
Abstract
The aim of the present study was to reveal how different microbial communities evolve in diesel fuel/crude oil-contaminated environments under aerobic and microaerobic conditions. To investigate this question, aerobic and microaerobic bacterial enrichments amended with a diesel fuel/crude oil mixture were established and analysed. The representative aerobic enrichment community was dominated by Gammaproteobacteria (64.5%) with high an abundance of Betaproteobacteriales (36.5%), followed by Alphaproteobacteria (8.7%), Actinobacteria (5.6%), and Candidatus Saccharibacteria (4.5%). The most abundant alkane monooxygenase (alkB) genotypes in this enrichment could be linked to members of the genus Rhodococcus and to a novel Gammaproteobacterium, for which we generated a high-quality draft genome using genome-resolved metagenomics of the enrichment culture. Contrarily, in the microaerobic enrichment, Gammaproteobacteria (99%) overwhelmingly dominated the microbial community with a high abundance of the genera Acinetobacter (66.3%), Pseudomonas (11%) and Acidovorax (11%). Under microaerobic conditions, the vast majority of alkB gene sequences could be linked to Pseudomonas veronii. Consequently, results shed light on the fact that the excellent aliphatic hydrocarbon degrading Rhodococcus species favour clear aerobic conditions, while oxygen-limited conditions can facilitate the high abundance of Acinetobacter species in aliphatic hydrocarbon-contaminated subsurface environments.
Similar content being viewed by others


Introduction
Petroleum hydrocarbons are still among the most common environmental contaminants. Accidental spills and leaks occurring during transport and storage of crude oil and refine products cause the majority of these contaminations. In terrestrial ecosystems, petroleum hydrocarbon pollutions considerably threaten subsurface water reservoirs, which are often the primary sources of drinking water. Soil contaminations can also cause serious damage in the affected ecosystem since the pollutants can accumulate in animal and plant tissues causing death or mutations. Saturated hydrocarbons (alkanes) are quantitatively the most abundant fraction among all petroleum hydrocarbons (Mbadinga et al. 2011). Although alkanes show relatively low reactivity, several microorganisms can use them as a sole source of carbon and energy coupled with the reduction of different electron acceptors (Mbadinga et al. 2011). Nevertheless, the most rapid biodegradation of alkanes can be observed under aerobic conditions due to the fact that under anaerobic conditions the chemical inertness of the carbon–carbon bond retards the degradation (Singh et al. 2012). For this reason, in case of bioremediation projects aiming the cleanup of large-scale terrestrial contaminations (e.g. the cleanup of pollutions at former Soviet military bases in Central and Eastern Europe), aerobic conditions are preferred (Sarlos and Gondár 1995; Kabelitz et al. 2009). However, aeration of such large-scale contaminated sites is a considerably costly and energy-consuming process. In the light of this fact, it would be beneficial to uncover the diversity of those aerobic alkane-degrading microbes, which are adapted to hypoxic/microaerobic conditions. Still, only a handful of studies have dealt with this question to date.
The initial step of aerobic alkane degradation is the incorporation of molecular oxygen into the hydrocarbon molecule by the activity of oxygenases (Mbadinga et al. 2011). Depending on the chain-length of the alkane molecule, different enzymes play a role in their initial oxidation. The C1–C4 alkanes are initially oxidized by methane-monooxygenase-like enzymes, the C5–C16 alkanes by cytochrome P450 or integral membrane non-heme iron (alkB) enzymes, while in case of longer alkanes essentially unknown enzyme systems play a role (van Beilen and Funhoff 2007). Either way, the presence of oxygen is necessary for the activity of these enzymes. Nevertheless, in subsurface ecosystems, the availability of oxygen is often restricted even in pristine environments. On the other hand, hydrocarbons are potential carbon and energy sources for several aerobic microorganisms. Therefore, contamination increases the microbial metabolism and consequently, the aerobic microbial respiration and accompanying biological processes decrease the dissolved oxygen concentration in the contaminated environments (Táncsics et al. 2013). In case of aromatic hydrocarbon degradation, it was observed that distinctly different microbial communities take part in the process under aerobic and oxygen-limited conditions (Martirani-Von Abercron et al. 2017; Benedek et al. 2018). A group of extradiol dioxygenases (in subfamily I.2.C of extradiol dioxygenases) was shown to be adapted to low substrate concentrations, thus function under hypoxic conditions as well (Kukor and Olsen 1996). Recently, it has been shown that Rhodocyclaceae bacteria harboring subfamily I.2.C-type extradiol dioxygenases played a key role in the microaerobic degradation of toluene in a BTEX-contaminated groundwater sediment (Táncsics et al. 2018; Bradford et al. 2018). Recently, a couple of studies have reported on alkane degradation under oxygen-limited conditions or alkane-degrading microbial communities in oxygen-limited environments (e.g. oil reservoirs) (Cyplik et al. 2011; Liu et al. 2018; Tribelli et al. 2018). Still, further research is needed to better understand the effect of decreased oxygen availability on the structure of contaminant-degrading bacterial communities. Thus, the aim of the present study was to reveal how different bacterial communities evolve in diesel fuel/crude oil-contaminated environments under aerobic and microaerobic conditions. Accordingly, aerobic and microaerobic (~ 0.5 g mL−1 dissolved oxygen concentration) bacterial enrichments amended with a diesel fuel/crude oil mixture were established and investigated.
Materials and methods
Enrichment setup
To reveal the effect of oxygen limitation on the structure of oil-degrading bacterial communities, fully aerobic (~ 7–8 mg L−1 O2) and microaerobic (≤ 0.5 mg L−1 O2) enrichment microcosms were set up in triplicates in a defined freshwater medium as described earlier (Benedek et al. 2018). To gain inoculum for enrichment cultures, biofilm sample was collected from groundwater well of a deeply investigated gasoline contaminated site of Hungary, which has already been described in some of our earlier studies (Benedek et al. 2016, 2018). Subsequently, 1 g (wet weight) of biofilm was added to 99 mL of physiological saline solution, and 5 mL of this solution was then used to inoculate each of the first enrichments. Enrichment cultures were set up in 100-mL crimp-sealed serum bottles, which contained 45 mL of the freshwater medium, 5 mL inoculum and 20 ppm diesel fuel/crude oil mixture (3:2, v/v) as a sole source of carbon and energy. Both crude oil and pure (additive-free) diesel fuel were obtained from the Hungarian Oil and Gas Plc. (MOL Plc.) Before inoculation of the microaerobic enrichments, microcosms were sparged aseptically with N2/CO2 (80:20, v/v) for 10 min. After that, the desired volume of sterile air (0.2-μm-pore-size-filtered) was injected into the bottles through butyl-rubber septa to set the 0.5 mg L−1 O2 concentration. Dissolved oxygen concentration in the liquid phase of both aerobic and microaerobic enrichments was measured and monitored non-invasively using a Fibox 3 trace v3 fibre optic oxygen meter with PSt3 sensor spots (PreSens). In the case of oxygen depletion, supplementation took place to maintain either clear aerobic or microaerobic conditions. Enrichments were incubated in a rotary incubator (28 °C, 150 rpm) for 1 week, and then 5 mL of each enrichment was transferred to 45 mL of fresh enrichment medium. Transfers were repeated for 5 consecutive weeks.
