
Abstract
Aims/hypothesis
Increased serum follicle-stimulating hormone (FSH) is correlated with fasting hyperglycaemia. However, the underlying mechanism remains unclear. Because excessive hepatic gluconeogenesis is a major cause of fasting hyperglycaemia the present study investigated whether FSH increases hepatic gluconeogenesis in mice.
Methods
Ovariectomised mice supplemented with oestradiol (E2) to maintain normal levels of serum E2 (OVX+E2 mice) were injected with low or high doses of FSH. We knocked out Crtc2, a crucial factor in gluconeogenesis, and Fshr to discern their involvement in FSH signalling. To evaluate the role of the G-protein-coupled receptor (GPCR) kinase 2 (GRK2), which could affect glucose metabolism and interact directly with non-GPCR components, a specific GRK2 inhibitor was used. The pyruvate tolerance test (PTT), quantification of PEPCK and glucose-6-phosphatase (G6Pase), key enzymes of gluconeogenesis, GRK2 and phosphorylation of AMP-activated protein kinase (AMPK) were examined to evaluate the level of gluconeogenesis in the liver. A nonphosphorylatable mutant of AMPK Ser485 (AMPK S485A) was transfected into HepG2 cells to evaluate the role of AMPK Ser485 phosphorylation.
Results
FSH increased fasting glucose (OVX+E2+high-dose FSH 8.18 ± 0.60 mmol/l vs OVX+E2 6.23 ± 1.33 mmol/l), the PTT results, and the transcription of Pepck (also known as Pck1; 2.0-fold increase) and G6pase (also known as G6pc; 2.5-fold increase) in OVX+E2 mice. FSH also enhanced the promoter luciferase activities of the two enzymes in HepG2 cells. FSH promoted the membrane translocation of GRK2, which is associated with increased AMPK Ser485 and decreased AMPK Thr172 phosphorylation, and enhanced the nuclear translocation of cyclic AMP-regulated transcriptional coactivator 2 (CRTC2). GRK2 could bind with AMPK and induce Ser485 hyperphosphorylation. Furthermore, either the GRK2 inhibitor or AMPK S485A blocked FSH-regulated AMPK Thr172 dephosphorylation and gluconeogenesis. Additionally, the deletion of Crtc2 or Fshr abolished the function of FSH in OVX+E2 mice.
Conclusions/interpretation
The results indicate that FSH enhances CRTC2-mediated gluconeogenesis dependent on AMPK Ser485 phosphorylation via GRK2 in the liver, suggesting an essential role of FSH in the pathogenesis of fasting hyperglycaemia.
Similar content being viewed by others


Introduction
Several metabolic disorders, including bone, glucose and lipid metabolic dysregulation, are observed in postmenopausal women. The postmenopausal state is associated with hyperglycaemia independent of age and certain metabolic factors (such as BMI and triacylglycerol levels) among women without diabetes [1]. Decreased oestrogen levels caused by ovarian failure could partly explain high glucose levels [2], but oestrogen has also been shown to increase glucose levels [3, 4], demonstrating a controversial effect of oestrogen and indicating the involvement of other mechanisms. In addition to the characteristic reduction in oestrogen levels postmenopause, follicle-stimulating hormone (FSH) levels are elevated. A clinical study of women with primary ovarian insufficiency indicated a positive association between FSH and fasting serum glucose [3]. However, the precise mechanism underlying the association remains unclear. FSH is a gonadotropin secreted by the pituitary gland that acts via the FSH receptor (FSHR), a G-protein-coupled receptor (GPCR) expressed exclusively in sexual glands [4]. Recently, multiple studies have found that extragonadal tissues, such as adipose tissue [5], the biliary epithelium [6], liver tissue [7] and bone [8], express functional FSHR.
To identify the mechanism underlying FSH regulation, we focused on GPCR kinase 2 (GRK2). Recent evidence suggests that GRK2 may play a role in regulating insulin signalling, mainly by directly interacting with non-GPCR components [9, 10]. This finding is in contrast to those of the classical definition of GRK2 function of desensitising GPCRs [11]. However, which of the GRK2 functions is dominant in FSHR signalling remains unknown.
