 2013). In contrast, IGF-1 was high in cerebrospinal fluid of antidepressant-treated patients (Schilling et al. 2011). These data suggest differential actions of IGF-1 in the periphery and the brain.
However, preclinical studies using conditional knockout mice showed that a decrease in either systemic or hippocampal IGF-1 levels could increase the susceptibility to develop depressive-like behavior (Mitschelen et al. 2011). In addition, in corticosterone-treated rats, a decrease of IGF-1 in both the hippocampus and serum has been reported. However, physical exercise using continuous running was not able to reverse this diminution while it did prevent depressive-like behavior (Yau et al. 2014). Interestingly, since IGF-1 can readily pass the blood-brain barrier (Pan and Kastin 2000) in contrast to BDNF, its effects on the brain can be achieved by direct injection into the blood. In line with this notion, when administrated chronically, a peripheral injection of IGF-1 in a mouse model of depression was shown to induce antidepressant-like behaviors, comparable to commonly used antidepressants (Duman et al. 2009). In addition, an increase of peripheral IGF-1 by direct injection of IGF-1 or inhibition of IGF-1 binding protein displayed anxiolytic and antidepressant effects in rodents (Duman et al. 2009; Hoshaw et al. 2005; Malberg et al. 2007; Park et al. 2011), which might be attributed, at least in part, to increased serotonin levels in the brain (Hoshaw et al. 2008). Of note, intranasal administration has been proposed in order to provide a shorter path for IGF-1 to enter the brain (Paslakis et al. 2012), avoiding unwanted effects of IGF-1 in peripheral tissues.
Nerve growth factor or NGF
General function
Nerve growth factor (NGF) is a growth factor first described as a neurite outgrowth factor (Olson 1967). Later, NGF proved to be involved in neuronal repair and survival (Kromer 1987; Shigeno et al. 1991; Sofroniew et al. 2001; Zhao et al. 2004). NGF has been implicated in proliferation and differentiation of neuronal stem cells (Cattaneo and McKay 1990) and more recently in neurogenesis in the striatum (Frielingsdorf et al. 2007; Zhu et al. 2011) and in regulating hippocampal plasticity (Conner et al. 2009).
NGF in MDD
To date, only a few studies have investigated the role of this trophic factor in MDD and only at peripheral level.
In the Flinders Sensitive Line (FSL) rat model of depression, ECT was found to increase NGF levels in the hippocampus (Angelucci et al. 2003a). In addition, subcutaneous NGF injections show antidepressant effects (Overstreet et al. 2010).
This—non-exhaustive—review of the literature further underlines the potential effects of the different members of the neurotrophin family in the alteration of neuroplasticity often observed in depression-like disorders, as summarized in Fig. 3.

Neurotrophins increase neuroplasticity through the activation of three main signaling pathways. Neurotrophins bind to their receptors in order to promote three main signaling pathways: the MAPK/ERK, the PI3-K, and the PLCγ signaling cascades. Once activated, they stimulate neuroplasticity, especially synaptic plasticity, neurotransmission and neuronal survival, growth, and differentiation. An increase of neuroplasticity is likely to induce antidepressant effects. BDNF, brain-derived neurotrophic factor; TrkB, tropomyosin-related kinase receptor B; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; GDNF, glial cell line-derived neurotrophic factor; GFRα1, GDNF-family receptor-α; IGF-1, insulin-like growth factor 1; IGF-1R, insulin-like growth factor 1 receptor; NGF, nerve growth factor; TrkA, tropomyosin-related kinase receptor A
Future directions: targeting growth factor signaling with synthetic small molecules, focus on the BDNF/TrkB pathway
Over the last decades, convergent studies suggested that the BDNF/TrkB signaling pathway was a main actor in the development and course of mood disorders, in particular depression, as well as in the action of currently available antidepressants. Because of the poor efficacy of antidepressants and their delayed therapeutic effect, there is a need to find novel and more efficacious compounds. The use of BDNF itself turned out to be rather difficult because of its unfavorable pharmacokinetic profile. Indeed, peripheral administered BDNF hardly crosses the blood-brain barrier and has a very short half-life. Interestingly, the recent data suggesting that antidepressant could also directly transactivated TrkB (Rantamaki et al. 2011) point to this receptor as a main molecular target for treating depression. Thus, attempts to develop new molecules that directly target TrkB signaling have been undertaken as proposed by Tsai (2007).
TrkB agonists
7,8-Dihydroxyflavone
The most extensive examined TrkB agonist so far is 7,8-dihydroxyflavone (DHF). It was first described as an antioxidant and later identified as a TrkB agonist after a screening of flavone derivatives (Choi et al. 2010, Liu et al. 2010). Many experiments have been performed and have revealed promising results regarding antidepressant-like properties. Recent studies show that DHF has antidepressant effects in mice displaying LPS-induced depressive-like behavior in the tail suspension test (TST) and the forced swim test (FST) (Zhang et al. 2014a). In the learned helplessness model of depression in rats, a single bilateral infusion of DHF in various subregions of the hippocampus and in the infralimbic medial PFC induced antidepressant effects (Shirayama et al. 2015).
Furthermore, DHF has been shown to normalize dendritic spine structure in an LPS model of depression (Zhang et al. 2014a) and to increase neurogenesis in the hippocampus of naïve mice (Liu et al. 2010). Besides its effect on depressive-like behavior, further studies have tested DHF on cognition. Indeed, restoration of memory deficits in 5XFAD and APP/PS1 mouse models of Alzheimer’s disease (AD) have been shown after both acute and chronic administration of DHF (Bollen et al. 2013; Devi and Ohno 2012; Zhang et al. 2014c), while it did not seem to exert this effect when injected chronically in APP23PS45 transgenic mice (Zhou et al. 2015). Because memory impairments were observed in MDD patients (Dere et al. 2010; Millan et al. 2012), the recovery of memory deficits in animal models of AD after administration of DHF could be expected. Liu et al. optimized the molecule by synthesizing various bioisosteric derivatives and showed an even more pronounced antidepressant effect in both the FST and TST requiring a lower dose for a more potent effect (Liu et al. 2010; Liu et al. 2012).
Other promising agonists
Gedunin is a tetranortriterpenoid isolated from the Indian neem tree. Its derivative, deoxygedunin, seems to be a promising selective TrkB agonist, showing protective effects against apoptosis, both in vitro, in hippocampal neurons incubated with a toxic dose of DMSO, and in vivo, in mice that received kainic acid (Jang et al. 2010). Antidepressant effects of deoxygedunin have also been shown after subchronic treatment, displaying reduced immobility in the FST (Jang et al. 2010).
LM22A-4 was designed to mimic the loop II domain of BDNF and proven to be a partial agonist. In vitro work showed that LM22A-4 has neuroprotective properties (Massa et al. 2010). It was first shown as an effective molecule to reverse respiratory abnormalities (Kron et al. 2014; Schmid et al. 2012) and to improve functional recovery after stroke (Han et al. 2012). Furthermore, it was also shown to reverse alcohol drinking in mice.
TDP6 and 29D7 have been shown to represent other promising BDNF mimetic molecules able to promote oligodendrocyte-mediated myelination in vitro (Wong et al. 2014) and to enhance neuronal survival and neurotic outgrowth in vitro and in vivo as well as to provide long-lasting neuroprotection against neonatal hypoxic-ischemic brain injury (Kim et al. 2014; Qian et al. 2006).
TAM-163, an antibody targeting TrkB, has been shown to be a partial TrkB agonist but only tested as an agonist agent for body weight regulatory disorders (Perreault et al. 2013).
Another way to activate Trk receptors is to potentiate their neurotrophic-mediated activation. BMS355349 has been described as a selective potentiator of NT-3 mediated TrkA and TrkB receptor activity and has proven to induce neurogenesis (Chen et al. 2009; Lewis et al. 2006).
To sum up, further investigations are required regarding the action of TrkB agonists in mood disorders, but their observed neuroprotective effects so far are promising.
TrkB antagonists
Knowing that, in a rat model of depression, increases in BDNF in the VTA-NAc (Shirayama et al. 2015; Zhang et al. 2015b) induced pro-depressive-like effects (Eisch et al. 2003), the use of partial antagonists as a treatment for diseases related to nervous system dysregulations has been considered. To our knowledge, only ANA-12 and cyclotraxin B, both described as selective antagonists for TrkB, have been tested so far.
ANA-12 is a selective partial agonist that was first developed by Carzola et al. in 2011 using a structure-based in silico screening (Cazorla et al. 2011). In their study, the authors identified a low-molecular weight antagonist of TrkB that could induce anxiolytic and antidepressant properties. While ANA-12 was mainly used as a tool to block the BDNF/TrkB complex in order to better understand the mechanism of action of TrkB signaling (Montalbano et al. 2013), two recent studies tried to use it as a therapeutic agent. When infused bilaterally in the NAc, ANA-12 showed antidepressant properties in the TST and FST in LPS-treated mice (Zhang et al. 2014a). Moreover, in mice that exhibit a reduced social interaction, ANA-12 infusion into the NAc has been shown to completely block social avoidance (Walsh et al. 2014). When administrated intraperitoneally and alone, ANA-12 could also decrease the immobility time in the TST and the FST in the same LPS-treated mice.
Cyclotraxin B has been mainly used to antagonize BDNF/TrkB signaling. Nevertheless, some behavioral studies have been performed. While one study has observed anxiolytic properties, this molecule did not seem to display clear antidepressant effects (Cazorla et al. 2010) but it may be suitable for the treatment of neuropathic pain (M’Dahoma et al. 2015). In spite of these interesting results, antagonists could also induce cell death, an issue that should be addressed when considering using these agents in chronic treatment regimens (Cazorla et al. 2011; Takemoto et al. 2015).