DNA isolation and T-RFLP fingerprinting
To isolate DNA from the enrichments, the microbial biomasses were harvested from 45 mL of the enrichments by centrifugation at 2360 g at 4 °C for 10 min using a Rotanta 460 R centrifuge (Hettich), and DNA was isolated from the pellets using the DNeasy UltraClean Microbial Kit (Qiagen) according to the instructions of the manufacturer. To isolate DNA from the biofilm inoculum, the NucleoSpin Soil Kit (Macherey–Nagel) was used by following the instructions of the manufacturer. For 16S rDNA-based community profiling, the VIC-labeled amplicons were generated using 27f-VIC (5′-AGA GTT TGA TCM TGG CTC AG-3′) and 1492r primers (5′-TACGGYTACCTTGTTACGACTT-3′), as described earlier (Benedek et al. 2016). For alkB-based T-RFLP, the VIC-labeled amplicons were generated using forward primer alkB 1f_deg-VIC (5′-AAY ACI GCI CAY GAR CTI GGI CAY AA-3′) and reverse primer alkB 1r_deg (5′-GCR TGR TGR TCI GAR TGI CGY TG-3′) developed by Kloos et al. (2006) and slightly modified by Pérez-de-Mora et al. (2011). The PCR cycle program was the same as it was described by Giebler et al. (2014). The PCR reaction mixture (final volume of 50 µL) included: 5 µL of 10 × DreamTaq Buffer (Thermo Fisher Scientific), 0.2 mM of each of the four dNTP, 0.3 μM of each primer, 0.25 µL of a 5-U μL−1 DreamTaq DNA Polymerase solution (Thermo Fisher Scientific), 20 ng template DNA and autoclaved MilliQ water up to 50 µL. All amplifications were performed in a ProFlex PCR System (Life Technologies). All amplification products were checked by electrophoresis on 1% agarose gels stained with ethidium bromide.
To gain T-RFLP electropherograms, the VIC-labelled 16S rDNA amplicons were digested with RsaI (Thermo Fisher Scientific) while the alkB amplicons were digested with HPyCH4V (New England BioLabs), as described earlier (Révész et al. 2006). After ethanol precipitation, fragments were separated on a Model 3130 Genetic Analyzer (Applied Biosystems), while primary evaluation of electropherograms was performed using GeneMapper 4.0 software (Applied Biosystems). T-RFLP data were handled as described earlier (Farkas et al. 2017). Cluster analysis (Jaccard and Bray–Curtis methods) of the T-RFLP electropherograms was performed using the PAST software package.
16S rDNA amplicon sequencing and data handling
To assess precisely the bacterial community composition of the initial biofilm sample as well as of the aerobic and microaerobic enrichments of the 5th week Illumina 16S rDNA amplicon sequencing was carried out. The variable V3 and V4 region of the 16S rDNA was amplified using the primers recommended by Klindworth et al. (2013). PCR amplification was carried out using the KAPA HiFi HotStart Ready Mix (KAPA Biosystems) according to the 16S metagenomics sequencing library preparation guide of Illumina. Paired-end fragment reads were generated on an Illumina MiSeq sequencer using MiSeq Reagent Kit v3 (600-cycle). Read numbers were the following: 195 745 for AER2, 174 767 for MIK1 and 222 988 for initial biofilm sample (BF). Primary data analysis (base-calling) was carried out with Bbcl2fastq^ software (v2.17.1.14, Illumina). Reads were quality and length trimmed in CLC Genomics Workbench Tool 9.5.1 using an error probability of 0.05 (Q13) and a minimum length of 50 nucleotides as threshold. Trimmed sequences were analysed, BLASTN alignment was performed using the most recent SILVA rRNA database. Downstream taxonomical analysis and visualization of the retained reads were performed by the MEGAN6 software (Huson et al. 2007). The 16S Percent Identity Filter item was used as an additional filter for assigning the 16S amplicon reads to a specific taxonomic level. The filter was set to a minimum of 97% for the genus level identification. To avoid sample size effect before comparisons and further analysis, the number of reads was normalized. The 16S rDNA amplicon sequence reads (raw data in FASTQ format) were deposited in the SRA under the study accession number SRP159162 (BioProject PRJNA488537).
Cloning of alkB amplicons, Sanger-sequencing and phylogenetic analysis
The alkB PCR amplicons generated with the primer set alkB 1f_deg/alkB 1r_deg were cloned and sequenced (Táncsics et al. 2012) from enrichment cultures AER2 and MIK1 (48 clones in case of each enrichment). The alkB sequences of each clone library were manually grouped into operational protein units (OPU) by applying a cutoff value of 0.03. Terminal restriction fragments (T-RFs) predicted in silico for representative clones of each of the OPUs were verified in vitro. Maximum-likelihood phylogenetic tree was reconstructed based on the deduced amino acid sequences using MEGA ver. 7.0 (Kumar et al. 2016). For tree reconstruction, the Jones–Taylor–Thornton model was used, gaps were treated by complete deletion, the number of bootstrap replications was set to 1000, while the substitution rates were set to be the same among the sites and lineages. All of the clone sequences obtained in the present study were deposited with GenBank and can be found under the accession numbers MK390373–MK390468.
Genome-resolved metagenomics and phylogenomics of organism with novel alkB
Metagenome DNA quality and integrity was analysed using an Agilent 2200 Tapestation system. Paired-end fragment reads (2 × 250 nucleotides) were generated using the MiSeq Reagent Kit v2 (500-cycles) with an Illumina MiSeq sequencer. The total cluster number was 8 890 097. Primary data analysis (base-calling) was carried out with “bcl2fastq” software (v.2.17.1.14, Illumina). Quality check of raw reads was performed using BBDuk (v. 37.09; https://sourceforge.net/projects/bbmap/) followed by SICKLE (https://github.com/najoshi/sickle) and subsequently assembled and scaffolded using metaSPADES (v 3.13; Nurk et al. 2017). Genes of scaffolds larger than 1 kb were predicted with prodigal in the meta mode (Hyatt et al. 2010) and annotated using diamond blast (Buchfink et al. 2015) against UniRef100 (Suzek et al. 2007). 16S rRNA gene sequences were predicted as described previously (Brown et al. 2015) and annotated against SILVA 132 (Quast et al. 2013). The taxonomy of the annotated genes was used to calculate a consensus taxonomy for each scaffold (Schulze-Makuch et al. 2018). Coverage of scaffolds was calculated with bowtie2 (mode-sensitive; Langmead and Salzberg 2012).
The novel alkB gene from T-RFLP analysis was used to identify the scaffold carrying the respective complete gene (100% identity) and the genome was binned using emergent self-organizing maps (ESOM) based on tetranucleotide frequencies (Dick et al. 2009). The obtained bin was cleaned using GC, coverage and taxonomy of scaffolds. Completeness estimation was based on the presence of 51 bacterial single copy genes.
