
Abstract
Summary
Janus kinase (JAK) inhibitors are used to treat rheumatoid arthritis (RA). We assessed the effects of tofacitinib on bone density and bone markers in association with clinical and laboratory parameters in RA. Tofacitinib stabilized bone density and resulted in a positive balance of bone turnover.
Introduction
Janus kinase (JAK) inhibitors emerged as new therapeutic options in rheumatoid arthritis (RA). We have little information on how it affects areal and volumetric bone mineral density (BMD) and bone turnover markers. The aim of this study was to assess the effects of 1-year tofacitinib therapy on bone metabolism in RA.
Methods
Thirty RA patients with active disease were treated with either 5 mg bid or 10 mg bid tofacitinib for 12 months. We determined DAS28, CRP, IgM rheumatoid factor (RF), and anti-cyclic citrullinated peptide (CCP) levels, as well as serum levels of sclerostin, osteocalcin (OC), P1NP, DKK-1, OPG, RANKL, and 25-hydroxy-vitamin D3. Areal and volumetric BMD were assessed by DXA and peripheral quantitative CT (QCT), respectively.
Results
Twenty-six patients (13 on each arm) completed the study. Tofacitinib was clinically effective by suppressing DAS28, CRP, and HAQ. This was accompanied by the attenuation of further bone loss. Tofacitinib therapy significantly increased OC, OPG, and vitamin D3, while decreased CTX levels (p < 0.05). Age and multiple bone markers (OC, CTX, P1NP, RANKL) inversely correlated with L2–4 and femoral neck BMD by DXA. CRP, DAS28, and RANKL inversely determined volumetric BMD by QCT. Age, CRP, anti-CCP, and DKK-1 influenced the effects of tofacitinib therapy on BMD changes.
Conclusions
One-year tofacitinib treatment stabilized BMD in RA patients and resulted in a positive balance of bone turnover as indicated by bone biomarkers. Further studies are needed to evaluate the potential beneficial effects of JAK inhibitors on inflammatory bone loss.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) has been associated with generalized bone loss (osteoporosis), as well as localized inflammatory bone resorption [1,2,3,4,5,6]. Low BMD compared to age- and sex-matched healthy individuals, as well as increased fracture risk, has been observed in about 60–70% of these patients [1, 3, 5, 6]. The receptor activator of nuclear factor κB (RANK)-RANK ligand (RANKL) system is the major driver of inflammatory bone resorption [7, 8]. Pro-inflammatory cytokines have been implicated in RA-related, RANKL-dependent bone loss [2, 7,8,9,10]. RANKL, osteoprotegerin (OPG), which is a soluble decoy receptor of RANKL, and the Wnt signaling pathways play important roles in osteoclast and osteoblast differentiation and activation [11]. Dickkopf-1 (DKK1) and sclerostin (SOST) are inhibitors of Wnt signaling. Activation of Wnt signaling pathway in osteoblasts induces OPG production, which reduces RANKL-induced bone resorption [11]. DKK1 and SOST may exert direct effects on each other, and inhibition of DKK1 suppresses the level of SOST in rodent models [4, 12, 13].
Before the biologic era, RA patients experienced significant annual bone loss. For example, Gough and Emery [14] assessed BMD in 148 early, active, DMARD-naive RA patients. The biannual BMD loss at different sites was between 5.5% and 10% [14]. Sambrook et al. [15] assessed the effects of low-dose corticosteroids on BMD in RA for 2 years in three patient groups. The mean rates of annual bone loss at the femoral neck were 2% in corticosteroid-treated, 1.9% in untreated RA patients, and 1.0% in controls [15].
As systemic inflammation and pro-inflammatory cytokines are major drivers of inflammatory bone loss, biologics may slow down both generalized osteoporosis and the development of periarticular erosions in RA [2, 4, 16,17,18]. Biologics may also influence bone turnover and the production of bone biomarkers. Anti-TNF-α agents were shown to increase serum osteocalcin (OC) and procollagen type I N-propeptide (P1NP) levels, which are markers of bone formation, and suppressed C-terminal telopeptide (CTX) and RANKL levels, which are markers of bone resorption in RA (reviewed in [2]). On the other hand, anti-TNF agents increased OPG/RANKL, OC/CTX, and P1NP/CTX ratios [19,20,21]. In most studies, changes in bone biomarkers by biologics were associated with improvements in disease activity and inflammatory markers (e.g., CRP) in RA [2].