The liver is a primary organ involved in gluconeogenesis, a main mechanism through which humans maintain blood glucose levels [12]. Excessive hepatic gluconeogenesis is a major contributor to hyperglycaemia observed in type 2 diabetes mellitus [13]. Nuclear translocation of cyclic AMP-regulated transcriptional coactivator 2 (CRTC2) can stimulate the transcription of two rate-limiting enzymes involved in gluconeogenesis, PEPCK and G6Pase (also known as G6PC), a key step in increasing hepatic gluconeogenesis [14, 15]. AMP-activated protein kinase (AMPK), an important intracellular energy sensor, inactivates CRTC2 and blocks its nuclear translocation [16]. Previous studies have shown that AMPK activity could be regulated by phosphorylation sites, such as Ser485 (inactivation) and Thr172 (activation) [17, 18]. In this study, we investigated whether FSH induces gluconeogenesis dysfunction in the liver and whether AMPK and CRTC2 are involved.
More specifically we explored the effect of FSH on hepatic gluconeogenesis. We hypothesised that FSH, via FSHR, targets GRK2 and thereby induces the CRTC2-mediated transcription of PEPCK and G6Pase, which is dependent on AMPK inactivation by Ser485 hyperphosphorylation.
Methods
Animals
Eight-week-old C57BL/6N female mice were obtained from Beijing Vital River Laboratory Animal Technology Company (Beijing, China). Mice were kept in a 12:12 h light:dark cycle at 25 ± 0.5°C and 50–60% (vol./vol.) humidity and fed a standard diet with water ad libitum. The Ethics Committee of Shandong Provincial Hospital affiliated to Shandong University approved the procedures for animal experiments.
Ovariectomised mice
Eight-week-old female mice were randomly divided into four groups: (1) sham-operation (sham), (2) bilateral ovariectomy (OVX) with diet-supplemented oestradiol (E2) (OVX+E2), (3) OVX+E2 with low-dose FSH (L-FSH; 30 U/kg body weight recombinant human follitropin alfa solution, Merck, Kenilworth, New Jersey, USA) (OVX+E2+L-FSH) and (4) OVX+E2 with high-dose FSH (H-FSH; 60 U/kg body weight) (OVX+E2+H-FSH). Groups (1) and (2) were injected with vehicle and used as controls. To exclude the effect of E2, the mice were placed on a hormone replacement diet supplemented with 2.6 ppm desiccated E2 powder (Bayer, Leverkusen, Germany). After E2 was added for 5 days, FSH was given i.p. daily for 2 weeks, until mice were killed.
Additionally, OVX+E2 mice were randomly divided into four groups (n = 6/group): control (saline [154 mmol/l NaCl]), H-FSH, GRK2 inhibitor (200 μg/kg body weight) and GRK2 inhibitor + FSH (see Electronic supplementary material (ESM) Methods for further details).
Fshr and Crtc2 knockout mice, and Fshr siRNA
Fshr+/− mice (Wuhan Kangweida Gene Technology Company, Wuhan, China) were intercrossed to produce Fshr−/− mice. Female Fshr−/− mice were fed a 2.6 ppm E2-supplemented diet from 3 weeks of age. Female wild-type (Fshr+/+) littermates were used as the controls. At 8–10 weeks, mice were ovariectomised (Fshr+/+ mice were then also fed 2.6 ppm of E2) and injected with H-FSH (see ESM Methods for details).
Additionally, OVX+E2 mice were injected with Fshr siRNA adenovirus and, 7 days later, treated with FSH or vehicle. Mice were killed on day 22 (see ESM Methods for details).
Crtc2 knockout mice (Crtc2−/−) and their Crtc2+/+ littermates were also ovariectomised, fed 2.6 ppm of E2 and injected with H-FSH or vehicle daily for 2 weeks (see ESM Methods for details).
All the samples were randomised using the random number table method. All group assignments and outcome assessments carried out in this study were blinded. Illnesses including dermatitis, dehydration, inflammation and weight loss were exclusion criteria for animals; however, no animals were excluded. No experimental data was excluded using the Grubbs outlier test.
Glucose metabolism
After treatment, mice underwent OGTTs, insulin tolerance tests (ITTs) and pyruvate tolerance tests (PTTs) (see ESM Methods for details).
Mice were fasted for 8 h or refed and then killed for blood and tissue collection. Blood was collected immediately before mice were killed, and glucose, FSH (Abnova ELISA Kit, #KA2330, Wuhan, China) and E2 (Demeditec Oestradiol-Sensitive ELISA Kit, #DE4399, Kiel, Germany) levels were assessed. Tissues were rapidly obtained after mice were killed and repetitive freeze–thaw cycles were avoided (see ESM Methods for details).