For community analysis, scaffolds carrying an annotated ribosomal protein S3 (rpS3) were extracted and the coverage of the respective scaffolds was used to calculate a rank abundance curve.
For calculating a phylogenetic tree, 16 ribosomal proteins (L2, L3, L4, L5, L6, L14, L16, L18, L22, L24, S3, S8, S10, S17 and S19) were extracted of the target organism’s genome (Hug et al. 2016), combined with previous datasets as described earlier (Probst et al. 2018) and aligned using MUSCLE (v3.8.31, Edgar 2004). To remove ambiguously aligned terminal regions, the aligned sequences were end-trimmed in the Geneious software (11.0.5). The resulting 16 protein alignments were then concatenated and all organisms, whose sequences spanned less than 50% of the AA positions in the concatenated alignment, were removed. The remaining sequences were used to build a tree using a maximum-likelihood approximation (Price et al. 2010). Visualization was performed in Dendroscope (v. 3.5.10). Average nucleotide identity (ANI) was calculated using EzBioCloud ANI calculator (Yoon et al. 2017).
The metagenome sequence reads (raw data in FASTQ format) were deposited in the SRA under the accession number SRR9332771. The reconstructed genome can be accessed under accession number VIAD00000000 (BioSample accession number SAMN12125306).
Results and discussion
Microbial community compositions as revealed by 16S rDNA amplicon sequencing
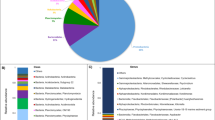
To reveal how different microbial communities evolve in petroleum hydrocarbon contaminated environments under aerobic and microaerobic conditions, an enrichment approach was used, which proved to be a powerful tool earlier to study the primary effect of oxygen limitation on aromatic hydrocarbon-degrading microbial community structure (Benedek et al. 2018). To establish the enrichment cultures, a biofilm sample was used as inoculum. The biofilm developed in a groundwater monitoring well of a gasoline contaminated site of Hungary, on the stainless steel surface of a submersible pump (Benedek et al. 2016). It was shown earlier that the microbial community of this biofilm has considerably high diversity and was used as inoculum in previous enrichment studies successfully (Benedek et al. 2016, 2018). The new biofilm material contained a microbial community overwhelmingly dominated by Betaproteobacteriales of the Gammaproteobacteria. The most abundant genus was Sulfuritalea (16% of total 16S rDNA sequence reads), followed by Azoarcus (4.8%), Acidovorax (2.6%), Simplicispira (0.9%), Thiobacillus (0.9%), Hydrogenophaga (0.7%), Thauera (0.6%), Zoogloea (0.6%) and Rhodoferax (0.5%) (Fig. 1). Although Alphaproteobacteria and Pseudomonadales-related species of the Gammaproteobacteria were also relatively abundant in the community, the typical petroleum hydrocarbon degraders such as the members of the genera Novosphingobium, Sphingobium and Pseudomonas were detected in considerably low amount (typically lower than 0.05% abundance). Overall, the community composition of the biofilm was highly similar to that of usually observed in petroleum hydrocarbon-contaminated subsurface environments in which hypoxic (oxygen-limited) and/or nitrate-reducing conditions prevail.

Class and genus level bacterial community structure of the biofilm inoculum, the AER2 aerobic enrichment and, the MIK1 microaerobic enrichment as revealed by Illumina paired-end 16S rDNA amplicon sequencing. All taxa contributing more than 1% abundance were depicted
Enrichment microbial communities were first subjected to 16S rDNA-based T-RFLP analysis to evaluate the level of similarity between the triplicate enrichments. Cluster analysis of the T-RFLP electropherograms by the Jaccard algorithm showed that microbial communities of clear aerobic (AER) and microaerobic (MIK) enrichments were markedly different and clustered separately (Online Resource 1). The cluster that was prepared using the Bray–Curtis algorithm, which takes into account the abundance of T-RFs as well, showed that microaerobic communities shared a high level of similarity. On the other hand, in case of clear aerobic enrichments only AER2 and AER3 communities were similar, while AER1 showed a bit outlying nature (Online Resource 1). Thus, microbial communities of clear aerobic enrichments showed higher variability than that of the microaerobic enrichments. This phenomenon can be explained by the fact that the aerobic enrichments were subjected to much larger disturbance due to the larger scale aeration. Based on the 16S rDNA T-RFLP results, the microbial communities of enrichments AER2 and MIK1 were analysed by 16S rDNA amplicon sequencing, using Illumina Next-Generation Sequencing Technology (Illumina NGS), which is a suitable tool to gain in-depth knowledge about phylogeny of contaminant-degrading microbial communities (Caporaso et al. 2012; Ławniczak et al. 2016; Szczepaniak et al. 2016).
The microbial community of the aerobic enrichment AER2 was dominated by Betaproteobacteriales (36.5% of sequence reads). The most abundant Betaproteobacteriales-related genus was Polaromonas (14%), followed by Acidovorax (6.7%) and Janthinobacterium (4%) (Fig. 1, Table 1). Several members of the genus Polaromonas have already been reported to be able to degrade petroleum hydrocarbons such as benzene and toluene (Sun et al. 2010; Xie et al. 2011) and their dominance was reported in a benzene contaminated aquifer (Aburto et al. 2009). Similarly, members of the genus Acidovorax are often reported as dominant members of petroleum hydrocarbon degrading microbial communities (Popp et al. 2006; Daghio et al. 2015), and especially the degradation of phenanthrene and chlorobenzene by Acidovorax strains is well documented (Nestler et al. 2007; Singleton et al. 2018). Members of the genus Janthinobacterium are often detected in aliphatic hydrocarbon-contaminated environments, but they are primarily considered petroleum hydrocarbon tolerant bacteria rather than degrading ones (Giebler et al. 2013; Pham et al. 2014). At the genus level, Pseudomonas proved to be the second most abundant group (10.5%). The role of this genus in petroleum hydrocarbon degradation is well known. Among Alphaproteobacteria (8.5% of sequence reads), the genus Sphingobium has to be noted since several members of this genus are involved in petroleum hydrocarbon degradation under aerobic conditions (Révész et al. 2018). Another well-known genus with alkane-degrading species is the Rhodococcus (class of Actinobacteria) which members were prominent representatives of the aerobic enrichment community. This was expected since alkB genes can be found in almost all members of this genus (Táncsics et al. 2015) and especially some species of the so-called “erythropolis” clade are highly efficient aerobic alkane degraders. It is supposed that members of this clade are generalist species of petroleum hydrocarbon contaminated soils, since they are the most commonly detectable hydrocarbon-degrading bacteria in contaminated soils, regardless of soil type or alkane profile of the contaminating petroleum hydrocarbon (e.g. crude oil, diesel or kerosene) (Hamamura et al. 2006, 2013). Some of them encode three or more different alkB genes (Whyte et al. 2002), and can produce biosurfactants (typically glycolipids) (Lang and Philp 1998). Interestingly, Candidatus Saccharibacteria-related 16S rDNA sequences were also found in relatively high abundance (4.5%). This group was formerly known as Candidate Division TM7, which members can be found in a wide variety of habitats. Until recently, this group has been known solely through 16S rDNA sequences and due to this it is often referred to as “microbial dark matter” (He et al. 2015). Nevertheless, subdivision 3 of Saccharibacteria (TM7-3) was found to be abundant in diesel fuel-contaminated soil (Winsley et al. 2014) and SIP studies revealed the possible role of TM7 bacteria in aerobic degradation of toluene and benzene (Luo et al. 2009; Xie et al. 2011). Last but not least, two genera of the Flavobacteriia, the Chryseobacterium and Flavobacterium have to be mentioned as notable members of the aerobic enrichment. Both genera contains species which were isolated from petroleum hydrocarbon-contaminated environments (Szoboszlay et al. 2008; Chaudhary and Kim 2018) and especially members of the genus Flavobacterium can have a remarkable role in the degradation of petroleum hydrocarbons in contaminated environments (Rahman et al. 2002).