It is often observed that in the presence of the endogenous ligand, a synthetic molecule presents antagonistic properties. This was, e.g., shown in a study where they screened several potential TrkB agonists in cells stably transfected with TrkB. When the cells were solely incubated with the synthetic molecules, an increase of TrkB phosphorylation was observed. However, in presence of BDNF, the molecules lost their agonistic effect and showed an antagonistic effect with a decrease of P-TrkB (Cardenas-Aguayo Mdel et al. 2013). Thus, the use of partial agonists or antagonists in order to treat mood disorders seems to be a promising avenue of research. However, whether we should agonize or antagonize the pathway is not yet well defined. Also, further investigations have to be achieved regarding the role of the BDNF/TrkB complex in mood disorders, in order to have a better knowledge of how to correct the dysregulation of this system in mental illness.
Concluding remarks
Growth factors and associated neurotrophic signaling play an essential role in the development and maintenance of the central nervous system (Anlar et al. 1999; Ford-Perriss et al. 2001; Greenberg et al. 2009). Accordingly, there is growing evidence that abnormal trophic support in cortico-limbic regions regulating mood and emotions may take part in the pathophysiology of depression (Krishnan and Nestler 2008). In addition, clean-cut evidence shows that antidepressants require neuroplasticity pathways to rescue the observed deficits in neuronal and synaptic plasticity often associated with mood disorders (Pittenger and Duman 2008). However, current knowledge makes it difficult to conclude whether neuroplasticity and neurogenesis represent a cause, a consequence (or both), or an epiphenomenon of the pathological processes associated with depression. Hence, future research should focus on elucidating the exact involvement of neurotrophic signaling in the onset and course of major depression. Furthermore, mounting evidence seems to indicate that neurogenesis might not be required for the therapeutic action of antidepressants (Bessa et al. 2009). In line with this hypothesis, the usage of N-methyl-d-aspartate (NMDA) receptor antagonists showed that acute induction of neuroplasticity pathways, e.g., increased BDNF signaling, was sufficient to produce a robust and prolonged antidepressant effect (Autry et al. 2011; Li et al. 2010). Hence, the rapid enhancement of hippocampal neuroplasticity—involving dendritic growth, spine density, and synaptic transmission—represents an original strategy to circumvent the delayed efficacy of current antidepressant drugs. Finally, the use of drugs that specifically target neurotrophic signaling should provide more insights in the role of neuroplasticity pathways in antidepressant responses. As growth factors and neurotrophins generally display a poor blood-brain barrier penetration and a short half-life in plasma (Nave et al. 1985; Ochs et al. 2000; Poduslo and Curran 1996), identification of small non-peptidic neurotrophin mimetics, albeit challenging on its own, may represent an interesting target for the development of a new class of therapeutic agents for mood-related disorders.
References
Aarse J, Herlitze S, Manahan-Vaughan D (2016) The requirement of BDNF for hippocampal synaptic plasticity is experience-dependent. Hippocampus 26:739–751
Åberg MA, Åberg ND, Hedbäcker H, Oscarsson J, Eriksson PS (2000) Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci 20:2896–2903
Aberg ND, Brywe KG, Isgaard J (2006) Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. ScientificWorld J 6:53–80
Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM (2008) Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry 63:642–649
Aggleton JP (1993) The contribution of the amygdala to normal and abnormal emotional states. Trends Neurosci 16:328–333
Alderman BL, Olson RL, Brush CJ, Shors TJ (2016) MAP training: combining meditation and aerobic exercise reduces depression and rumination while enhancing synchronized brain activity. Transl Psychiatry 6:e726
Allen AP, Naughton M, Dowling J, Walsh A, Ismail F, Shorten G, Scott L, McLoughlin DM, Cryan JF, Dinan TG, Clarke G (2015) Serum BDNF as a peripheral biomarker of treatment-resistant depression and the rapid antidepressant response: a comparison of ketamine and ECT. J Affect Disord 186:306–311
Alonso M, Medina JH, Pozzo-Miller L (2004) ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem 11:172–178
Altar CA, Whitehead RE, Chen R, Wortwein G, Madsen TM (2003) Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry 54:703–709
Anacker C, Hen R (2017) Adult hippocampal neurogenesis and cognitive flexibility—linking memory and mood. Nat Rev Neurosci 18:335–346
Anderson MF, Åberg MA, Nilsson M, Eriksson PS (2002) Insulin-like growth factor-I and neurogenesis in the adult mammalian brain. Develop Brain Res 134:115–122
Angelucci F, Aloe L, Jimenez-Vasquez P, Mathe AA (2003a) Electroconvulsive stimuli alter nerve growth factor but not brain-derived neurotrophic factor concentrations in brains of a rat model of depression. Neuropeptides 37:51–56
Angelucci F, Aloe L, Jimenez-Vasquez P, Mathe AA (2003b) Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in a rat model of depression. Int J Neuropsychopharmacol 6:225–231
Angst F, Stassen HH, Clayton PJ, Angst J (2002) Mortality of patients with mood disorders: follow-up over 34-38 years. J Affect Disord 68:167–181
Anlar B, Sullivan KA, Feldman EL (1999) Insulin-like growth factor-I and central nervous system development. Horm Metab Res 31:120–125
Arnold JF, Zwiers MP, Fitzgerald DA, van Eijndhoven P, Becker ES, Rinck M, Fernandez G, Speckens AE, Tendolkar I (2012) Fronto-limbic microstructure and structural connectivity in remission from major depression. Psychiatry Res 204:40–48
Arsenijevic Y, Weiss S (1998) Insulin-like growth factor-I is a differentiation factor for postmitotic CNS stem cell-derived neuronal precursors: distinct actions from those of brain-derived neurotrophic factor. J Neurosci 18:2118–2128
Autry AE, Monteggia LM (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64:238–258
Autry AE, Adachi M, Cheng P, Monteggia LM (2009) Gender-specific impact of brain-derived neurotrophic factor signaling on stress-induced depression-like behavior. Biol Psychiatry 66:84–90
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95
Balu DT, Hoshaw BA, Malberg JE, Rosenzweig-Lipson S, Schechter LE, Lucki I (2008) Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res 1211:37–43
Baquet ZC, Gorski JA, Jones KR (2004) Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J.Neurosci 24:4250–4258
Bartsch T, Wulff P (2015) The hippocampus in aging and disease: from plasticity to vulnerability. Neuroscience 309:1–16
Bath KG, Jing DQ, Dincheva I, Neeb CC, Pattwell SS, Chao MV, Lee FS, Ninan I (2012) BDNF Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology 37:1297–1304
Beck KD, Powell-Braxtont L, Widmer H-R, Valverde J, Hefti F (1995) Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 14:717–730
Bellani M, Baiano M, Brambilla P (2011) Brain anatomy of major depression II. Focus on amygdala. Epidemiol Psychiatr Sci 20:33–36
Bergmann O, Spalding KL, Frisen J (2015) Adult neurogenesis in humans. Cold Spring Harb Perspect Biol 7:a018994
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ (2006) Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311:864–868
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, Almeida OF, Sousa N (2009) The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 14:764–773, 739
Bewernick BH, Hurlemann R, Matusch A, Kayser S, Grubert C, Hadrysiewicz B, Axmacher N, Lemke M, Cooper-Mahkorn D, Cohen MX, Brockmann H, Lenartz D, Sturm V, Schlaepfer TE (2010) Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol Psychiatry 67:110–116
Bewernick BH, Kayser S, Sturm V, Schlaepfer TE (2012) Long-term effects of nucleus accumbens deep brain stimulation in treatment-resistant depression: evidence for sustained efficacy. Neuropsychopharmacology 37:1975–1985
Bliss TV, Cooke SF (2011) Long-term potentiation and long-term depression: a clinical perspective. Clinics (Sao Paulo) 66(Suppl 1):3–17
Boku S, Nakagawa S, Takamura N, Kato A, Takebayashi M, Hisaoka-Nakashima K, Omiya Y, Inoue T, Kusumi I (2013) GDNF facilitates differentiation of the adult dentate gyrus-derived neural precursor cells into astrocytes via STAT3. Biochem Biophys Res Commun 434:779–784
Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, Arango V (2009) Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34:2376–2389
Boldrini M, Hen R, Underwood MD, Rosoklija GB, Dwork AJ, Mann JJ, Arango V (2012) Hippocampal angiogenesis and progenitor cell proliferation are increased with antidepressant use in major depression. Biol Psychiatry 72:562–571
Boldrini M, Santiago AN, Hen R, Dwork AJ, Rosoklija GB, Tamir H, Arango V, John Mann J (2013) Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology 38:1068–1077
Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, Hen R, Mann JJ (2018) Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell 22:589–599 e5
Bollen E, Vanmierlo T, Akkerman S, Wouters C, Steinbusch HM, Prickaerts J (2013) 7,8-Dihydroxyflavone improves memory consolidation processes in rats and mice. Behav Brain Res 257:8–12
Bot M, Milaneschi Y, Penninx BW, Drent ML (2016) Plasma insulin-like growth factor I levels are higher in depressive and anxiety disorders, but lower in antidepressant medication users. Psychoneuroendocrinology 68:148–155
Boulle F, Kenis G, Cazorla M, Hamon M, Steinbusch HW, Lanfumey L, van den Hove DL (2012) TrkB inhibition as a therapeutic target for CNS-related disorders. Prog Neurobiol 98:197–206