The Janus kinase (JAK) family of tyrosine kinases are transducers of cytokine signaling [22, 23]. JAK inhibitors including tofacitinib have demonstrated efficacy and safety in patients with RA [18, 22, 24, 25]. In clinical trials, tofacitinib inhibited radiographic progression (localized bone resorption) [2, 26,27,28,29,30]. There have been relatively few studies on the effects of tofacitinib on bone metabolism in arthritis. Tofacitinib acts on osteoclast-mediated bone resorption by inhibiting RANKL expression within the joints [27]. Another JAK inhibitor, baricitinib, also inhibits RANKL-mediated osteoclast activity [31]. Tofacitinib and baricitinib enhanced osteoblast function and improved bone mass in murine models [32]. These JAKinibs resulted in increased stabilization of β-catenin and expression of anabolic bone markers, such as osteocalcin and Wnt proteins [32]. In the rat adjuvant-induced arthritis model, at the tissue level, tofacitinib increased bone cortical and trabecular hardness after 22 days. Tofacitinib decreased RANKL expression and increased OPG/RANKL ratio in the serum. However, this compound was unable to reverse the effects of inflammation on the cortical and trabecular bone structure and on mechanical properties. Authors concluded that longer exposure to tofacitinib may be needed in order to see bone effects [33]. Tofacitinib may promote bone healing by recruiting human mesenchymal stromal cells, inducing osteogenic differentiation and reducing osteoclast activity [34]. In an MRI study, tofacitinib treatment, both in monotherapy and in combination with methotrexate, resulted in an early reduction of inflammation, bone edema, and that of the progression of structural damage [28].
BMD is traditionally assessed by dual-energy X-ray absorptiometry (DXA). Peripheral quantitative computed tomography (QCT) is a useful and sensitive tool to study bone microarchitecture, as well as various compartments, such as trabecular and cortical bone. While DXA is able to assess areal (2-dimensional) BMD, QCT determines volumetric (3-dimensional) BMD. Whereas QCT, similarly to DXA, is usually used to evaluate BMD of the lumbar spine or hip, peripheral QCT assesses BMD of peripheral parts of the body, such as the forearms or legs [35,36,37,38]. In a previous study, we compared QCT and DXA in RA patients and healthy individuals [39].
To our best knowledge, there have been no prospective studies assessing bone status in RA patients undergoing tofacitinib therapy. Therefore we conducted a 1-year study in order to assess the effects on this compound on bone density and metabolism as determined by DXA, QCT, and bone biochemical markers.
Patients and methods
Patients and study design
Thirty RA patients in need for targeted therapy are consecutively recruited. Patient characteristics are seen in Table 1. The cohort included 27 women and 3 men with mean age of 52.8 ± 10.0 (range: 27–69) years. Altogether 7 women in the 5 mg bid and 6 patients in the 10 mg bid subset were in postmenopausal status. The mean disease duration was 7.7 ± 5.0 (range: 1–21) years. Patients with active disease were recruited prior to initiating tofacitinib therapy. Inclusion criteria included definitive diagnosis of RA according to the 2010 EULAR/ACR classification criteria for RA [40]; moderate–high disease activity (DAS28 > 3.2) at baseline; and clinical indication of targeted therapy. Patients were either naive to any targeted therapies (n = 16) or initiated tofacitinib after stopping a bDMARD followed by an appropriate washout period (n = 14). Exclusion criteria included inflammatory disease other than RA, acute/recent infection, standard contraindications to JAK inhibition, chronic renal or liver failure, malignancy within 10 years, and the current use of anti-osteoporotic drugs (bisphosphonates, denosumab, teriparatide, calcitonin). None of the patients had known primary osteoporosis prior to the diagnosis of RA. Altogether 10 patients in the 5 mg bid and 6 in the 10 mg bid groups had received vitamin D supplementation prior to the study (Table 1). However, the dose of vitamin D remained unchanged throughout the study. Although most patients may have received corticosteroids prior to the study, none of the patients were on corticosteroids for at least 3 months prior to and during the study. The 30 enrolled patients were randomly assigned in a 1:1 ratio to either 5 mg or 10 mg of tofacitinib twice daily (bid) treatment arms. All patients received tofacitinib in combination with either methotrexate (n = 23) or leflunomide (n = 7). Clinical assessments were performed at baseline and after 6 and 12 months of therapy. Eventually 4 patients (2 on each arm) completed the 6-month follow-up but did not complete the 1-year treatment. Patients completing the 1-year treatment period were included in the data analysis.
The study was approved by the Hungarian Scientific Research Council Ethical Committee (approval No. 56953-0/2015-EKL). A written informed consent was obtained from each patient and assessments were carried out according to the Declaration of Helsinki.