The microbial community of the microaerobic enrichment MIK1 was considerably simpler than that of the aerobic enrichment. It was overwhelmingly dominated by Gammaproteobacteria (98.8%) with high abundance of Pseudomonadales-related species, while Actinobacteria, Flavobacteriia and Alphaproteobacteria were only minor members of the community (< 0.5% abundance) (Fig. 1). At the genus level, Acinetobacter-related sequence reads were the most abundant (66.3%). Members of this genus are frequently reported as prominent alkane and aromatic hydrocarbon degraders under aerobic conditions (Lal and Khanna 1996; Di Cello et al. 1997; Margesin et al. 2003; Czarny et al. 2019) and some of them are adapted to the degradation of long-chain non-branched alkanes (Tani et al. 2001; Rojo 2009). Acinetobacter species can be considerably abundant in deep subsurface oil reservoirs, and most probably they are introduced into this environment by injection water (Orphan et al. 2000; Zhao et al. 2012). Oil reservoirs, especially deep subsurface ones are considered oxygen-limited or anaerobic, thus provide excellent environment for studying anaerobic alkane degradation. Nevertheless, shallow subsurface oil reservoirs can receive oxygen by surface recharge of meteoric waters (Jones et al. 2007), while deep subsurface oil reservoirs receive oxygen with the injection water during production, thus niches with trace amounts of oxygen may be present in these environments. Due to these facts, it was assumed that biodegradation of petroleum hydrocarbons in subsurface oil reservoirs is a joint achievement of aerobic and anaerobic microbes (da Cruz et al. 2011). Overall, it can be speculated that members of the genus Acinetobacter became dominant in the microaerobic enrichments due to their possible ability to cope with oxygen limitation and outcompeted other aerobic alkane degraders. The second most abundant Gammaproteobacterial genus was the Pseudomonas (11%). Recently, it has been reported by Tribelli et al. (2018) that in P. extremaustralis strain 14–3T expression of genes involved in alkane degradation (e.g. the alkB gene itself) were up-regulated under low-oxygen conditions and the strain was able to use diesel fuel as sole source of carbon under microaerophilic conditions. The possible genetic background of this phenomenon was revealed, and it was shown that similar mechanism may be present in other alkane-degrading Pseudomonas strains (Tribelli et al. 2018) and can be important degraders in oxygen-limited petroleum hydrocarbon-contaminated environments. Betaproteobacteriales-related sequence reads belonged mainly to the genera Acidovorax (11%), Variovorax (1.9%) and Simplicispira (1.5%). Some members of the genera Acidovorax and Variovorax are known aromatic hydrocarbon degraders (Sydow et al. 2016; Posman et al. 2017; Singleton et al. 2018). Moreover, Acidovorax-related bacteria can be highly abundant in BTEX-degrading enrichment cultures under hypoxic conditions (Benedek et al. 2018). Although members of the genus Simplicispira can be isolated from petroleum hydrocarbon-contaminated subsurface environments (Benedek et al. 2016), and their high abundance was shown in benzene-degrading enrichment cultures under nitrate-reducing conditions (Keller et al. 2018), their role in these environments is still unclear.
The predictable role in petroleum hydrocarbon degradation of the above mentioned, most notable genera (containing cultivable species and showing > 1% abundance) detected either in the aerobic or microaerobic enrichments is summarized in Table 1.
Diversity and phylogenetic analysis of alkB genes in the enrichment cultures
To reveal and compare alkB gene diversity in the aerobic and microaerobic enrichment cultures a T-RFLP assay was used, described by Giebler et al. (2014) as the most appropriate T-RFLP protocol to analyze alkane-degrading bacterial communities. Results of the cluster analysis of the alkB gene T-RFLP electropherograms were in accordance with results of the 16S rDNA-based analysis (Online Resource 2). Accordingly, the alkB T-RFLP patterns of the parallel microaerobic enrichments were highly similar and could be characterized with the 523-bp long T-RF (Fig. 2). The alkB diversity of the aerobic enrichments showed higher variability than that of the microaerobic enrichments. In the enrichments AER2 and AER3, the 89-bp long T-RF was the characteristic T-RF, but it was missing in case of AER1. To link sequence information to the detected T-RFs, alkB clone libraries were generated and analysed in case of enrichments AER2 and MIK1.

Abundance of alkB gene T-RFs in triplicates of the enrichment microcosms at the fifth week of the enrichment procedure. For clone library-based alkB gene diversity analysis and for the identification of T-RFs samples, AER2 and MIK1 were used
In the alkB clone library of the aerobic enrichment AER2, the sequences could be divided into six OPUs (Fig. 3). In case of the largest cluster (AER-OPU 1, 63% of clones), sequences showed considerably low similarity to known alkB genotypes, therefore, it was not possible to link them to any cultivated bacterium. The most similar nucleotide sequences (with a similarity level of 76–80%) were retrieved from Agitococcus lubricus DSM 5822T, and from environmental samples, e.g. crude oil-contaminated seawater (Wang et al. 2014) and from soil samples of the sub-Antarctic Macquarie Island (Powell et al. 2010). T-RFLP analysis of individual clones showed that these sequences could be characterized with the 89-bp long T-RF. Most of the other clusters (AER-OPU 2, 3, 4 and 6) contained Pseudomonas (P. putida and P. chlororaphis subsp. aureofaciens) and Rhodococcus-related alkB gene sequences, except AER-OPU 5 which contained sequences most closely related to AER-OPU 1.