Boulle F, Massart R, Stragier E, Paizanis E, Zaidan L, Marday S, Gabriel C, Mocaer E, Mongeau R, Lanfumey L (2014) Hippocampal and behavioral dysfunctions in a mouse model of environmental stress: normalization by agomelatine. Transl Psychiatry 4:e485
Boulle F, Velthuis H, Koedam K, Steinbusch HW, van den Hove DL, Kenis G, Gabriel C, Mocaer E, Franc B, Rognan D, Mongeau R, Lanfumey L (2016) Behavioral and neurochemical characterization of TrkB-dependent mechanisms of agomelatine in glucocorticoid receptor-impaired mice. Eur Neuropsychopharmacol 26:65–77
Bramham CR (2007) Control of synaptic consolidation in the dentate gyrus: mechanisms, functions. and therapeutic implications Prog Brain Res 163:453–471
Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS (2000) Hippocampal volume reduction in major depression. Am J Psychiatry 157:115–118
Bremner JD, Vythilingam M, Vermetten E, Nazeer A, Adil J, Khan S, Staib LH, Charney DS (2002) Reduced volume of orbitofrontal cortex in major depression. Biol Psychiatry 51:273–279
Bruchim-Samuel M, Lax E, Gazit T, Friedman A, Ahdoot H, Bairachnaya M, Pinhasov A, Yadid G (2016) Electrical stimulation of the vmPFC serves as a remote control to affect VTA activity and improve depressive-like behavior. Exp Neurol 283:255–263
Bubl E, Kern E, Ebert D, Riedel A, Tebartz van Elst L, Bach M (2015) Retinal dysfunction of contrast processing in major depression also apparent in cortical activity. Eur Arch Psychiatry Clin Neurosci 265:343–350
Canli T, Cooney RE, Goldin P, Shah M, Sivers H, Thomason ME, Whitfield-Gabrieli S, Gabrieli JD, Gotlib IH (2005) Amygdala reactivity to emotional faces predicts improvement in major depression. Neuroreport 16:1267–1270
Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, During MJ (2004) VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet 36:827–835
Cao X, Liu Z, Xu C, Li J, Gao Q, Sun N, Xu Y, Ren Y, Yang C, Zhang K (2012) Disrupted resting-state functional connectivity of the hippocampus in medication-naive patients with major depressive disorder. J Affect Disord 141:194–203
Cardenas-Aguayo Mdel C, Kazim SF, Grundke-Iqbal I, Iqbal K (2013) Neurogenic and neurotrophic effects of BDNF peptides in mouse hippocampal primary neuronal cell cultures. PLoS One 8:e53596
Carmeliet P, Ruiz de Almodovar C (2013) VEGF ligands and receptors: implications in neurodevelopment and neurodegeneration. Cell Mol Life Sci 70:1763–1778
Castren E, Hen R (2013) Neuronal plasticity and antidepressant actions. Trends Neurosci 36:259–267
Castren E, Kojima M (2017) Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol Dis 97:119–126
Cattaneo E, McKay R (1990) Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature 347:762–765
Cavanaugh JE, Ham J, Hetman M, Poser S, Yan C, Xia Z (2001) Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci 21:434–443
Cazorla M, Jouvenceau A, Rose C, Guilloux JP, Pilon C, Dranovsky A, Premont J (2010) Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS One 5:e9777
Cazorla M, Premont J, Mann A, Girard N, Kellendonk C, Rognan D (2011) Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest 121:1846–1857
Chacón-Fernández P, Säuberli K, Colzani M, Moreau T, Ghevaert C, Barde YA (2016) Brain-derived neurotrophic factor in megakaryocytes. J Biol Chem 291:9872–9881
Chao MV, Hempstead BL (1995) p75 and Trk: a two-receptor system. Trends Neurosci 18:321–326
Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT (2001) Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 50:260–265
Chen Y, Ai Y, Slevin JR, Maley BE, Gash DM (2005) Progenitor proliferation in the adult hippocampus and substantia nigra induced by glial cell line-derived neurotrophic factor. Exp Neurol 196:87–95
Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS (2006) Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314:140–143
Chen D, Brahimi F, Angell Y, Li YC, Moscowicz J, Saragovi HU, Burgess K (2009) Bivalent peptidomimetic ligands of TrkC are biased agonists and selectively induce neuritogenesis or potentiate neurotrophin-3 trophic signals. ACS Chem Biol 4:769–781
Chen F, Madsen TM, Wegener G, Nyengaard JR (2010) Imipramine treatment increases the number of hippocampal synapses and neurons in a genetic animal model of depression. Hippocampus 20:1376–1384
Chen YT, Huang MW, Hung IC, Lane HY, Hou CJ (2014) Right and left amygdalae activation in patients with major depression receiving antidepressant treatment, as revealed by fMRI. Behav Brain Funct 10:36
Choi DC, Maguschak KA, Ye K, Jang SW, Myers KM, Ressler KJ (2010) Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc Natl Acad Sci U S A 107:2675–2680
Clapp WC, Kirk IJ, Hamm JP, Shepherd D, Teyler TJ (2005a) Induction of LTP in the human auditory cortex by sensory stimulation. Eur J Neurosci 22:1135–1140
Clapp WC, Zaehle T, Lutz K, Marcar VL, Kirk IJ, Hamm JP, Teyler TJ, Corballis MC, Jancke L (2005b) Effects of long-term potentiation in the human visual cortex: a functional magnetic resonance imaging study. Neuroreport 16:1977–1980
Clark-Raymond A, Halaris A (2013) VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res 47:1080–1087
Cole J, Costafreda SG, McGuffin P, Fu CH (2011) Hippocampal atrophy in first episode depression: a meta-analysis of magnetic resonance imaging studies. J Affect Disord 134:483–487
Colle R, Cury C, Chupin M, Deflesselle E, Hardy P, Nasser G, Falissard B, Ducreux D, Colliot O, Corruble E (2016) Hippocampal volume predicts antidepressant efficacy in depressed patients without incomplete hippocampal inversion. Neuroimage Clin 12:949–955
Conner JM, Franks KM, Titterness AK, Russell K, Merrill DA, Christie BR, Sejnowski TJ, Tuszynski MH (2009) NGF is essential for hippocampal plasticity and learning. J Neurosci 29:10883–10889
Cook SC, Wellman CL (2004) Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol 60:236–248
Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP (2002) Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex 12:386–394
Cramer SC, Sur M, Dobkin BH, O'Brien C, Sanger TD, Trojanowski JQ, Rumsey JM, Hicks R, Cameron J, Chen D, Chen WG, Cohen LG, deCharms C, Duffy CJ, Eden GF, Fetz EE, Filart R, Freund M, Grant SJ, Haber S, Kalivas PW, Kolb B, Kramer AF, Lynch M, Mayberg HS, McQuillen PS, Nitkin R, Pascual-Leone A, Reuter-Lorenz P, Schiff N, Sharma A, Shekim L, Stryker M, Sullivan EV, Vinogradov S (2011) Harnessing neuroplasticity for clinical applications. Brain 134:1591–1609
Crutcher KA (1986) The role of growth factors in neuronal development and plasticity. Crit Rev Clin Neurobiol 2:297–333
D'Arcangelo G, Triossi T, Buglione A, Melchiorri G, Tancredi V (2017) Modulation of synaptic plasticity by short-term aerobic exercise in adult mice. Behav Brain Res 332:59–63
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R (2009) Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62:479–493
Delaveau P, Jabourian M, Lemogne C, Guionnet S, Bergouignan L, Fossati P (2011) Brain effects of antidepressants in major depression: a meta-analysis of emotional processing studies. J Affect Disord 130:66–74
Deltheil T, Guiard BP, Cerdan J, David DJ, Tanaka KF, Reperant C, Guilloux JP, Coudore F, Hen R, Gardier AM (2008) Behavioral and serotonergic consequences of decreasing or increasing hippocampus brain-derived neurotrophic factor protein levels in mice. Neuropharmacology 55:1006–1014
Deltheil T, Tanaka K, Reperant C, Hen R, David DJ, Gardier AM (2009) Synergistic neurochemical and behavioural effects of acute intrahippocampal injection of brain-derived neurotrophic factor and antidepressants in adult mice. Int J Neuropsychopharmacol 12:905–915
Dempsey RJ, Sailor KA, Bowen KK, Tureyen K, Vemuganti R (2003) Stroke-induced progenitor cell proliferation in adult spontaneously hypertensive rat brain: effect of exogenous IGF-1 and GDNF. J Neurochem 87:586–597
Dere E, Pause BM, Pietrowsky R (2010) Emotion and episodic memory in neuropsychiatric disorders. Behav Brain Res 215:162–171
Devi L, Ohno M (2012) 7,8-Dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology 37:434–444
Di Liberto V, Mudo G, Fuxe K, Belluardo N (2014) Interactions between cholinergic and fibroblast growth factor receptors in brain trophism and plasticity. Curr Protein Pept Sci 15:691–702
Diniz BS, Teixeira AL, Machado-Vieira R, Talib LL, Gattaz WF, Forlenza OV (2012a) Reduced serum nerve growth factor in patients with late-life depression. Am J Geriatr Psychiatry 21:493–496
Diniz BS, Teixeira AL, Miranda AS, Talib LL, Gattaz WF, Forlenza OV (2012b) Circulating glial-derived neurotrophic factor is reduced in late-life depression. J Psychiatr Res 46:135–139
Djavadian RL (2004) Serotonin and neurogenesis in the hippocampal dentate gyrus of adult mammals. Acta Neurobiol Exp 64:189–200
D'Mello SR, Galli C, Ciotti T, Calissano P (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A 90:10989–10993
Dokter M, Busch R, Poser R, Vogt MA, von Bohlen Und Halbach V, Gass P, Unsicker K, von Bohlen Und Halbach O (2015) Implications of p75NTR for dentate gyrus morphology and hippocampus-related behavior revisited. Brain Struct Func 220: 1449–1462
Drevets WC (2000) Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res 126:413–431
Drevets WC (2003) Neuroimaging abnormalities in the amygdala in mood disorders. Ann N Y Acad Sci 985:420–444
Duman RS, Aghajanian GK (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72
Duman CH, Schlesinger L, Terwilliger R, Russell DS, Newton SS, Duman RS (2009) Peripheral insulin-like growth factor-I produces antidepressant-like behavior and contributes to the effect of exercise. Behav Brain Res 198:366–371
Dunham JS, Deakin JFW, Miyajima F, Payton A, Toro CT (2009) Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains. J Psychiatr Res 43:1175–1184
During MJ, Cao L (2006) VEGF, a mediator of the effect of experience on hippocampal neurogenesis. Curr Alzheimer Res 3:29–33
Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN (2003) Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60:804–815