Clinical assessment
First, detailed medical history was taken. We inquired for history of fragility fractures in all patients (Table 1). Further clinical assessments including physical examination were performed at baseline and after 6 and 12 months of therapy.
Bone densitometry and radiographic assessments
DXA examination in order to determine areal bone mineral density (BMD) was performed using the LUNAR Prodigy (GE-Lunar Corp., Madison, WI, USA) densitometer by a single technician during the study period. The coefficient of variation (CV) of the technique at our institute was 0.8% using the anatomical spine phantom measured daily and there was absence of machine drift during the study. The short-term in vivo precision error for L2–L4 lumbar spine is 0.012 g/cm2 (LSC = 0.034 g/cm2 at 95% confidence level) and femur neck is 0.013 g/cm2 (LSC = 0.035 g/cm2 at 95% confidence level).
In order to determine volumetric (3D) BMD, single-slice quantitative computed tomography (QCT) assessments of the ultra-distal region of the dominant forearm were performed using a Stratec XCT-2000 instrument (Stratec Medizintechnik GmbH, Pforzheim, Germany) as described before [39]. Distal sites at 4% of the radius length contain mainly trabecular bone. QCT can differentiate between cortical and trabecular bone. Total, trabecular, and cortical BMD values as determined by QCT are expressed as mg/cm3. Setting to acquire the image were 0.59 mm voxel. Analysis was completed using the XCT6.00B software (Stratec Medizintechnik GmbH, Pforzheim, Germany) with measuring mask set to radius and threshold density to 269 mg/mm3 to define trabecular bone.
Most patients underwent spine radiography in order to assess fragility fractures.
Laboratory measurements and assessment of disease activity and function
Blood samples were drawn from fasting patients in the morning into vacutainer tubes with clot activator. After centrifugation, the serum was separated, aliquoted, and stored at − 70 °C until use. Blood samples were taken at baseline, as well as after 6 and 12 months of tofacitinib treatment.
Serum high-sensitivity C-reactive protein (hsCRP; normal: ≤ 5 mg/l) and IgM rheumatoid factor (RF; normal: ≤ 14 IU/ml) were measured by quantitative turbidimetry (Cobas e 601-Roche Diagnostics). ACPA (anti-CCP) autoantibodies were detected in serum samples using a second-generation Immunoscan-RA CCP2 ELISA test (Euro Diagnostica; normal: ≤ 25 U/ml). The assay was performed according to the manufacturer’s instructions. Disease activity of RA was calculated as DAS28-CRP (3 variables). Functional capacity of the patients was determined by health assessment questionnaire (HAQ).
Bone biomarkers
Serum calcium (Ca; Roche Diagnostics; normal: 2.1–2.6 mmol/l) and phosphate (P; Roche Diagnostics; normal: 0.8–1.45 mmol/l); parathyroid hormone (PTH; Roche Diagnostics; normal: 1.6–6.9 pmol/l); 25-hydroxy-vitamin D3 (25OHVITD3; DiaSorin; normal: ≥ 75 nmol/l); osteocalcin (OC; Roche Diagnostics; normal: < 41 μg/l), procollagen 1 N-terminal propeptide (P1NP; Roche Diagnostics; normal: < 75 μg/l), C-terminal collagen crosslinks (CTX; Roche Diagnostics; normal: < 0.57 μg/l), sclerostin (SOST; Biomedica; median: 24.14 pmol/l), and cathepsin K (CATHK; Biomedica; median: 8.7 pmol/l) were determined by ELISA. Osteoprotegerin (OPG; normal: 515.3–1964.2 pg/ml), soluble RANKL (normal: < 1067.4 pg/ml), and Dickkopf-1 (DKK1; normal: 46.5–2225.7 pg/ml) levels were determined by flow cytometry using a custom multiplex bead immunoassay kit (LEGENDplex, BioLegend) and analyzed by a LEGENDplex software (verison 8.0) [41]. All measurements were performed at baseline, as well as 6 and 12 months after treatment initiation.
Statistical analysis
Statistical analysis was performed using SPSS version 22.0 (IBM) software. Data are expressed as the mean ± SD for continuous variables and percentages for categorical variables. The distribution of continuous variables was evaluated by Kolmogorov-Smirnov test. Continuous variables were evaluated by paired two-tailed t test and Wilcoxon test. Nominal variables were compared between groups using the chi-squared or Fisher’s exact test, as appropriate. Correlations were determined by Pearson’s analysis. Univariable and multivariable regression analysis using the stepwise method were applied to investigate independent associations between BMD as determined by DXA or QCT (dependent variables) and other clinical and laboratory parameters (independent variables). The β standardized linear coefficients showing linear correlations between two parameters were determined. The B (+ 95% CI) regression coefficient indicated independent associations between dependent and independent variables during changes. General linear model (GLM) repeated measures analysis of variance (RM-ANOVA) was performed in order to determine the additional effects of multiple parameters including therapy on 12-month changes of DXA and QCT BMD. In this analysis, partial η2 is given as indicator of effect size, with values of 0.01 suggesting small, 0.06 medium, and 0.14 large effects. P values < 0.05 were considered significant.