Maximum-likelihood tree showing the phylogenetic position of alkB amino acid sequences retrieved from the aerobic enrichment AER2 (red color) and the microaerobic enrichment MIK1 (blue color). Bootstrap values from 1000 resamplings are indicated with black circles for values of 95–100% and gray circles for values between 50 and 94%. OPUs were determined using a distance cutoff of 0.03 (97% sequence similarity). The tree was rooted with a xylene monooxygenase (hydroxylase component) amino acid sequence of TOL plasmid pDK1 (Pseudomonas putida) (color figure online)
In the alkB clone library of the microaerobic enrichment MIK1, the sequences could be divided into five OPUs (Fig. 3). The vast majority of sequences (71% of clones, 238/523 bp T-RF) belonged to MIK-OPU 1, and showed high similarity (98.4% at nucleotide level) with alkB gene sequences of Pseudomonas veronii strains. The closest relative of P. veronii is P. extremaustralis, which bacterium is capable of degrading aliphatic hydrocarbons under microaerophilic conditions (Tribelli et al. 2018). Although P. veronii strains have already been reported to degrade BTEX-compounds and alkyl methyl ketones, still little is known about their role in alkane degradation (Morales et al. 2016; Onaca et al. 2007). Our results indicate that P. veronii, similarly to P. extremaustralis may also prefer microaerobic conditions for growth on alkanes. Despite to the fact that Acinetobacter species were considerably more abundant in enrichment MIK1 than members of the genus Pseudomonas, Acinetobacter-related alkB sequences were found only in low amount (MIK-OPU 2, 10% of clones, 376-bp T-RF) and showed the highest similarity (97.8% at nucleotide level) with alkB gene of A. calcoaceticus strain CA16. However, it has to be noted here that the PCR primers used in the present study for T-RFLP and cloning purposes do not amplify all types of alkB genes encoded by Acinetobacter species (Jurelevicius et al. 2013). Moreover, several alkane-degrading Acinetobacter strains harbor alkM-type alkane hydroxylases, rather than alkB-type ones. Clone sequences belonging to MIK-OPU 3 and 4 showed high similarity to Pseudomonas-related alkB sequences (10% and 6% of clone sequences, 548 and 238-bp T-RF). It has to be noted here that clone sequences of MIK-OPU 3 and AER-OPU 2 were identical. The least abundant MIK-OPU 5 contained a single-clone sequence showing similarity to Rhodococcus-related alkB gene sequences (and to sequences of AER-OPU 3).
Metagenome sequencing of enrichment AER2: phylogenetic affiliation of the abundant alkB genotype by genome binning
Among the alkB genotypes recovered in this study, only those remained unaffiliated, which belonged to AER-OPU 1 and AER-OPU 5. Since AER-OPU 1 contained the most abundant alkB gene sequences in case of the aerobic enrichment AER2, genome-resolved metagenomics was used to link them to a bacterial lineage. This novel and abundant alkB gene was successfully reconstructed in metagenomic assemblies of the aerobic enrichment culture AER2. The gene was on a scaffold with the length of 554 kb, which was confidentially classified as Gammaproteobacterial based on the consensus taxonomy of 505 predicted and annotated proteins. Using tetranucleotide frequencies, which work reliably well on long scaffolds as given here, we generated an ESOM map (Dick et al. 2009), which is depicted in Fig. 4a. The manually curated genome bin had a GC content of 44%, an average coverage of 116, a completeness of > 99% and 0% detectable contamination. Based on the ribosomal protein S3-based rank abundance curve from the metagenomic data, we provide evidence that the genome belonged to the third most abundant organism in the sample (Fig. 4b). Since no 16S rRNA gene sequence could be binned along with the genome, we investigated the ribosomal proteins of the binned genome. Based on blastp against NR, the organism is closely related to Agitococcus lubricus (Franzmann and Skerman 1981), a Firmicute based on NCBI taxonomy, which did not agree with the predicted taxonomy of the bin. To investigate correct placement of the organism and Agitococcus lubricus on the tree of life, we generated a phylogenetic tree based on concatenated ribosomal proteins, placing both organisms’ genomes into the clade of Moraxellaceae of the Gammaproteobacteria (Fig. 4c). With an ANI of 74.36% over a 1.1 Mbps, the recovered Gammaproteobacteria genome is closely related to A. lubricus, however, the organism represents at least a novel species (Kim et al. 2014). Agitococcus lubricus, which was misclassified in the NCBI taxonomy, grows on Tween (80, 40 and 20) (Franzmann and Skerman 1981), a complex hydrocarbon, suggesting catabolic similarity between the two organisms. Analysis of the alkB gene containing cluster revealed that in upstream position to the alkB gene an AraC family transcriptional regulator coding gene can be found (in opposite orientation) (Fig. 5), which had been designated earlier as alkR gene in case of Acinetobacter sp. strain ADP1. It was found that this gene also plays crucial role in alkane degradation (Ratajczak et al. 1998). Consequently, the combined metagenome-cultivation approach revealed a novel organism, possibly capable of alkane degradation.

Genome-resolved metagenomics for identification of the genome, to which the novel alkB gene from TRFLP analysis belongs. a ESOM of sample AER2, highlighting in red the bin carrying the scaffold with the alkB gene, named Gammaproteobacteria_44_116 based on its taxonomy, GC, and coverage in the metagenome. Escherichia coli K12 and Streptomyces griseus NBRC13350, which were used as controls for constructing the ESOM, are also shown. b Rank-abundance curve of sample AER2 based on ribosomal protein S3. Red column corresponds to Gammaproteobacteria_44_116. c Phylogenetic tree for Gammaproteobacteria_44_116, constructed using 16 concatenated ribosomal proteins. The shown tree is an excerpt of a tree encompassing 3618 genomes, which is provided Online Resource 3. For details, please see methods (color figure online)

Schematic representation of the alkB gene-containing cluster located in the genome of Gammaproteobacteria_44_116. ORF1 tRNA (cytidine(34)-2′-O)-methyltransferase, ORF2 alpha/beta fold hydrolase, ORF3 alkane-1 monooxygenase, ORF4 AraC family transcriptional regulator, ORF5 short subunit dehydrogenase, ORF6 oxygen-independent coproporphyrinogen III oxidase. Arrows indicate the orientation of the ORFs
Conclusion and outlook
Overall, the observed differences between the bacterial community compositions of clear aerobic and microaerobic enrichments can be instructive regarding bioremediation of crude oil/diesel fuel-contaminated environments. Results have shown that members of the genus Rhodococcus were abundant members of the enrichment communities only under clear aerobic conditions. This observation is important, since rhodococci are frequently used to treat hydrocarbon-contaminated sites due to their enormous metabolic diversity (Kuyukina and Ivshina 2010; Kis et al. 2017). On the other hand, microaerobic conditions caused the overwhelming dominance of Acinetobacter and Pseudomonas genera-related bacteria. Based on the alkB gene diversity analyses, it was also observable that among Pseudomonas-related bacteria, the P. veronii-lineage was dominant in the microaerobic enrichment cultures. This result together with the fact that the closely related P. extremaustralis preferably degrades alkanes under microaerobic conditions enable to assume that a certain group of Pseudomonas species is adapted to these conditions and may have an important role in alkane degradation in subsurface ecosystems. Last but not least, a yet unknown but abundant alkB genotype was recovered from the aerobic enrichment and was linked to a yet uncultivated member of the family Moraxellaceae by genome binning. Thus, the present study provides new evidence that the known diversity of alkane-degrading bacteria is still incomplete.