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR (2003) The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112:257–269
Eisch AJ, Petrik D (2012) Depression and hippocampal neurogenesis: a road to remission? Science 338:72–75
Eisch AJ, Bolanos CA, de Wit J, Simonak RD, Pudiak CM, Barrot M, Verhaagen J, Nestler EJ (2003) Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol Psychiatry 54:994–1005
Elfving B, Wegener G (2012) Electroconvulsive seizures stimulate the vegf pathway via mTORC1. Synapse 66:340–345
Elsayed M, Banasr M, Duric V, Fournier NM, Licznerski P, Duman RS (2012) Antidepressant effects of fibroblast growth factor-2 in behavioral and cellular models of depression. Biol Psychiatry 72:258–265
Elvsashagen T, Moberget T, Boen E, Boye B, Englin NO, Pedersen PO, Andreassen OA, Dietrichs E, Malt UF, Andersson S (2012) Evidence for impaired neocortical synaptic plasticity in bipolar II disorder. Biol Psychiatry 71:68–74
Encinas JM, Vaahtokari A, Enikolopov G (2006) Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci U S A 103:8233–8238
Epstein J, Pan H, Kocsis JH, Yang Y, Butler T, Chusid J, Hochberg H, Murrough J, Strohmayer E, Stern E, Silbersweig DA (2006) Lack of ventral striatal response to positive stimuli in depressed versus normal subjects. Am J Psychiatry 163:1784–1790
Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4:1313–1317
Ernfors P, Wetmore C, Olson L, Persson H (1990) Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron 5:511–526
Ernfors P, Lee KF, Jaenisch R (1994) Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368:147–150
Evans SJ, Choudary PV, Neal CR, Li JZ, Vawter MP, Tomita H, Lopez JF, Thompson RC, Meng F, Stead JD, Walsh DM, Myers RM, Bunney WE, Watson SJ, Jones EG, Akil H (2004) Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci U S A 101:15506–15511
Falowski SM, Sharan A, Reyes BA, Sikkema C, Szot P, Van Bockstaele EJ (2011) An evaluation of neuroplasticity and behavior after deep brain stimulation of the nucleus accumbens in an animal model of depression. Neurosurgery 69:1281–1290
Fitzgerald PB, Daskalakis ZJ (2011) The effects of repetitive transcranial magnetic stimulation in the treatment of depression. Expert Rev Med Devices 8:85–95
Fitzgerald PB, Laird AR, Maller J, Daskalakis ZJ (2008) A meta-analytic study of changes in brain activation in depression. Hum Brain Mapp 29:683–695
Ford-Perriss M, Abud H, Murphy M (2001) Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol 28:493–503
Fried EI, Kievit RA (2016) The volumes of subcortical regions in depressed and healthy individuals are strikingly similar: a reinterpretation of the results by Schmaal et al. Mol Psychiatry 21:724–725
Friedman A, Friedman Y, Dremencov E, Yadid G (2008) VTA dopamine neuron bursting is altered in an animal model of depression and corrected by desipramine. J Mol Neurosci 34:201–209
Frielingsdorf H, Simpson DR, Thal LJ, Pizzo DP (2007) Nerve growth factor promotes survival of new neurons in the adult hippocampus. Neurobiol Dis 26:47–55
Frodl T, O'Keane V (2013) How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiol Dis 52:24–37
Frodl T, Meisenzahl EM, Zetzsche T, Born C, Jager M, Groll C, Bottlender R, Leinsinger G, Moller HJ (2003) Larger amygdala volumes in first depressive episode as compared to recurrent major depression and healthy control subjects. Biol Psychiatry 53:338–344
Frodl T, Schule C, Schmitt G, Born C, Baghai T, Zill P, Bottlender R, Rupprecht R, Bondy B, Reiser M, Moller HJ, Meisenzahl EM (2007) Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch Gen Psychiatry 64:410–416
Frodl T, Jager M, Smajstrlova I, Born C, Bottlender R, Palladino T, Reiser M, Moller HJ, Meisenzahl EM (2008) Effect of hippocampal and amygdala volumes on clinical outcomes in major depression: a 3-year prospective magnetic resonance imaging study. J Psychiatry Neurosci 33:423–430
Frysak Z, Schovanek J, Iacobone M, Karasek D (2015) Insulin-like growth factors in a clinical setting: review of IGF-I. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 159:347–351
Fu CH, Steiner H, Costafreda SG (2013) Predictive neural biomarkers of clinical response in depression: a meta-analysis of functional and structural neuroimaging studies of pharmacological and psychological therapies. Neurobiol Dis 52:75–83
Furman DJ, Hamilton JP, Gotlib IH (2011) Frontostriatal functional connectivity in major depressive disorder. Biol Mood Anx Dis 1:11
Gaughran F, Payne J, Sedgwick PM, Cotter D, Berry M (2006) Hippocampal FGF-2 and FGFR1 mRNA expression in major depression, schizophrenia and bipolar disorder. Brain Res Bull 70:221–227
Giacobbe P, Mayberg HS, Lozano AM (2009) Treatment resistant depression as a failure of brain homeostatic mechanisms: implications for deep brain stimulation. Exp Neurol 219:44–52
Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, Morrison JH (2009) Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 164:798–808
Greenberg ME, Xu B, Lu B, Hempstead BL (2009) New insights in the biology of BDNF synthesis and release: implications in CNS function. J Neurosci 29:12764–12767
Greene J, Banasr M, Lee B, Warner-Schmidt J, Duman RS (2009) Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment: pharmacological and cellular characterization. Neuropsychopharmacology 34:2459–2468
Groenewold NA, Opmeer EM, de Jonge P, Aleman A, Costafreda SG (2013) Emotional valence modulates brain functional abnormalities in depression: evidence from a meta-analysis of fMRI studies. Neurosci Biobehav Rev 37:152–163
Hamilton JP, Gotlib IH (2008) Neural substrates of increased memory sensitivity for negative stimuli in major depression. Biol Psychiatry 63:1155–1162
Hamilton JP, Siemer M, Gotlib IH (2008) Amygdala volume in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Mol Psychiatry 13:993–1000
Hamilton JP, Etkin A, Furman DJ, Lemus MG, Johnson RF, Gotlib IH (2012) Functional neuroimaging of major depressive disorder: a meta-analysis and new integration of base line activation and neural response data. Am J Psychiatry 169:693–703
Han J, Pollak J, Yang T, Siddiqui MR, Doyle KP, Taravosh-Lahn K, Cekanaviciute E, Han A, Goodman JZ, Jones B, Jing D, Massa SM, Longo FM, Buckwalter MS (2012) Delayed administration of a small molecule tropomyosin-related kinase B ligand promotes recovery after hypoxic-ischemic stroke. Stroke 43:1918–1924
Harrisberger F, Smieskova R, Schmidt A, Lenz C, Walter A, Wittfeld K, Grabe HJ, Lang UE, Fusar-Poli P, Borgwardt S (2015) BDNF Val66Met polymorphism and hippocampal volume in neuropsychiatric disorders: a systematic review and meta-analysis. Neurosci Biobehav Rev 55:107–118
Heine VM, Zareno J, Maslam S, Joels M, Lucassen PJ (2005) Chronic stress in the adult dentate gyrus reduces cell proliferation near the vasculature and VEGF and Flk-1 protein expression. Eur J Neurosci 21:1304–1314
Herring MP, Puetz TW, O'Connor PJ, Dishman RK (2012) Effect of exercise training on depressive symptoms among patients with a chronic illness: a systematic review and meta-analysis of randomized controlled trials. Arch Intern Med 172:101–111
Hetman M, Hsuan SL, Habas A, Higgins MJ, Xia Z (2002) ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. J Biol Chem 277:49577–49584
Hisaoka K, Nishida A, Koda T, Miyata M, Zensho H, Morinobu S, Ohta M, Yamawaki S (2001) Antidepressant drug treatments induce glial cell line-derived neurotrophic factor (GDNF) synthesis and release in rat C6 glioblastoma cells. J Neurochem 79:25–34
Ho NF, Hooker JM, Sahay A, Holt DJ, Roffman JL (2013) In vivo imaging of adult human hippocampal neurogenesis: progress, pitfalls and promise. Mol Psychiatry 18:404–416
Hosang GM, Shiles C, Tansey KE, McGuffin P, Uher R (2014) Interaction between stress and the BDNF Val66Met polymorphism in depression: a systematic review and meta-analysis. BMC Med 12:7
Hoshaw BA, Malberg JE, Lucki I (2005) Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res 1037:204–208
Hoshaw BA, Hill TI, Crowley JJ, Malberg JE, Khawaja X, Rosenzweig-Lipson S, Schechter LE, Lucki I (2008) Antidepressant-like behavioral effects of IGF-I produced by enhanced serotonin transmission. Eur J Pharmacol 594:109–116
Huang Y, Coupland NJ, Lebel RM, Carter R, Seres P, Wilman AH, Malykhin NV (2013) Structural changes in hippocampal subfields in major depressive disorder: a high-field magnetic resonance imaging study. Biol Psychiatry 74:62–68
Jacobsen JP, Mork A (2004) The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels. Brain Res 1024:183–192
Jang SW, Liu X, Chan CB, France SA, Sayeed I, Tang W, Lin X, Xiao G, Andero R, Chang Q, Ressler KJ, Ye K (2010) Deoxygedunin, a natural product with potent neurotrophic activity in mice. PLoS One 5:e11528
Jaworska N, Yang XR, Knott V, MacQueen G (2015) A review of fMRI studies during visual emotive processing in major depressive disorder. World J Biol Psychiatry 16:448–471
Jeanneteau F, Garabedian MJ, Chao MV (2008) Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci U S A 105:4862–4867
Jiang P, Dang RL, Li HD, Zhang LH, Zhu WY, Xue Y, Tang MM (2014) The impacts of swimming exercise on hippocampal expression of neurotrophic factors in rats exposed to chronic unpredictable mild stress. Evid-Based Complement Altern Med 2014:729827
Jin KL, Mao XO, Greenberg DA (2000) Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U S A 97:10242–10247
Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA (2002) Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A 99:11946–11950
Jin K, Sun Y, Xie L, Batteur S, Mao XO, Smelick C, Logvinova A, Greenberg DA (2003) Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell 2:175–183
Joshi SH, Espinoza RT, Pirnia T, Shi J, Wang Y, Ayers B, Leaver A, Woods RP, Narr KL (2016) Structural plasticity of the hippocampus and amygdala induced by electroconvulsive therapy in major depression. Biol Psychiatry 79:282–292
Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, Lepack A, Majik MS, Jeong LS, Banasr M, Son H, Duman RS (2012) Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med 18:1413–1417
Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry J-M (2002) Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res 109:143–148
Karege F, Bondolfi G, Gervasoni N, Schwald M, Aubry JM, Bertschy G (2005) Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol Psychiatry 57:1068–7102
Keers R, Uher R (2012) Gene-environment interaction in major depression and antidepressant treatment response. Curr Psychiatry Rep 14:129–137
Kempton MJ, Salvador Z, Munafo MR, Geddes JR, Simmons A, Frangou S, Williams SC (2011) Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry 68:675–690
Kendler KS, Kessler RC, Walters EE, MacLean C, Neale MC, Heath AC, Eaves LJ (1995) Stressful life events, genetic liability, and onset of an episode of major depression in women. Am J Psychiatry 152:833–842
Khaibullina AA, Rosenstein JM, Krum JM (2004) Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res Dev Brain Res 148:59–68
Kim JJ, Diamond DM (2002) The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 3:453–462
Kim GS, Cho S, Nelson JW, Zipfel GJ, Han BH (2014) TrkB agonist antibody pretreatment enhances neuronal survival and long-term sensory motor function following hypoxic ischemic injury in neonatal rats. PLoS One 9:e88962
Kishi T, Yoshimura R, Ikuta T, Iwata N (2017) Brain-derived neurotrophic factor and major depressive disorder: evidence from meta-analyses. Front Psychiatry 8:308
Klein AB, Williamson R, Santini MA, Clemmensen C, Ettrup A, Rios M, Knudsen GM, Aznar S (2011) Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol 14:347–353
Klempin F, Beis D, Mosienko V, Kempermann G, Bader M, Alenina N (2013) Serotonin is required for exercise-induced adult hippocampal neurogenesis. J Neurosci 33:8270–8275
Knaepen K, Goekint M, Heyman EM, Meeusen R (2010) Neuroplasticity—exercise-induced response of peripheral brain-derived neurotrophic factor: a systematic review of experimental studies in human subjects. Sports Med 40:765–801
Kobayashi T, Ahlenius H, Thored P, Kobayashi R, Kokaia Z, Lindvall O (2006) Intracerebral infusion of glial cell line-derived neurotrophic factor promotes striatal neurogenesis after stroke in adult rats. Stroke 37:2361–2367
Kopczak A, Stalla GK, Uhr M, Lucae S, Hennings J, Ising M, Holsboer F, Kloiber S (2015) IGF-I in major depression and antidepressant treatment response. Eur Neuropsychopharmacol 25:864–872
Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455:894–902
Kromer LF (1987) Nerve growth factor treatment after brain injury prevents neuronal death. Science 235:214–216
Kron M, Lang M, Adams IT, Sceniak M, Longo F, Katz DM (2014) A BDNF loop-domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis Model Mech 7:1047–1055
Kronenberg G, Tebartz van Elst L, Regen F, Deuschle M, Heuser I, Colla M (2009) Reduced amygdala volume in newly admitted psychiatric in-patients with unipolar major depression. J Psychiatr Res 43:1112–1117
Lange C, Irle E (2004) Enlarged amygdala volume and reduced hippocampal volume in young women with major depression. Psychol Med 34:1059–1064
LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23:155–184
Lee J, Duan W, Mattson MP (2002) Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem 82:1367–1375
Lewis MA, Hunihan L, Franco D, Robertson B, Palmer J, Laurent DR, Balasubramanian BN, Li Y, Westphal RS (2006) Identification and characterization of compounds that potentiate NT-3-mediated Trk receptor activity. Mol Pharmacol 69:1396–1404
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964
Li M, Chang H, Xiao X (2016) BDNF Val66Met polymorphism and bipolar disorder in European populations: a risk association in case-control, family-based and GWAS studies. Neurosci Biobehav Rev 68:218–233
Licht T, Eavri R, Goshen I, Shlomai Y, Mizrahi A, Keshet E (2010) VEGF is required for dendritogenesis of newly born olfactory bulb interneurons. Development 137:261–271
Lichtenwalner R, Forbes M, Bennett S, Lynch C, Sonntag W, Riddle D (2001) Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience 107:603–613
Lim LW, Janssen ML, Kocabicak E, Temel Y (2015) The antidepressant effects of ventromedial prefrontal cortex stimulation is associated with neural activation in the medial part of the subthalamic nucleus. Behav Brain Res 279:17–21
Limon A, Mamdani F, Hjelm BE, Vawter MP, Sequeira A (2016) Targets of polyamine dysregulation in major depression and suicide: activity-dependent feedback, excitability, and neurotransmission. Neurosci Biobehav Rev 66:80–91
Liu Q, Yu J, Mao-Ying QL, Mi WL, Li B, Wang YQ, Wang J, Wu GC (2008) Repeated clomipramine treatment reversed the inhibition of cell proliferation in adult hippocampus induced by chronic unpredictable stress. Pharmacogen J 8:375–383
Liu X, Chan CB, Jang SW, Pradoldej S, Huang J, He K, Phun LH, France S, Xiao G, Jia Y, Luo HR, Ye K (2010) A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J Med Chem 53:8274–8286
Liu X, Chan CB, Qi Q, Xiao G, Luo HR, He X, Ye K (2012) Optimization of a small tropomyosin-related kinase B (TrkB) agonist 7,8-dihydroxyflavone active in mouse models of depression. J Med Chem 55:8524–8537
Lopez AD, Murray CC (1998) The global burden of disease, 1990-2020. Nat Med 4:1241–1243
Lorenzetti V, Allen NB, Fornito A, Yücel M (2009) Structural brain abnormalities in major depressive disorder: a selective review of recent MRI studies. J Affect Dis 117:1–17
Lu B, Nagappan G, Lu Y (2014) BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol 220:223–250
Lucassen PJ, Stumpel MW, Wang Q, Aronica E (2010) Decreased numbers of progenitor cells but no response to antidepressant drugs in the hippocampus of elderly depressed patients. Neuropharmacology 58:940–949
Lyttle K, Ohmura Y, Konno K, Yoshida T, Izumi T, Watanabe M, Yoshioka M (2015) Repeated fluvoxamine treatment recovers juvenile stress-induced morphological changes and depressive-like behavior in rats. Brain Res 1616:88–100
Ma M, Ren Q, Yang C, Zhang JC, Yao W, Dong C, Ohgi Y, Futamura T, Hashimoto K (2016) Adjunctive treatment of brexpiprazole with fluoxetine shows a rapid antidepressant effect in social defeat stress model: role of BDNF-TrkB signaling. Sci Rep 6:39209
MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, Nahmias C, Young LT (2003) Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci U S A 100:1387–1392
Magarinos AM, McEwen BS, Flugge G, Fuchs E (1996) Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci 16:3534–3540
Magarinos AM, Li CJ, Gal Toth J, Bath KG, Jing D, Lee FS, McEwen BS (2011) Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus 21:253–264
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20:9104–9110
Malberg JE, Platt B, Rizzo SJS, Ring RH, Lucki I, Schechter LE, Rosenzweig-Lipson S (2007) Increasing the levels of insulin-like growth factor-I by an IGF binding protein inhibitor produces anxiolytic and antidepressant-like effects. Neuropsychopharmacology 32:2360–2368
Malone DA Jr, Dougherty DD, Rezai AR, Carpenter LL, Friehs GM, Eskandar EN, Rauch SL, Rasmussen SA, Machado AG, Kubu CS, Tyrka AR, Price LH, Stypulkowski PH, Giftakis JE, Rise MT, Malloy PF, Salloway SP, Greenberg BD (2009) Deep brain stimulation of the ventral capsule/ventral striatum for treatment-resistant depression. Biol Psychiatry 65:267–275
Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM (2010) Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest 120:1774–1785
Matthews SC, Strigo IA, Simmons AN, Yang TT, Paulus MP (2008) Decreased functional coupling of the amygdala and supragenual cingulate is related to increased depression in unmedicated individuals with current major depressive disorder. J Affect Disord 111:13–20
McKinnon MC, Yucel K, Nazarov A, MacQueen GM (2009) A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. J Psychiatry Neurosci 34:41–54
M'Dahoma S, Barthelemy S, Tromilin C, Jeanson T, Viguier F, Michot B, Pezet S, Hamon M, Bourgoin S (2015) Respective pharmacological features of neuropathic-like pain evoked by intrathecal BDNF versus sciatic nerve ligation in rats. Eur Neuropsychopharmacol 25:2118–2130
Michel TM, Frangou S, Camara S, Thiemeyer D, Jecel J, Tatschner T, Zoechling R, Grunblatt E (2008) Altered glial cell line-derived neurotrophic factor (GDNF) concentrations in the brain of patients with depressive disorder: a comparative post-mortem study. Eur Psychiatry 23:413–420
Millan MJ, Agid Y, Brune M, Bullmore ET, Carter CS et al (2012) Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov 11:141–168
Miller BR, Hen R (2015) The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol 30:51–58
Miller CH, Hamilton JP, Sacchet MD, Gotlib IH (2015) Meta-analysis of functional neuroimaging of major depressive disorder in youth. JAMA Psychiatry 72:1045–1053
Milne AM, MacQueen GM, Hall GB (2012) Abnormal hippocampal activation in patients with extensive history of major depression: an fMRI study. J Psychiatry Neurosci 37:28–36
Mitschelen M, Yan H, Farley JA, Warrington JP, Han S, Herenu CB, Csiszar A, Ungvari Z, Bailey-Downs LC, Bass CE, Sonntag WE (2011) Long-term deficiency of circulating and hippocampal insulin-like growth factor I induces depressive behavior in adult mice: a potential model of geriatric depression. Neuroscience 185:50–60
Mitterschiffthaler MT, Kumari V, Malhi GS, Brown RG, Giampietro VP, Brammer MJ, Suckling J, Poon L, Simmons A, Andrew C, Sharma T (2003) Neural response to pleasant stimuli in anhedonia: an fMRI study. Neuroreport 14:177–182
Molendijk ML, Bus BA, Spinhoven P, Penninx BW, Kenis G, Prickaerts J, Voshaar RC, Elzinga BM (2011) Serum levels of brain-derived neurotrophic factor in major depressive disorder: state-trait issues, clinical features and pharmacological treatment. Mol Psychiatry 16:1088–1095