Results
Osteoporosis among the patients
In the 5-mg bid tofacitinib group, none had osteoporosis (T-score < − 2.5) and 7 had osteopenia (T-score < − 1) at the lumbar spine (L2–4 vertebrae) as determined by DXA. At the femoral neck region, one had osteoporosis and 4 had osteopenia. On the 10-mg bid arm, none of the patients had L2–4 and 2 had femoral neck osteoporosis. Three patients had L2–4 and 4 had femoral neck osteopenia. In the full cohort, none of the patients had L2–4 and 3 patients had femoral neck osteoporosis, while 10 patients had L2–4 and 8 had femoral neck osteopenia. Two patients in the 5-mg bid subset had previous vertebral fragility fractures. Interestingly, these two patients did not have osteopenia or osteoporosis (Table 1).
Clinical response to tofacitinib therapy
Eventually a total of 4 patients, 2-2 each treatment arms, dropped out after 6 months of treatment but before the end of the study. Out of the 4 patients, 2 had inefficacy, one had significantly elevated transaminases, and one moved abroad. Thus, 13-13 patients on each arm completed the study and were eligible for further data analysis.
JAK inhibition was highly effective. In the full cohort (n = 26), tofacitinib treatment resulted in significant decreases in DAS28 after 6 months (3.31 ± 0.91; p < 0.001) and 12 months of treatment (3.32 ± 1.12; p < 0.001) compared to baseline (5.05 ± 0.77) (data not shown). Similar observations were made in the 5-mg bid and 10-mg bid subsets. On the 5-mg bid arm, DAS28 at baseline, after 6 and 12 months, was 4.80 ± 0.69, 3.23 ± 0.54 (p < 0.001) and 3.05 ± 0.77 (p < 0.001). In the 10-mg bid subset, DAS28 changed from 5.29 ± 0.79 at baseline to 3.39 ± 1.19 after 6 (p < 0.001) and 3.58 ± 1.36 after 12 months (p < 0.001) (data not shown).
Clinical improvement was associated with the suppression of systemic inflammation as indicated by CRP. In the full cohort, CRP decreased from 14.8 ± 14.9 mg/l at baseline to 5.3 ± 5.3 mg/l after 6 months (p < 0.001) and 7.4 ± 7.7 mg/l after 12 months (p = 0.001). The respective CRP values were 13.3 ± 9.7 mg/l, 5.3 ± 3.7 mg/l (p = 0.002), and 7.1 ± 4.0 mg/l (p = 0.022) in the 5-mg bid group, as well as 16.3 ± 18.9 mg/l, 5.2 ± 6.7 mg/l (p = 0.016), and 7.7 ± 10.3 mg/l (p = 0.014) in the 10-mg bid group (data not shown).
With respect to functional capacity as determined by HAQ, in the full cohort, HAQ significantly improved from baseline (1.38 ± 0.58) to 6 months (1.02 ± 0.67; p = 0.001) and 12 months (1.02 ± 0.71; p = 0.001). Similar changes were observed in the 10-mg bid group: HAQ improved from 1.59 ± 0.50 at baseline to 1.10 ± 0.74 at 6 months (p = 0.010) and to 1.15 ± 0.73 at 12 months (p = 0.005). A non-significant tendency of HAQ improvement was seen in the 5-mg bid group (data not shown).
Effects of tofacitinib therapy on DXA and QCT bone mineral density
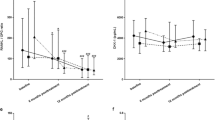
One-year tofacitinib treatment inhibited further generalized bone loss. When L2–4 vertebral (DXAL24BMD) and femoral neck areal BMD (DXAFNBMD) were measured by DXA, there were no differences between baseline and 12-month BMD in the full cohort, as well as the 5-mg bid and 10-mg bid subsets (p = NS) (Fig. 1A). Similarly, total (QCTTOTBMD), trabecular (QCTTRABBMD), and cortical volumetric BMD (QCTCORTBMD) determined by QCT was unchanged from baseline (− 0) to 12 months (− 12) in the full, 5-mg bid, and 10-mg bid groups (p = NS) (Fig. 1B).