References
Aburto A, Peimbert M (2011) Degradation of a benzene–toluene mixture by hydrocarbon-adapted bacterial communities. Ann Microbiol 61:553–562
Aburto A, Fahy A, Coulon F, Lethbridge G, Timmis KN, Ball AS, McGenity TJ (2009) Mixed aerobic and anaerobic microbial communities in benzene-contaminated groundwater. J Appl Microbiol 106:317–328
Benedek T, Táncsics A, Szabó I, Farkas M, Szoboszlay S, Fábián K, Maróti G, Kriszt B (2016) Polyphasic analysis of an Azoarcus-Leptothrix-dominated bacterial biofilm developed on a stainless steel surface in a gasoline-contaminated hypoxic groundwater. Environ Sci Pollut Res Int 23:9019–9035
Benedek T, Szentgyörgyi F, Szabó I, Kriszt B, Révész F, Radó J, Maróti G, Táncsics A (2018) Aerobic and oxygen-limited enrichment of BTEX-degrading biofilm bacteria: dominance of Malikia versus Acidovorax species. Environ Sci Pollut Res Int 25:32178–32195
Bradford LM, Vestergaard G, Táncsics A, Zhu B, Schloter M, Lueders T (2018) Transcriptome-stable isotope probing provides targeted functional and taxonomic insights into microaerobic pollutant-degrading aquifer microbiota. Front Microbiol 9:2696
Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, Wilkins MJ, Wrighton KC, Williams KH, Banfield JF (2015) Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523:208–2011
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60
Caporaso JG, Lauber CL, Walters WA et al (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Chaudhary DK, Kim J (2018) Flavobacterium naphthae sp. nov., isolated from oil-contaminated soil. Int J Syst Evol Microbiol 68:305–309
Chaudhary DK, Jeong SW, Kim J (2017) Sphingobium naphthae sp. nov., with the ability to degrade aliphatic hydrocarbons, isolated from oil-contaminated soil. Int J Syst Evol Microbiol 67:2986–2993
Cyplik P, Schmidt M, Szulc A, Marecik R, Lisiecki P, Heipieper HJ, Owsianiak M, Vainshtein M, Chrzanowski Ł (2011) Relative quantitative PCR to assess bacterial community dynamics during biodegradation of diesel and biodiesel fuels under various aeration conditions. Bioresour Technol 102:4347–4352
Czarny J, Staninska-Pięta J, Piotrowska-Cyplik A, Juzwa W, Wolniewicz A, Marecik R, Ławniczak Ł, Chrzanowski Ł (2019) Acinetobacter sp. as the key player in diesel oil degrading community exposed to PAHs and heavy metals. J Hazard Mater 383:121168. https://doi.org/10.1016/j.jhazmat.2019.121168
da Cruz GF, de Vasconcellos SP, Angolini CFF, Dellagnezze BM, Garcia INS, de Oliveira VM, dos Santos Neto EV, Marsaioli AJ (2011) Could petroleum biodegradation be a joint achievement of aerobic and anaerobic microorganisms in deep sea reservoirs? AMB Express 1:47
Daghio M, Tatangelo V, Franzetti A, Gandolfi I, Papacchini M, Careghini A, Sezenna E, Saponaro S, Bestetti G (2015) Hydrocarbon degrading microbial communities in bench scale aerobic biobarriers for gasoline contaminated groundwater treatment. Chemosphere 130:34–39
Di Cello F, Pepi M, Baldi F, Fani R (1997) Molecular characterization of an n-alkane-degrading bacterial community and identification of a new species, Acinetobacter venetianus. Res Microbiol 148:237–249
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF (2009) Community-wide analysis of microbial genome sequence signatures. Genome Biol 10:R85
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Farkas M, Szoboszlay S, Benedek T, Révész F, Veres PG, Kriszt B, Táncsics A (2017) Enrichment of dissimilatory Fe(III)-reducing bacteria from groundwater of the Siklós BTEX-contaminated site (Hungary). Folia Microbiol 62:63–71
Felföldi T, Székely AJ, Gorál R, Barkács K, Scheirich G, András J, Rácz A, Márialigeti K (2010) Polyphasic bacterial community analysis of an aerobic activated sludge removing phenols and thiocyanate from coke plant effluent. Bioresour Technol 101:3406–3414
Franzmann PD, Skerman VBD (1981) Agitococcus lubricus gen. nov. sp. nov., a lipolytic, twitching coccus from freshwater. Int J Syst Bacteriol 31:177–183
Gałązka A, Król M, Perzyński A (2012) The efficiency of rhizosphere bioremediation with Azospirillum sp. and Pseudomonas stutzeri in soils freshly contaminated with PAHs and diesel fuel. Pol J Environ Stud 21:345–353
Giebler J, Wick LY, Chatzinotas A, Harms H (2013) Alkane-degrading bacteria at the soil-litter interface: comparing isolates with T-RFLP-based community profiles. FEMS Microbiol Ecol 86:45–58
Giebler J, Wick LY, Harms H, Chatzinotas A (2014) Evaluating T-RFLP protocols to sensitively analyze the genetic diversity and community changes of soil alkane degrading bacteria. Eur J Soil Biol 65:107–113
Hamamura N, Olson SH, Ward DM, Inskeep WP (2006) Microbial population dynamics associated with crude-oil biodegradation in diverse soils. Appl Environ Microbiol 72:6316–6324
Hamamura N, Ward DM, Inskeep WP (2013) Effects of petroleum mixture types on soil bacterial population dynamics associated with the biodegradation of hydrocarbons in soil environments. FEMS Microbiol Ecol 85:168–178
He X, McLean JS, Edlund A, Yooseph S, Hall AP, Liu SY, Dorrestein PC, Esquenazi E, Hunter RC, Cheng G, Nelson KE, Lux R, Shi W (2015) Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. PNAS 112:244–249
Hug LA, Baker BJ, Anantharaman K et al (2016) A new view of the tree of life. Nat Microbiol 1:16048
Huson DH, Auch AF, Qi J, Schuster SC (2007) MEGAN analysis of metagenomic data. Genome Res 17:377–386
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform 11:119
Jeon CO, Park W, Ghiorse WC, Madsen EL (2004) Polaromonas naphthalenivorans sp. nov., a naphthalene-degrading bacterium from naphthalene-contaminated sediment. Int J Syst Evol Microbiol 54:93–97
Jones DM, Head IM, Gray ND, Adams JJ, Rowan AK, Aitken CM, Bennett B, Huang H, Brown A, Bowler BFJ, Oldenburg T, Erdmann M, Larter SR (2007) Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 451:176–180
Jurelevicius D, Alvarez VM, Peixoto R, Rosado AS, Seldin L (2013) The use of combination of alkB primers to better characterize the distribution of alkane-degrading bacteria. PLoS ONE 8:e66565
Kabelitz N, Machackova J, Imfeld G, Brennerova M, Pieper DH, Heipieper HJ, Junca H (2009) Enhancement of the microbial community biomass and diversity during air sparging bioremediation of a soil highly contaminated with kerosene and BTEX. Appl Microbiol Biotechnol 82:565–577