Molendijk ML, Spinhoven P, Polak M, Bus BA, Penninx BW, Elzinga BM (2014) Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations (N = 9484). Mol Psychiatry 19:791–800
Montalbano A, Baj G, Papadia D, Tongiorgi E, Sciancalepore M (2013) Blockade of BDNF signaling turns chemically-induced long-term potentiation into long-term depression. Hippocampus 23:879–889
Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ (2004) Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A 101:10827–10832
Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ (2007) Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry 61:187–197
Morais M, Patricio P, Mateus-Pinheiro A, Alves ND, Machado-Santos AR, Correia JS, Pereira J, Pinto L, Sousa N, Bessa JM (2017) The modulation of adult neuroplasticity is involved in the mood-improving actions of atypical antipsychotics in an animal model of depression. Transl Psychiatry 7:e1146
Mostany R, Valdizán EM, Pazos A (2008) A role for nuclear β-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Neuropharmacology 55:18–26
Mudo G, Bonomo A, Di Liberto V, Frinchi M, Fuxe K, Belluardo N (2009) The FGF-2/FGFRs neurotrophic system promotes neurogenesis in the adult brain. J Neural Transm 116:995–1005
Nakamura K, Ito M, Liu Y, Seki T, Suzuki T, Arai H (2013) Effects of single and repeated electroconvulsive stimulation on hippocampal cell proliferation and spontaneous behaviors in the rat. Brain Res 1491:88–97
Nave KA, Probstmeier R, Schachner M (1985) Epidermal growth factor does not cross the blood-brain barrier. Cell Tissue Res 241:453–457
Nestler EJ, Carlezon WA Jr (2006) The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59:1151–1159
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34:13–25
Nibuya M, Morinobu S, Duman RS (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15:7539–7547
Ninan I, Bath KG, Dagar K, Perez-Castro R, Plummer MR, Lee FS, Chao MV (2010) The BDNF Val66Met polymorphism impairs NMDA receptor-dependent synaptic plasticity in the hippocampus. J Neurosci 30:8866–8870
Nordanskog P, Dahlstrand U, Larsson MR, Larsson EM, Knutsson L, Johanson A (2010) Increase in hippocampal volume after electroconvulsive therapy in patients with depression: a volumetric magnetic resonance imaging study. J ECT 26:62–67
Nordanskog P, Larsson MR, Larsson EM, Johanson A (2014) Hippocampal volume in relation to clinical and cognitive outcome after electroconvulsive therapy in depression. Acta Psychiatr Scand 129:303–311
Normann C, Schmitz D, Furmaier A, Doing C, Bach M (2007) Long-term plasticity of visually evoked potentials in humans is altered in major depression. Biol Psychiatry 62:373–380
Nowacka MM, Obuchowicz E (2012) Vascular endothelial growth factor (VEGF) and its role in the central nervous system: a new element in the neurotrophic hypothesis of antidepressant drug action. Neuropeptides 46:1–10
Obrietan K, Gao X-B, van den Pol AN (2002) Excitatory actions of GABA increase BDNF expression via a MAPK-CREB-dependent mechanism—a positive feedback circuit in developing neurons. J Neurophysiol 88:1005–1015
Ochs G, Penn RD, York M, Giess R, Beck M, Tonn J, Haigh J, Malta E, Traub M, Sendtner M, Toyka KV (2000) A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:201–206
O'Kusky JR, Ye P, D'Ercole AJ (2000) Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J Neurosci 20:8435–8442
Olson L (1967) Outgrowth of sympathetic adrenergic neurons in mice treated with a nerve-growth factor (NGF). Zeitschrift fur Zellforschung und mikroskopische Anatomie (Vienna, Austria : 1948) 81:155–173
Ongur D, Price JL (2000) The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex 10:206–219
Öngür D, Drevets WC, Price JL (1998) Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci 95:13290–13295
Ota KT, Duman RS (2013) Environmental and pharmacological modulations of cellular plasticity: role in the pathophysiology and treatment of depression. Neurobiol Dis 57:28–37
Overstreet DH, Fredericks K, Knapp D, Breese G, McMichael J (2010) Nerve growth factor (NGF) has novel antidepressant-like properties in rats. Pharmacol Biochemi Behav 94:553–560
Paizanis E, Renoir T, Lelievre V, Saurini F, Melfort M, Gabriel C, Barden N, Mocaer E, Hamon M, Lanfumey L (2010) Behavioural and neuroplastic effects of the new-generation antidepressant agomelatine compared to fluoxetine in glucocorticoid receptor-impaired mice. Int J Neuropsychopharmacol 13:759–774
Pallavi P, Sagar R, Mehta M, Sharma S, Subramanium A, Shamshi F, Sengupta U, Qadri R, Pandey RM, Mukhopadhyay AK (2013) Serum neurotrophic factors in adolescent depression: gender difference and correlation with clinical severity. J Affect Disord 150:415–423
Pan W, Kastin AJ (2000) Interactions of IGF-1 with the blood-brain barrier in vivo and in situ. Neuroendocrinology 72:171–178
Pandey GN, Ren X, Rizavi HS, Conley RR, Roberts RC, Dwivedi Y (2008) Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int J Neuropsychopharmacol 11:1047–1061
Park SE, Dantzer R, Kelley KW, McCusker RH (2011) Central administration of insulin-like growth factor-I decreases depressive-like behavior and brain cytokine expression in mice. J Neuroinflammation 8:12
Park SW, Nhu le H, Cho HY, Seo MK, Lee CH, Ly NN, Choi CM, Lee BJ, Kim GM, Seol W, Lee JG, Kim YH (2016) p11 mediates the BDNF-protective effects in dendritic outgrowth and spine formation in B27-deprived primary hippocampal cells. J Affect Disord 196:1–10
Paslakis G, Blum WF, Deuschle M (2012) Intranasal insulin-like growth factor I (IGF-I) as a plausible future treatment of depression. Med Hypotheses 79:222–225
Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I (2012) The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci 32:2410–2421
Pechnick RN, Zonis S, Wawrowsky K, Cosgayon R, Farrokhi C, Lacayo L, Chesnokova V (2011) Antidepressants stimulate hippocampal neurogenesis by inhibiting p21 expression in the subgranular zone of the hippocampus. PLoS One 6:e27290
Peluso MA, Glahn DC, Matsuo K, Monkul ES, Najt P, Zamarripa F, Li J, Lancaster JL, Fox PT, Gao JH, Soares JC (2009) Amygdala hyperactivation in untreated depressed individuals. Psychiatry Res 173:158–161
Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C, Spitzer G, Santarelli L, Scharf B, Hen R, Rosoklija G, Sackeim HA, Dwork AJ (2007) Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J Neurosci 27:4894–4901
Perreault M, Feng G, Will S, Gareski T, Kubasiak D, Marquette K, Vugmeyster Y, Unger TJ, Jones J, Qadri A, Hahm S, Sun Y, Rohde CM, Zwijnenberg R, Paulsen J, Gimeno RE (2013) Activation of TrkB with TAM-163 results in opposite effects on body weight in rodents and non-human primates. PLoS One 8:e62616
Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, Egan MF, Mattay VS, Hariri AR, Weinberger DR (2005) 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci 8:828–834
Pilar-Cuellar F, Vidal R, Diaz A, Castro E, dos Anjos S, Pascual-Brazo J, Linge R, Vargas V, Blanco H, Martinez-Villayandre B, Pazos A, Valdizan EM (2013) Neural plasticity and proliferation in the generation of antidepressant effects: hippocampal implication. Neural Plast 2013:537265
Pincus HA, Pettit AR (2001) The societal costs of chronic major depression. J Clin Psychiatry 62(Suppl 6):5–9
Pittenger C (2013) Disorders of memory and plasticity in psychiatric disease. Dialogues Clin Neurosci 15:455–463
Pittenger C, Duman RS (2008) Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33:88–109
Pizzagalli DA, Holmes AJ, Dillon DG, Goetz EL, Birk JL, Bogdan R, Dougherty DD, Iosifescu DV, Rauch SL, Fava M (2009) Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am J Psychiatry 166:702–710
Poduslo JF, Curran GL (1996) Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res 36:280–286
Polacchini A, Metelli G, Francavilla R, Baj G, Florean M, Mascaretti LG, Tongiorgi E (2015) A method for reproducible measurements of serum BDNF: comparison of the performance of six commercial assays. Sci Rep 5:17989
Polyakova M, Stuke K, Schuemberg K, Mueller K, Schoenknecht P, Schroeter ML (2015) BDNF as a biomarker for successful treatment of mood disorders: a systematic & quantitative meta-analysis. J Affect Disord 174:432–440
Price JL (1999) Prefrontal cortical networks related to visceral function and mood. Ann N Y Acad Sci 877:383–396
Qian MD, Zhang J, Tan XY, Wood A, Gill D, Cho S (2006) Novel agonist monoclonal antibodies activate TrkB receptors and demonstrate potent neurotrophic activities. J Neurosci 26:9394–9403
Radahmadi M, Hosseini N, Alaei H (2016) Effect of exercise, exercise withdrawal, and continued regular exercise on excitability and long-term potentiation in the dentate gyrus of hippocampus. Brain Res 1653:8–13
Rai KS, Hattiangady B, Shetty AK (2007) Enhanced production and dendritic growth of new dentate granule cells in the middle-aged hippocampus following intracerebroventricular FGF-2 infusions. Eur J Neurosci 26:1765–1779
Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A (2004) Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci 24:3471–3479
Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA (1999) Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry 45:1085–1098
Rantamaki T, Hendolin P, Kankaanpaa A, Mijatovic J, Piepponen P, Domenici E, Chao MV, Mannisto PT, Castren E (2007) Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology 32:2152–2162
Rantamaki T, Vesa L, Antila H, Di Lieto A, Tammela P, Schmitt A, Lesch KP, Rios M, Castren E (2011) Antidepressant drugs transactivate TrkB neurotrophin receptors in the adult rodent brain independently of BDNF and monoamine transporter blockade. PLoS One 6:e20567
Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA (2010) Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci 30:1739–1749
Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, Lesch KP (2006) Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry 11:514–522
Rosenstein JM, Mani N, Khaibullina A, Krum JM (2003) Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J Neurosci 23:11036–11044
Russo SJ, Nestler EJ (2013) The brain reward circuitry in mood disorders. Nat Rev Neurosci 14:609–625
Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, Agerman K, Haapasalo A, Nawa H, Aloyz R, Ernfors P, Castren E (2003) Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci 23:349–357
Sackeim HA, Decina P, Prohovnik I, Malitz S, Resor SR (1983) Anticonvulsant and antidepressant properties of electroconvulsive therapy: a proposed mechanism of action. Biol Psychiatry 18:1301–1310
Sahay A, Hen R (2007) Adult hippocampal neurogenesis in depression. Nat Neurosci 10:1110–1115
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301:805–809
Schiavon AP, Milani H, Romanini CV, Foresti ML, Castro OW, Garcia-Cairasco N, de Oliveira RM (2010) Imipramine enhances cell proliferation and decreases neurodegeneration in the hippocampus after transient global cerebral ischemia in rats. Neurosci Lett 470:43–48
Schilling C, Blum WF, Heuser I, Paslakis G, Wudy SA, Deuschle M (2011) Treatment with antidepressants increases insulin-like growth factor-I in cerebrospinal fluid. J Clin Psychopharmacol 31:390–392
Schlaepfer TE, Cohen MX, Frick C, Kosel M, Brodesser D, Axmacher N, Joe AY, Kreft M, Lenartz D, Sturm V (2008) Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology 33:368–377
Schloesser RJ, Orvoen S, Jimenez DV, Hardy NF, Maynard KR, Sukumar M, Manji HK, Gardier AM, David DJ, Martinowich K (2015) Antidepressant-like effects of electroconvulsive seizures require adult neurogenesis in a neuroendocrine model of depression. Brain Stimul 8:862–867
Schmaal L, Veltman DJ, van Erp TG, Samann PG, Frodl T et al (2016) Subcortical brain alterations in major depressive disorder: findings from the ENIGMA major depressive disorder working group. Mol Psychiatry 21:806–812
Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, Katz DM (2012) A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci 32:1803–1810
Schmuckermair C, Gaburro S, Sah A, Landgraf R, Sartori SB, Singewald N (2013) Behavioral and neurobiological effects of deep brain stimulation in a mouse model of high anxiety- and depression-like behavior. Neuropsychopharmacology 38:1234–1244
Schuch FB, Deslandes AC, Stubbs B, Gosmann NP, Silva CT, Fleck MP (2016) Neurobiological effects of exercise on major depressive disorder: a systematic review. Neurosci Biobehav Rev 61:1–11
Segi-Nishida E, Warner-Schmidt JL, Duman RS (2008) Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc Natl Acad Sci U S A 105:11352–11357
Sen S, Duman R, Sanacora G (2008) Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry 64:527–532
Settell ML, Testini P, Cho S, Lee JH, Blaha CD, Jo HJ, Lee KH, Min HK (2017) Functional circuitry effect of ventral tegmental area deep brain stimulation: imaging and neurochemical evidence of mesocortical and mesolimbic pathway modulation. Front Neurosci 11:104
Sharma AN, da Costa ESBF, Soares JC, Carvalho AF, Quevedo J (2016) Role of trophic factors GDNF, IGF-1 and VEGF in major depressive disorder: a comprehensive review of human studies. J Affect Disord 197:9–20
Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW (1996) Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A 93:3908–3913
Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA (2001) Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry 50:651–658
Sheline YI, Gado MH, Kraemer HC (2003) Untreated depression and hippocampal volume loss. Am J Psychiatry 160:1516–1518
Shigeno T, Mima T, Takakura K, Graham DI, Kato G, Hashimoto Y, Furukawa S (1991) Amelioration of delayed neuronal death in the hippocampus by nerve growth factor. J Neurosci 11:2914–2919
Shirayama Y, Chaki S (2006) Neurochemistry of the nucleus accumbens and its relevance to depression and antidepressant action in rodents. Curr Neuropharmacol 4:277–291
Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS (2002) Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 22:3251–3261
Shirayama Y, Yang C, Zhang JC, Ren Q, Yao W, Hashimoto K (2015) Alterations in brain-derived neurotrophic factor (BDNF) and its precursor proBDNF in the brain regions of a learned helplessness rat model and the antidepressant effects of a TrkB agonist and antagonist. Eur Neuropsychopharmacol 25:2449–2458
Siegle GJ, Konecky RO, Thase ME, Carter CS (2003) Relationships between amygdala volume and activity during emotional information processing tasks in depressed and never-depressed individuals: an fMRI investigation. Ann N Y Acad Sci 985:481–484
Silverman WF, Krum JM, Mani N, Rosenstein JM (1999) Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience 90:1529–1541
Sirianni RW, Olausson P, Chiu AS, Taylor JR, Saltzman WM (2010) The behavioral and biochemical effects of BDNF containing polymers implanted in the hippocampus of rats. Brain Res 1321:40–50
Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM (1997) Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav 56:131–137
Smoski MJ, Felder J, Bizzell J, Green SR, Ernst M, Lynch TR, Dichter GS (2009) fMRI of alterations in reward selection, anticipation, and feedback in major depressive disorder. J Affect Disord 118:69–78
Sofroniew MV, Howe CL, Mobley WC (2001) Nerve growth factor signaling, neuroprotection, and neural repair. Ann Rev Neurosci 24:1217–1281
Sondell M, Lundborg G, Kanje M (1999) Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci 19:5731–5740
Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, Chang EF, Gutierrez AJ, Kriegstein AR, Mathern GW, Oldham MC, Huang EJ, Garcia-Verdugo JM, Yang Z, Alvarez-Buylla A (2018) Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555:377–381
Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, Possnert G, Mash DC, Druid H, Frisen J (2013) Dynamics of hippocampal neurogenesis in adult humans. Cell 153:1219–1227
Stewart CA, Reid IC (2000) Repeated ECS and fluoxetine administration have equivalent effects on hippocampal synaptic plasticity. Psychopharmacology 148:217–223
Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, Uylings HB, Friedman L, Rajkowska G (2004) Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry 56:640–650
Stoy M, Schlagenhauf F, Sterzer P, Bermpohl F, Hagele C, Suchotzki K, Schmack K, Wrase J, Ricken R, Knutson B, Adli M, Bauer M, Heinz A, Strohle A (2012) Hyporeactivity of ventral striatum towards incentive stimuli in unmedicated depressed patients normalizes after treatment with escitalopram. J Psychopharmacol 26:677–688
Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest 111:1843–1851
Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA (2006) Vascular endothelial growth factor-B (VEGFB) stimulates neurogenesis: evidence from knockout mice and growth factor administration. Dev Biol 289:329–335
Sun R, Li N, Li T (2012) VEGF regulates antidepressant effects of lamotrigine. Eur Neuropsychopharmacol 22:424–430
Surget A, Tanti A, Leonardo ED, Laugeray A, Rainer Q, Touma C, Palme R, Griebel G, Ibarguen-Vargas Y, Hen R, Belzung C (2011) Antidepressants recruit new neurons to improve stress response regulation. Mol Psychiatry 16:1177–1188
Szczesny E, Slusarczyk J, Glombik K, Budziszewska B, Kubera M, Lason W, Basta-Kaim A (2013) Possible contribution of IGF-1 to depressive disorder. Pharmacol Rep 65:1622–1631
Tabassum H, Frey JU (2013) The effect of acute swim stress and training in the water maze on hippocampal synaptic activity as well as plasticity in the dentate gyrus of freely moving rats: revisiting swim-induced LTP reinforcement. Hippocampus 23:1291–1298
Takemoto T, Ishihara Y, Ishida A, Yamazaki T (2015) Neuroprotection elicited by nerve growth factor and brain-derived neurotrophic factor released from astrocytes in response to methylmercury. Environ Toxicol Pharmacol 40:199–205
Taliaz D, Stall N, Dar DE, Zangen A (2010) Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry 15:80–92
Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M (2006) Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmacol Rev 58:115–134
Tendulkar RD, Hunter GK, Reddy CA, Stephans KL, Ciezki JP, Abdel-Wahab M, Stephenson AJ, Klein EA, Mahadevan A, Kupelian PA (2013) Causes of mortality after dose-escalated radiation therapy and androgen deprivation for high-risk prostate cancer. Int J Radiat Oncol Biol Phys 87:94–99
Toki S, Okamoto Y, Onoda K, Matsumoto T, Yoshimura S, Kunisato Y, Okada G, Shishida K, Kobayakawa M, Fukumoto T, Machino A, Inagaki M, Yamawaki S (2014) Hippocampal activation during associative encoding of word pairs and its relation to symptomatic improvement in depression: a functional and volumetric MRI study. J Affect Disord 152-154:462–467
Tsai SJ (2007) TrkB partial agonists: potential treatment strategy for major depression. Med Hypotheses 68:674–676
Tsai TH, Chen SL, Chiang YH, Lin SZ, Ma HI, Kuo SW, Tsao YP (2000) Recombinant adeno-associated virus vector expressing glial cell line-derived neurotrophic factor reduces ischemia-induced damage. Exp Neurol 166:266–275
Turner CA, Akil H, Watson SJ, Evans SJ (2006) The fibroblast growth factor system and mood disorders. Biol Psychiatry 59:1128–1135
Turner CA, Calvo N, Frost DO, Akil H, Watson SJ (2008a) The fibroblast growth factor system is downregulated following social defeat. Neurosci Lett 430:147–150
Turner CA, Gula EL, Taylor LP, Watson SJ, Akil H (2008b) Antidepressant-like effects of intracerebroventricular FGF2 in rats. Brain Res 1224:63–68
Turner CA, Watson SJ, Akil H (2012) The fibroblast growth factor family: neuromodulation of affective behavior. Neuron 76:160–174
Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, Suzuki T, Miyata N, Watanabe Y (2011) Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69:359–372