Effects of 1-year tofacitinib therapy on areal and volumetric BMD, total, 5-mg bid, and 10-mg bid subsets. (A) Baseline and 12-month L2–4 vertebral and femoral neck BMD as determined by DXA. (B) Total, trabecular, and cortical volumetric BMD changes as determined by QCT
Table S1 shows the percentages on bone loss during tofacitinib treatment in the full cohort, as well as in the 5-mg bid and 10-mg bid subsets. One-year changes in areal BMD were between − 0.9% and 0.7% in the full cohort, − 1.5% and 0.1% in the 5-mg bid, and − 0.2% and 1.4% in the 10-mg bid subset (Table S1). Changes in volumetric BMD were between − 4.9% and 6.6% in the full cohort, − 8.1% and 8.2% in the 5-mg bid, and − 1.5% and 4.9% in the 10-mg bid subset (Table S1).
Effects of tofacitinib on bone biomarkers
In the full cohort, serum OC significantly increased after 6 months compared to baseline (p = 0.013), but only non-significantly after 12 months (Fig. 2A). CTX significantly decreased after 6 months (p = 0.009) and 12 months (p = 0.003) versus baseline (Fig. 2B). Tofacitinib also increased OPG levels after 6 (p = 0.006) and 12 months (p = 0.004) (Fig. 2C), as well as 25OHVITD3 after 6 (p = 0.017) and 12 months (p = 0.009) (Fig. 2D). In the 5-mg bid group, OC also increased after 6 months (p = 0.027) (Fig. 2A), OPG increased after 6 (p = 0.005) and 12 months (p = 0.002) (Fig. 2C), and vitamin D3 also increased after 6 (p = 0.001) and 12 months (p = 0.004) (Fig. 2D). With respect to the 10-mg bid subset, CTX decreased after 6 (p = 0.005) and 12 months (p = 0.007) (Fig. 2B) and OPG increased after 6 (p = 0.047) and 12 months (p = 0.029) (Fig. 2C). Tofacitinib did not change PTH, P1NP, RANKL, DKK1, and SOST significantly (data not shown).

Effects of 1-year tofacitinib therapy on (A) osteocalcin, (B) CTX, (C) osteoprotegerin, and (D) 25-hydroxy-vitamin D3 levels. Total, 5-mg bid, and 10-mg bid subsets. *p < 0.05
Table S2 shows the above presented changes in bone biomarker levels in the full cohort, as well as in the 5-mg bid and 10-mg bid subsets. Only those markers are included in Table S2, which show significant changes.
Correlations between bone mineral density and laboratory biomarkers
In the simple Pearson’s correlation analysis, there have been multiple correlations between areal/volumetric BMD data and bone biomarkers at baseline or after 12 months of tofacitinib treatment. All these correlations are presented in Table S3.
The univariable regression analysis suggested that CTX-0 may be a negative determinant of DXAL24BMD-0, while CTX-0, RANKL-0, CTX-12, and P1NP12 may inversely determine DXAL2BMD-12 (p < 0.05) (Table 2). Similarly, age, OC-0 and CTX-0 were negative determinants of DXAFNBMD-0 and DXAFNBMD-12. Moreover, OC-12 and P1NP-12 also negatively associated with DXAFNBMD-12 (p < 0.05) (Table 2). Among the QCT parameters, QCTTOTBMD-12 was negatively determined by CRP-12, QCTTRABBMD-0 by DAS28-0, while QCTCORTBMD-12 by RANKL-0 and CRP-12 (p < 0.05) (Table 2).
The multivariable analysis confirmed negative associations between CTX-0 and DXAL24BMD-12, age and OC-0 with DXAFNBMD-0, age, OC-0 and CTX-0 with DXAFNBMD-12, as well as RANKL-0 and CRP-12 with QCTCORTBMD-12 (p < 0.005) (Table 2).
Finally, RM-ANOVA was performed in order to assess independent determinants of 12-month changes in areal and volumetric BMD as dependent variables. Tofacitinib treatment exerted combined effects with either lower CCP-0 or lower DKK1-0 on 12-month changes in DXAL24BMD. Similarly, tofacitinib therapy in combination with whether lower age or lower CRP-0 significantly determined 1-year changes in QCTCORTBMD (p < 0.05) (Table 3).
Discussion
To our best knowledge, this may be the first 1-year, prospective study assessing bone status including areal and volumetric BMD, as well as bone biomarkers in RA patients undergoing tofacitinib therapy. We also compared 5-mg bid and 10-mg bid dosing.