Keller AH, Kleinsteuber S, Vogt C (2018) Anaerobic benzene mineralization by nitrate-reducing and sulfate-reducing microbial consortia enriched from the same site: comparison of community composition and degradation characteristics. Microbiol Ecol 75:941–953
Kim M, Oh HS, Park SC, Chun J (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351
Kis ÁE, Laczi K, Zsíros S, Kós P, Tengölics R, Bounedjoum N, Kovács T, Rákhely G, Perei K (2017) Characterization of the Rhodococcus sp. MK1 strain and its pilot application for bioremediation of diesel oil-contaminated soil. Acta Microbiol Immunol Hung 64:463–482
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1
Kloos K, Munch JC, Schloter M (2006) A new method for the detection of alkane monooxygenase homologous genes (alkB) in soils based on PCR-hybridization. J Microbiol Methods 66:486–496
Kukor JJ, Olsen RH (1996) Catechol 2,3-dioxygenases functional in oxygen-limited (hypoxic) environments. Appl Environ Microbiol 62:1728–1740
Kumar S, Stetcher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kuyukina MS, Ivshina IB (2010) Application of Rhodococcus in bioremediation of contaminated environments. In: Alvarez HM (ed) Biology of Rhodococcus. Springer, Berlin, pp 231–262
Lal B, Khanna S (1996) Degradation of crude oil by Acinetobacter calcoaceticus and Alcaligenes odorans. J Appl Microbiol 81:355–362
Lang S, Philp JC (1998) Surface-active lipids in rhodococci. Antonie Van Leeuwenhoek 74:59–70
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Larkin MJ, Kulakov LA, Allen CCR (2005) Biodegradation and Rhodococcus—masters of catabolic versatility. Curr Opin Biotechnol 16:282–290
Ławniczak L, Syguda A, Borkowski A, Cyplik P, Marcinkowska K, Wolko Ł, Praczyk T, Chrzanowski Ł, Pernak J (2016) Influence of oligomeric herbicidal ionic liquids with MCPA and Dicamba anions on the community structure of autochthonic bacteria present in agricultural soil. Sci Total Environ 563–564:247–255
Liu YF, Galzerani DD, Mbadinga SM, Zaramela LS, Gu JD, Mu BZ, Zengler K (2018) Metabolic capability and in situ activity of microorganisms in an oil reservoir. Microbiome 6:5
Luo C, Xie S, Sun W, Li X, Cupples AM (2009) Identification of a novel toluene-degrading bacterium from the candidate phylum TM7, as determined by DNA stable isotope probing. Appl Environ Microbiol 75:4644–4647
Maeda AH, Kunihiro M, Ozeki Y, Nogi Y, Kanaly RA (2015) Sphingobium barthaii sp. nov., a high molecular weight polycyclic aromatic hydrocarbon-degrading bacterium isolated from cattle pasture soil. Int J Syst Evol Microbiol 65:2919–2924
Margesin R, Labbé D, Schinner F, Greer CW, Whyte LG (2003) Characterization of hydrocarbon-degrading microbial populations in contaminated and pristine alpine soils. Appl Environ Microbiol 69:3085–3092
Martirani-Von Abercron SM, Marín P, Solsona-Ferraz M, Castañeda-Cataña MA, Marqués S (2017) Naphthalene biodegradation under oxygen-limiting conditions: community dynamics and the relevance of biofilm-forming capacity. Microb Biotechnol 10:1781–1796
Mbadinga SM, Wang LY, Zhou L, Liu JF, Gu JD, Mu BZ (2011) Microbial communities involved in anaerobic degradation of alkanes. Int Biodeterior Biodegrad 65:1–13
Morales M, Sentchilo V, Bertelli C et al (2016) The genome of the toluene-degrading Pseudomonas veronii strain 1YdBTEX2 and its differential gene expression in contaminated sand. PLoS ONE 11:e0165850
Nestler H, Kiesel B, Kaschabek SR, Mau M, Schlömann M (2007) Biodegradation of chlorobenzene under hypoxic and mixed hypoxic-denitrifying conditions. Biodegradation 18:755–767
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA (2017) metaSPAdes: a new versatile metagenomics assembler. Genome Res 27:824–834
Onaca C, Kieninger M, Engesser KH, Altenbuchner J (2007) Degradation of alkyl methyl ketones by Pseudomonas veronii MEK700. J Bacteriol 189:3759–3767
Orphan VJ, Taylor LT, Hafenbradl D, Delong EF (2000) Culture-dependent and culture-independent characterization of microbial assemblages associated with high-temperature petroleum reservoirs. Appl Environ Microbiol 66:700–711
Pérez-de-Mora A, Engel M, Schloter M (2011) Abundance and diversity of n-alkane-degrading bacteria in a forest soil co-contaminated with hydrocarbons and metals: a molecular study on alkB homologous genes. Microb Ecol 62:959–972
Pham VHT, Kim J, Jeong SW (2014) Enhanced isolation and culture of highly efficient psychrophilic oil-degrading bacteria from oil-contaminated soils in South Korea. J Environ Biol 35:1145–1149
Popp N, Schlömann M, Mau M (2006) Bacterial diversity in the active stage of a bioremediation system for mineral oil hydrocarbon-contaminated soils. Microbiology 152:3291–3304
Posman KM, DeRito CM, Madsen EL (2017) Benzene degradation by a Variovorax species within a coal tar-contaminated groundwater microbial community. Appl Environ Microbiol 83:e02658–e2716
Powell SM, Bowman JP, Ferguson SH, Snape I (2010) The importance of soil characteristics to the structure of alkane-degrading bacterial communities on sub-Antarctic Macquarie Island. Soil Biol Biochem 42:2012–2021
Price MN, Dehal PS, Arkin AP (2010) FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490
Probst AJ, Ladd B, Jarett JK et al (2018) Differential depth distribution of microbial function and putative symbionts through sediment-hosted aquifers in the deep terrestrial subsurface. Nat Microbiol 3:328–336
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
Rahman KSM, Thahira-Rahman J, Lakshmanaperumalsamy P, Banat IM (2002) Towards efficient crude oil degradation by a mixed bacterial consortium. Bioresour Technol 85:257–261
Ratajczak A, Geissdörfer W, Hillen W (1998) Alkane hydroxylase from Acinetobacter sp. strain ADp1 is encoded by alkM and belongs to a new family of bacterial integral-membrane hydrocarbon hydroxylases. Appl Environ Microbiol 64:1175–1179
Révész S, Sipos R, Kende A, Rikker T, Romsics C, Mészáros E, Mohr A, Táncsics A, Márialigeti K (2006) Bacterial community changes in TCE biodegradation detected in microcosm experiments. Int Biodeterior Biodegrad 58:239–247
Révész F, Tóth EM, Kriszt B, Bóka K, Benedek T, Sárkány O, Nagy Z, Táncsics A (2018) Sphingobium aquiterrae sp. nov., a toluene, meta- and paraxylene- degrading bacterium isolated from petroleum hydrocarbon-contaminated groundwater. Int J Syst Evol Microbiol 68:2807–2812