Van Bokhoven P, Oomen CA, Hoogendijk WJ, Smit AB, Lucassen PJ, Spijker S (2011) Reduction in hippocampal neurogenesis after social defeat is long-lasting and responsive to late antidepressant treatment. Eur J Neurosci 33:1833–1840
Vassilopoulou K, Papathanasiou M, Michopoulos I, Boufidou F, Oulis P, Kelekis N, Rizos E, Nikolaou C, Pantelis C, Velakoulis D, Lykouras L (2013) A magnetic resonance imaging study of hippocampal, amygdala and subgenual prefrontal cortex volumes in major depression subtypes: melancholic versus psychotic depression. J Affect Disord 146:197–204
Veeraiah P, Noronha JM, Maitra S, Bagga P, Khandelwal N, Chakravarty S, Kumar A, Patel AB (2014) Dysfunctional glutamatergic and gamma-aminobutyric acidergic activities in prefrontal cortex of mice in social defeat model of depression. Biol Psychiatry 76:231–238
Victor TA, Furey ML, Fromm SJ, Ohman A, Drevets WC (2010) Relationship between amygdala responses to masked faces and mood state and treatment in major depressive disorder. Arch Gen Psychiatry 67:1128–1138
von Bohlen Und Halbach O, von Bohlen Und Halbach V (2018) BDNF effects on dendritic spine morphology and hippocampal function. Cell Tissue Res Feb 15
von Bohlen und Halbach O, Minichiello L, Unsicker K (2003) Haploinsufficiency in trkB and/or trkC neurotrophin receptors causes structural alterations in the aged hippocampus and amygdala. Eur J Neurosci 18:2319–2325
Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S (2002) Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 22:6810–6818
Vyas A, Jadhav S, Chattarji S (2006) Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience 143:387–393
Walsh JJ, Friedman AK, Sun H, Heller EA, Ku SM, Juarez B, Burnham VL, Mazei-Robison MS, Ferguson D, Golden SA, Koo JW, Chaudhury D, Christoffel DJ, Pomeranz L, Friedman JM, Russo SJ, Nestler EJ, Han MH (2014) Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci 17:27–29
Wang JW, David DJ, Monckton JE, Battaglia F, Hen R (2008) Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. J Neurosci 28:1374–1384
Wang L, Xia M, Li K, Zeng Y, Su Y, Dai W, Zhang Q, Jin Z, Mitchell PB, Yu X, He Y, Si T (2015a) The effects of antidepressant treatment on resting-state functional brain networks in patients with major depressive disorder. Hum Brain Mapp 36:768–778
Wang XL, Du MY, Chen TL, Chen ZQ, Huang XQ, Luo Y, Zhao YJ, Kumar P, Gong QY (2015b) Neural correlates during working memory processing in major depressive disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 56:101–108
Warner-Schmidt JL, Duman RS (2007) VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci U S A 104:4647–4652
Warner-Schmidt JL, Duman RS (2008) VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol 8:14–19
Watanabe Y, Gould E, McEwen BS (1992) Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res 588:341–345
Wellman CL (2001) Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol 49:245–253
Werner S, Unsicker K, von Bohlen Und Halbach O (2011) Fibroblast growth factor-2 deficiency causes defects in adult hippocampal neurogenesis, which are not rescued by exogenous fibroblast growth factor-2. J Neurosci Res 89:1605–1617
Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jonsson B, Olesen J, Allgulander C, Alonso J, Faravelli C, Fratiglioni L, Jennum P, Lieb R, Maercker A, van Os J, Preisig M, Salvador-Carulla L, Simon R, Steinhausen HC (2011) The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol 21:655–679
Wong AW, Giuffrida L, Wood R, Peckham H, Gonsalvez D, Murray SS, Hughes RA, Xiao J (2014) TDP6, a brain-derived neurotrophic factor-based trkB peptide mimetic, promotes oligodendrocyte myelination. Mol Cell Neurosci 63:132–140
Woodbury ME, Ikezu T (2014) Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration. J NeuroImmune Pharmacol 9:92–101
Woolley CS, Gould E, McEwen BS (1990) Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res 531:225–231
Xu L, Anwyl R, Rowan MJ (1997) Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387:497–500
Xu H, Chen Z, He J, Haimanot S, Li X, Dyck L, Li XM (2006) Synergetic effects of quetiapine and venlafaxine in preventing the chronic restraint stress-induced decrease in cell proliferation and BDNF expression in rat hippocampus. Hippocampus 16:551–559
Yadid G, Friedman A (2008) Dynamics of the dopaminergic system as a key component to the understanding of depression. In: Giuseppe Di Giovann VDM, Ennio E (eds) Prog brain res. Elsevier, pp 265–286
Yamada MK (2016) Angiogenesis in refractory depression: a possible phenotypic target to avoid the blood brain barrier. Drug Discov Ther 10:74–78
Yan T, Wang L, Kuang W, Xu J, Li S, Chen J, Yang Y (2014) Brain-derived neurotrophic factor Val66Met polymorphism association with antidepressant efficacy: a systematic review and meta-analysis. Asia Pac Psychiatry 6:241–251
Yang RJ, Mozhui K, Karlsson RM, Cameron HA, Williams RW, Holmes A (2008) Variation in mouse basolateral amygdala volume is associated with differences in stress reactivity and fear learning. Neuropsychopharmacology 33:2595–2604
Yasuda S, Yoshida M, Yamagata H, Iwanaga Y, Suenaga H, Ishikawa K, Nakano M, Okuyama S, Furukawa Y, Furukawa S, Ishikawa T (2014) Imipramine ameliorates pain-related negative emotion via induction of brain-derived neurotrophic factor. Cell Mol Neurobiol 34:1199–1208
Yau SY, Li A, Zhang ED, Christie BR, Xu A, Lee TM, So KF (2014) Sustained running in rats administered corticosterone prevents the development of depressive behaviors and enhances hippocampal neurogenesis and synaptic plasticity without increasing neurotrophic factor levels. Cell Transplant 23:481–492
Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR (2002) Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22:1532–1540
Youssef MM, Underwood MD, Huang YY, Hsiung SC, Liu Y, Simpson NR, Bakalian MJ, Rosoklija GB, Dwork AJ, Arango V, Mann JJ (2018) Association of BDNF Val66Met polymorphism and brain BDNF levels with major depression and suicide. Int J Neuropsychopharmacol 21:528–538
Zhang X, Zhang Z, Xie C, Xi G, Zhou H, Zhang Y, Sha W (2008) Effect of treatment on serum glial cell line-derived neurotrophic factor in depressed patients. Prog Neuro-Psychopharmacol Biol Psychiatry 32:886–890
Zhang WN, Chang SH, Guo LY, Zhang KL, Wang J (2013) The neural correlates of reward-related processing in major depressive disorder: a meta-analysis of functional magnetic resonance imaging studies. J Affect Disord 151:531–539
Zhang JC, Wu J, Fujita Y, Yao W, Ren Q, Yang C, Li S-x, Shirayama Y, Hashimoto K (2014a) Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int J Neuropsychopharmacol 18(4):pyu077
Zhang Y, Mao RR, Chen ZF, Tian M, Tong DL, Gao ZR, Huang M, Li X, Xu X, Zhou WH, Li CY, Wang J, Xu L, Qiu Z (2014b) Deep-brain magnetic stimulation promotes adult hippocampal neurogenesis and alleviates stress-related behaviors in mouse models for neuropsychiatric disorders. Mol Brain 7:11
Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K (2014c) 7,8-Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology 39:638–650
Zhang H, Dong W, Dang W, Quan W, Tian J, Chen R, Zhan S, Yu X (2015a) Near-infrared spectroscopy for examination of prefrontal activation during cognitive tasks in patients with major depressive disorder: a meta-analysis of observational studies. Psychiatry Clin Neurosci 69:22–33
Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, Han M, Hashimoto K (2015b) Comparison of ketamine, 7,8-dihydroxyflavone, and ANA-12 antidepressant effects in the social defeat stress model of depression. Psychopharmacology 232:4325–4335
Zhao HM, Liu XF, Mao XW, Chen CF (2004) Intranasal delivery of nerve growth factor to protect the central nervous system against acute cerebral infarction. Chin Med Sci J 19:257–261
Zhao M, Li D, Shimazu K, Zhou YX, Lu B, Deng CX (2007) Fibroblast growth factor receptor-1 is required for long-term potentiation, memory consolidation, and neurogenesis. Biol Psychiatry 62:381–390
Zhong X, Pu W, Yao S (2016) Functional alterations of fronto-limbic circuit and default mode network systems in first-episode, drug-naive patients with major depressive disorder: a meta-analysis of resting-state fMRI data. J Affect Disord 206:280–286
Zhou W, Li X, Huang D, Zhou W, Li T, Song W (2015) No significant effect of 7,8-dihydroxyflavone on APP processing and Alzheimer-associated phenotypes. Curr Alzheimer Res 12:47–52
Zhu Y, Jin K, Mao XO, Greenberg DA (2003) Vascular endothelial growth factor promotes proliferation of cortical neuron precursors by regulating E2F expression. FASEB J 17:186–193
Zhu W, Cheng S, Xu G, Ma M, Zhou Z, Liu D, Liu X (2011) Intranasal nerve growth factor enhances striatal neurogenesis in adult rats with focal cerebral ischemia. Drug Delivery 18:338–343
Zou YF, Ye DQ, Feng XL, Su H, Pan FM, Liao FF (2010) Meta-analysis of BDNF Val66Met polymorphism association with treatment response in patients with major depressive disorder. Eur Neuropsychopharmacol 20:535–544
Zung S, Souza-Duran FL, Soeiro-de-Souza MG, Uchida R, Bottino CM, Busatto GF, Vallada H (2016) The influence of lithium on hippocampal volume in elderly bipolar patients: a study using voxel-based morphometry. Transl Psychiatry 6:e846
Funding
Funding was provided by the Institut National de la Santé et de la Recherche Médicale (France), ANR (2011-BSV-017-01), and ISAO/LECMA 12530.
Author information
Authors and Affiliations
Contributions
ML and FB wrote this review paper. HWS, DH, GK, and LL edited it. All authors reviewed the paper and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Levy, M.J.F., Boulle, F., Steinbusch, H.W. et al. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology 235, 2195–2220 (2018). https://doi.org/10.1007/s00213-018-4950-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-018-4950-4