RA has been associated with both generalized osteoporosis, as well as localized bone resorption leading to periarticular erosions [1,2,3,4]. There have been numerous studies indicating that biologics inhibit the development of osteoporosis and periarticular erosions in RA [2, 4, 16,17,18]. We and others have reported that biologics may also influence bone turnover and the production of bone biomarkers [2, 19,20,21]. JAK inhibitors including tofacitinib have emerged as potent targeted synthetic DMARDs with efficacy and safety comparable with biologics [18, 22, 24, 25]. Tofacitinib inhibits localized bone loss as in clinical trials this compound inhibited radiographic progression [2, 26,27,28,29,30]. Tofacitinib suppresses RANKL expression and osteoclast-dependent bone resorption [27, 33] and it also stimulates osteoblast function and stabilizes Wnt-dependent bone formation [32].
Many of our patients had osteopenia and some also had osteoporosis. Eventually 24 out of 30 patients completed the study. Tofacitinib in both doses resulted in significant clinical improvement and suppression of systemic inflammation. In parallel, one-year tofacitinib therapy attenuated the further development of osteoporosis, as neither areal nor volumetric BMD changed over time. These findings may be explained by the bone metabolic effects of JAK inhibition described above [27, 32, 33]. However, we could not compare our data on BMD with any other reports as no prospective studies have been carried out on the effects of tofacitinib on BMD changes. On the other hand, before the era of targeted therapies, RA patients experienced an annual BMD loss of about 2–5% [14, 15]. In our study, in general, the percentage of areal bone loss at different sites during tofacitinib treatment was between 0.2% and 1.5%. Increased BMD upon treatment could be observed at some sites.
With respect to bone turnover, we assessed 12 bone biomarkers described above. One-year tofacitinib treatment increased OC, OPG, and 25OHVITD3 and decreased CTX levels. Thus, markers of bone formation and 25OHVITD3 increased, while the marker of bone resorption decreased resulting in positive balance of bone turnover. Increased 25OHVITD3 levels upon tofacitinib treatment may be, at least in part, associated with the usual improvement of functional capacity and physical activity and when comparing the two dosages, 10 mg tofacitinib bid increased OPG and decreased CTX also resulting in a positive balance. However, we did not see these effects in the 5-mg bid subset. In other studies, tofacitinib decreased RANKL production [27, 33] and increased OPG/RANKL ratio [33]. This compound also stabilized the anabolic Wnt proteins, β-catenin, and OC [32]. However, there have been no studies assessing multiple biomarkers of bone resorption and bone formation.
In the correlation analyses, there have been numerous associations between areal or volumetric BMD at baseline and after 12 months of treatment and multiple bone biomarkers especially OC, p1NP, CTX, RANKL, and PTH. Interestingly, areal BMD assessed by DXA inversely correlated with OC, P1NP, CTX, and RANKL, while volumetric BMD as determined by QCT was inversely associated with PTH and RANKL but not with the other biomarkers. The univariable and multiple regression analyses supported the associations between some of these bone markers (OC, CTX, P1NP, RANKL) and BMD. Thus, these biomarkers may be the most important in determining areal and volumetric BMD. Moreover, some of the biomarkers at baseline correlated with DXA or QCT BMD after 12 months indicating that these bone markers may predict BMD after 12 months.
In the univariable and multivariable analyses, not only bone markers but also age, DAS28, and CRP correlated with BMD. Age determined femoral neck but not lumbar spine BMD. In addition, age at baseline not only associated with DXAFNBMD at baseline but also predicted FN BMD after 12 months. Disease activity (DAS28) and marker of systemic inflammation (CRP) negatively correlated with volumetric BMD suggesting that the dampening of systemic inflammation by tofacitinib treatment may be a major reason for the improvement of bone status in RA.
RM-ANOVA is a useful tool in determining the combined effects of tofacitinib treatment and other biomarkers on BMD changes. Tofacitinib therapy together with lower anti-CCP antibody or DKK1 levels predicted the change of DXA L2-4 vertebral BMD, while therapy in combination with lower age or CRP levels determined QCT cortical BMD change. These results suggest that age, autoimmunity, as well as inflammatory and bone markers may influence the effects of tofacitinib treatment on 12-month BMD changes.
With respect to the two doses, we did not find major differences between the 5-mg bid and 10-mg bid subsets in terms of BMD or bone biomarker changes. As the 10-mg bid dose may have more safety issues and this dose is not registered for the treatment of RA in the EU, 5-mg bid tofacitinib could be recommended for the preservation of bone status in RA.