Rojo F (2009) Degradation of alkanes by bacteria. Environ Microbiol 11:2477–2490
Saimmai A, Kaewrueng J, Maneerat S (2012) Used lubricating oil degradation and biosurfactant production by SC-9 consortia obtained from oil-contaminated soil. Ann Microbiol 62:1757–1767
Sarlos TT, Gondár K (1995) Bioremediation of Four Former Soviet Military Bases in Hungary: experience useful for future decision-making. In: Herndon RC, Moerlins JE, Kuperberg JM, Richter PI, Biczó IL (eds) Clean-up of former Soviet military installations, vol 1. Springer, Berlin, pp 7–20
Scheps D, Malca SH, Hoffmann H, Hauer B (2011) Regioselective ω-hydroxylation of medium-chain n-alkanes and primary alcohols by CYP153 enzymes from Mycobacterium marinum and Polaromonas sp. strain JS666. Org Biomol Chem 9:6727–6733
Schulze-Makuch D, Wagner D, Kounavas SP et al (2018) Transitory microbial habitat in the hyperarid Atacama Desert. Proc Natl Acad Sci USA 115:2670–2675
Singh SN, Kumari B, Mishra S (2012) Microbial degradation of alkanes. In: Singh S (ed) Microbial degradation of xenobiotics. Environmental science and engineering. Springer, Berlin, pp 439–469
Singleton DR, Lee J, Dickey AN, Stroud A, Scholl EH, Wright FA, Aitken MD (2018) Polyphasic characterization of four soil-derived phenanthrene-degrading Acidovorax strains and proposal of Acidovorax carolinensis sp. nov. Syst Appl Microbiol 41:460–472
Sun WM, Xie SG, Luo CL, Cupples AM (2010) Direct link between toluene degradation in contaminated-site microcosms and a Polaromonas strain. Appl Environ Microbiol 76:956–959
Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH (2007) UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23:1282–1288
Sydow M, Owsianiak M, Szczepaniak Z, Framski G, Smets FB, Ławniczak Ł, Lisiecki P, Szulc A, Cyplik P, Chrzanowski Ł (2016) Evaluating robustness of a diesel-degrading bacterial consortium isolated from contaminated soil. New Biotechnol 33:852–859
Szczepaniak Z, Czarny J, Staninska-Pięta J et al (2016) Influence of soil contamination with PAH on microbial community dynamics and expression level of genes responsible for biodegradation of PAH and production of rhamnolipids. Environ Sci Pollut Res Int 23:23043–23056
Szoboszlay S, Atzél B, Kukolya J, Tóth EM, Márialigeti K, Schumann P, Kriszt B (2008) Chryseobacterium hungaricum sp. nov., isolated from hydrocarbon-contaminated soil. Int J Syst Evol Microbiol 58:2748–2754
Táncsics A, Szoboszlay S, Szabó I, Farkas M, Kovács B, Kukolya J, Mayer Z, Kriszt B (2012) Quantification of subfamily I.2.C catechol 2,3-dioxygenase mRNA transcripts in groundwater samples of an oxygen-limited BTEX-contaminated site. Environ Sci Technol 46:232–240
Táncsics A, Farkas M, Szoboszlay S, Szabó I, Kukolya J, Vajna B, Kovács B, Benedek T, Kriszt B (2013) One-year monitoring of meta-cleavage dioxygenase gene expression and microbial community dynamics reveals the relevance of subfamily I.2.C extradiol dioxygenases in hypoxic, BTEXcontaminated groundwater. Syst Appl Microbiol 36:339–350
Táncsics A, Benedek T, Szoboszlay S, Veres PG, Farkas M, Márialigeti K, Kukolya J, Lányi S, Kriszt B (2015) The detection and phylogenetic analysis of the alkane 1-monooxygenase gene of members of the genus Rhodococcus. Syst Appl Microbiol 38:1–7
Táncsics A, Szalay AR, Farkas M, Benedek T, Szoboszlay S, Szabó I, Lueders T (2018) Stable isotope probing of hypoxic toluene degradation at the Siklós aquifer reveals prominent role of Rhodocyclaceae. FEMS Microbiol Ecol 94:fiy088
Tani A, Ishige T, Sakai Y, Kato N (2001) Gene structures and regulation of the alkane hydroxylase complex in Acinetobacter sp. strain M-1. J Bacteriol 183:1819–1823
Tribelli PM, Rossi L, Ricardi MM, Gomez-Lozano M, Molin S, Raiger Iustman LJ, Lopez NI (2018) Microaerophilic alkane degradation in Pseudomonas extremaustralis: a transcriptomic and physiological approach. J Ind Microbiol Biotechnol 45:15–23
van Beilen JB, Funhoff EG (2007) Alkane hydroxylases involved in microbial alkane degradation. Appl Microbiol Biotechnol 74:13–21
Wang W, Zhong R, Shan D, Shao Z (2014) Indigenous oil-degrading bacteria in crude oil-contaminated seawater of the Yellow sea, China. Appl Microbiol Biotechnol 98:7253–7269
Whyte LG, Bourbonniére L, Greer CW (1997) Biodegradation of petroleum hydrocarbons by psychrotrophic Pseudomonas strains possessing both alkane (alk) and naphthalene (nah) catabolic pathways. Appl Environ Microbiol 63:3719–3723
Whyte LG, Smits TH, Labbé D, Witholt B, Greer CW, van Beilen JB (2002) Gene cloning and characterization of multiple alkane hydroxylase systems in Rhodococcus. Appl Environ Microbiol 68:5933–5942
Winsley TJ, Snape I, McKinlay J, Stark J, van Dorst JM, Ji M, Ferrari BC, Siciliano SD (2014) The ecological controls on the prevalence of candidate division TM7 in polar regions. Front Microbiol 5:345
Xie S, Sun W, Luo C, Cupples AM (2011) Novel aerobic benzene degrading microorganisms identified in three soils by stable isotope probing. Biodegradation 22:71–81
Yoon SH, Ha SM, Lim J, Kwon S, Chun J (2017) A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leewenhoek 110:1281–1286
Zhao L, Ma T, Gao M, Gao P, Cao M, Zhu X, Li G (2012) Characterization of microbial diversity and community in water flooding oil reservoirs in China. World J Microbiol Biotechnol 28:3039–3052
Acknowledgements
Open access funding provided by Szent István University (SZIE). This research was supported by the Higher Education Institutional Excellence Program (NKFIH-1159-6/2019) awarded by the Ministry of Human Capacities within the framework of water related researches of Szent István University. Fruzsina Révész was supported by the UNKP-18-3 New National Excellence Program of the Ministry of Human Capacities of Hungary. PAFG and AJP were supported by the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfahlen (“Nachwuchsgruppe Dr. Alexander Probst”).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Révész, F., Figueroa-Gonzalez, P.A., Probst, A.J. et al. Microaerobic conditions caused the overwhelming dominance of Acinetobacter spp. and the marginalization of Rhodococcus spp. in diesel fuel/crude oil mixture-amended enrichment cultures. Arch Microbiol 202, 329–342 (2020). https://doi.org/10.1007/s00203-019-01749-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01749-2