Our study has certain advantages and limitations. To our knowledge, this is the first study that longitudinally assesses the effects of tofacitinib on BMD in association with disease activity and bone markers. We indeed determined a high number of bone biomarkers in these patients. We were able to compare two doses of tofacitinib and had only 4 dropout cases. The possible limitations of this study include the relatively low number of patients. However, we assessed BMD, as well as a great number of biomarkers in a prospective manner, which would have been more difficult in a larger patient cohort.
In summary, tofacitinib therapy slowed down bone loss in RA. Independent predictors of areal BMD were age, OC, and CTX, while those of volumetric BMD were CRP and RANKL. Age, CRP, ACPA, and DKK-1 influenced the effects of tofacitinib therapy on BMD changes. Further studies are needed to evaluate the potential beneficial effects of tofacitinib and other JAK inhibitors on inflammatory bone loss.
References
Lems WF, Dijkmans BA (1998) Should we look for osteoporosis in patients with rheumatoid arthritis? Ann Rheum Dis 57:325–327
Szentpetery A, Horvath A, Gulyas K, Petho Z, Bhattoa HP, Szanto S, Szucs G, FitzGerald O, Schett G, Szekanecz Z (2017) Effects of targeted therapies on the bone in arthritides. Autoimmun Rev 16:313–320
Deal C (2012) Bone loss in rheumatoid arthritis: systemic, periarticular, and focal. Curr Rheumatol Rep 14:231–237
Schett G, Gravallese E (2012) Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol 8:656–664
Rotta D, Fassio A, Rossini M, Giollo A, Viapiana O, Orsolini G, Bertoldo E, Gatti D, Adami G (2020) Osteoporosis in inflammatory arthritides: new perspective on pathogenesis and treatment. Front Med 7:613720
Raterman HG, Bultink IE, Lems WF (2020) Osteoporosis in patients with rheumatoid arthritis: an update in epidemiology, pathogenesis, and fracture prevention. Expert Opin Pharmacother 21:1725–1737
Schett G, Hayer S, Zwerina J, Redlich K, Smolen JS (2005) Mechanisms of disease: the link between RANKL and arthritic bone disease. Nat Clin Pract Rheumatol 1:47–54
Takayanagi H (2009) Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol 5:667–676
Daoussis D, Andonopoulos AP, Liossis SN (2010) Wnt Pathway and IL-17: novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK-RANKL-OPG axis. Semin Arthritis Rheum 39:369–383
McInnes IB, Buckley CD, Isaacs JD (2016) Cytokines in rheumatoid arthritis - shaping the immunological landscape. Nat Rev Rheumatol 12:63–68
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423:337–342
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, van der Heide D, Landewe R, Lacey D, Richards WG, Schett G (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13:156–163
Schett G, Zwerina J, David JP (2008) The role of Wnt proteins in arthritis. Nat Clin Pract Rheumatol 4:473–480
Gough AK, Lilley J, Eyre S, Holder RL, Emery P (1994) Generalised bone loss in patients with early rheumatoid arthritis. Lancet 344:23–27
Sambrook PN, Cohen ML, Eisman JA, Pocock NA, Champion GD, Yeates MG (1989) Effects of low dose corticosteroids on bone mass in rheumatoid arthritis: a longitudinal study. Ann Rheum Dis 48:535–538
Gulyas K, Horvath A, Vegh E et al (2020) Effects of 1-year anti-TNF-alpha therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin Rheumatol 39:167–175
Seriolo B, Paolino S, Sulli A, Ferretti V, Cutolo M (2006) Bone metabolism changes during anti-TNF-alpha therapy in patients with active rheumatoid arthritis. Ann N Y Acad Sci 1069:420–427
Smolen JS, Landewe RBM, Bijlsma JWJ et al (2020) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis 79:685–699
Vis M, Havaardsholm EA, Haugeberg G, Uhlig T, Voskuyl AE, van de Stadt RJ, Dijkmans BA, Woolf AD, Kvien TK, Lems WF (2006) Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NFkappaB ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis 65:1495–1499
Chopin F, Garnero P, le Henanff A, Debiais F, Daragon A, Roux C, Sany J, Wendling D, Zarnitsky C, Ravaud P, Thomas T (2008) Long-term effects of infliximab on bone and cartilage turnover markers in patients with rheumatoid arthritis. Ann Rheum Dis 67:353–357
Woo JH, Lee HJ, Sung IH, Kim TH (2007) Changes of clinical response and bone biochemical markers in patients with ankylosing spondylitis taking etanercept. J Rheumatol 34:1753–1759
O'Shea JJ (2004) Targeting the Jak/STAT pathway for immunosuppression. Ann Rheum Dis 63(Suppl 2):ii67–ii71
Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ (2015) Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol 194:21–27
Roskoski R Jr (2020) Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol Res 152:104609
Winthrop KL (2017) The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol 13:234–243
van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, Tegzová D, Wyman BT, Gruben D, Benda B, Wallenstein G, Krishnaswami S, Zwillich SH, Bradley JD, Connell CA, and the ORAL Scan Investigators (2013) Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum 65:559–570
Labranche TP, Jesson MI, Radi ZA et al (2012) JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum 64:3531–3542
Conaghan PG, Ostergaard M, Bowes MA et al (2016) Comparing the effects of tofacitinib, methotrexate and the combination, on bone marrow oedema, synovitis and bone erosion in methotrexate-naive, early active rheumatoid arthritis: results of an exploratory randomised MRI study incorporating semiquantitative and quantitative techniques. Ann Rheum Dis 75:1024–1033
Orsolini G, Bertoldi I, Rossini M (2020) Osteoimmunology in rheumatoid and psoriatic arthritis: potential effects of tofacitinib on bone involvement. Clin Rheumatol 39:727–736
van der Heijde D, Strand V, Tanaka Y et al (2019) Tofacitinib in combination with methotrexate in patients with rheumatoid arthritis: clinical efficacy, radiographic, and safety outcomes from a twenty-four-month, phase III study. Arthritis Rheum 71:878–891
Murakami K, Kobayashi Y, Uehara S, Suzuki T, Koide M, Yamashita T, Nakamura M, Takahashi N, Kato H, Udagawa N, Nakamura Y (2017) A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS One 12:e0181126
Adam S, Simon N, Steffen U, Andes FT, Scholtysek C, Müller DIH, Weidner D, Andreev D, Kleyer A, Culemann S, Hahn M, Schett G, Krönke G, Frey S, Hueber AJ (2020) JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci Transl Med 12:eaay4447
Vidal B, Cascao R, Finnila MAJ, Lopes IP, da Gloria VG, Saarakkala S, Zioupos P, Canhao H, Fonseca JE (2018) Effects of tofacitinib in early arthritis-induced bone loss in an adjuvant-induced arthritis rat model. Rheumatology (Oxford) 57:1461–1471
Gaber T, Brinkman ACK, Pienczikowski J, Diesing K, Damerau A, Pfeiffenberger M, Lang A, Ohrndorf S, Burmester GR, Buttgereit F, Hoff P (2020) Impact of Janus kinase inhibition with tofacitinib on fundamental processes of bone healing. Int J Mol Sci 21:865
Genant HK, Ettinger B, Cann CE, Reiser U, Gordan GS, Kolb FO (1985) Osteoporosis: assessment by quantitative computed tomography. Orthop Clin N Am 16:557–568
Genant HK, Block JE, Steiger P, Glueer CC, Smith R (1987) Quantitative computed tomography in assessment of osteoporosis. Semin Nucl Med 17:316–333
Engelke K, Adams JE, Armbrecht G, Augat P, Bogado CE, Bouxsein ML, Felsenberg D, Ito M, Prevrhal S, Hans DB, Lewiecki EM (2008) Clinical use of quantitative computed tomography and peripheral quantitative computed tomography in the management of osteoporosis in adults: the 2007 ISCD Official Positions. J Clin Densitom 11:123–162
Tam LS (2016) The role of high-resolution peripheral quantitative computed tomography as a biomarker for joint damage in inflammatory arthritis. J Rheumatol 43:1911–1913
Juhasz B, Gulyas K, Horvath A et al (2017) Comparison of peripheral quantitative computed tomography forearm bone density versus DXA in rheumatoid arthritis patients and controls. Osteoporos Int 28:1271–1277
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G (2010) 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 69:1580–1588
Elshal MF, McCoy JP (2006) Multiplex bead array assays: performance evaluation and comparison of sensitivity to ELISA. Methods 38:317–323
Funding
Open access funding provided by University of Debrecen. This research was supported by the European Union and the State of Hungary and co-financed by the European Social Fund in the framework of TAMOP-4.2.4.A/2-11/1-2012-0001 “National Excellence Program” (Z.S.); by the European Union grant GINOP-2.3.2-15-2016-00015 (G.P., G.T. and Z.S.); and by the Pfizer Investigator Initiated Research Grant no. WI188341 (Z.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 19 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hamar, A., Szekanecz, Z., Pusztai, A. et al. Effects of one-year tofacitinib therapy on bone metabolism in rheumatoid arthritis. Osteoporos Int 32, 1621–1629 (2021). https://doi.org/10.1007/s00198-021-05871-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-021-05871-0