H&E and PAS staining
Paraffin-embedded liver tissues were sectioned and stained with haematoxylin and eosin (H&E). For hepatic glycogen staining, samples were stained with periodic acid–Schiff (PAS) according to the manufacturer’s instructions (Yili Company, Beijing, China).
Immunofluorescence
Optimal cutting temperature compound (OCT)-embedded liver tissues were incubated with rabbit anti-GRK2 antibodies (1:200, Santa Cruz, Dallas, TX, USA) and then with fluorescein (tetramethylrhodamine [TRITC])-conjugated goat anti-rabbit IgG (1:300, Invitrogen, Beijing, China). Nuclei were stained with DAPI. Confocal fluorescence microscopy (LSM 780, Zeiss, Oberkochen, Germany) was used for imaging (see ESM Methods).
Hepatic cell lines, mouse primary hepatocyte cultures
HepG2 cells (Cell Library of the Chinese Academy of Sciences, Shanghai, China), mice hepatocyte NCTC 1469 cells (Xiehe Cell Library, Beijing, China) and mouse primary hepatocytes were used. HepG2 cells were treated with FSH (0, 10, 50 and 100 ng/ml) for 6 h or 24 h, or insulin (100 nmol/l) for 4 h or 6 h. Other regents including glucagon (100 nmol/l), protein kinase A (PKA) inhibitor (PKI; 10 μmol/l) and 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR; 1 mmol/l) were added 2 h before FSH. HepG2 and NCTC 1469 cells were authenticated by the cell library from which they were sourced and were not contaminated with mycoplasma.
NCTC cells were infected with Grk2 shRNA or scrambled shRNA and collected for protein analysis.
HepG2 cells were transfected with AMPK siRNA (targeting human AMPKα1, AMPKα2, AMPKβ1 and AMPKγ1) (see ESM Methods for details).
Glucose production measurements
After treatment with FSH for 22 h or insulin for 2 h, HepG2 cells were incubated for 2 h in glucose-free and phenol red-free DMEM media (Gibco, Grand Island, NY, USA) supplemented with 10 mmol/l lactate and 1 mmol/l pyruvate (and FSH/insulin). Glucose was measured in media using a glucose detection kit (Applygen Technologies, Beijing, China). Data were analysed relative to the controls (see ESM Methods).
Plasmid construction and transfection and dual luciferase activity assays
The plasmid encoding wild-type GRK2 (a gift from J. Staňková [19]), Firefly luciferase reporter plasmids containing wild-type PEPCK and G6Pase promoters (provided by H.S. Choi [20]), plasmids encoding green fluorescent protein (GFP)-tagged wild-type AMPK and a nonphosphorylatable mutant of AMPK substituted with alanine (AMPK S485A; Shanghai Genechem, Shanghai, China), and the Firefly luciferase reporter plasmid containing the CRTC2-CREB complex-binding site (CRE)-mutant PEPCK promoter (Hanbio Biotechnology, Shanghai, China) were used for transfection. A Renilla luciferase plasmid (Promega, Madison, WI, USA) served as an internal control. Luciferase activity was measured using the dual luciferase reporter assay (Promega), according to the manufacturer’s protocol. Firefly luciferase activity was normalised to Renilla luciferase activity. Data were analysed relative to controls (see ESM Methods).
RNA isolation and real-time quantitative RT-PCR
Total RNA from cells and mice liver tissue was isolated and real-time quantitative RT-PCR was used to determine relative mRNA expression of mouse Pepck (also known as Pck1), G6pase (also known as G6pc), Crtc2, Pgc1a (also known as Ppargc1a), Gck, Pfkfb1, Pygl, Gys2 and β-actin (Actb), and human PEPCK (also known as PCK1), G6Pase, GLUT2 (also known as SLC2A2), GLUT4 (also known as SLC2A4), AMPK (also known as PRKAA1) and β-actin (ACTB) (see ESM Methods for further details and primers are listed in ESM Table 1).
Protein extraction and western blotting
Protein was extracted from hepatocytes and tissues, separated on 10% (wt/vol.) SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Merck Millipore, Darmstadt, Germany). Anti-p-Thr172 AMPKα (1:1000), p-Ser485 AMPKα (1:1000), AMPKα (1:1000), AMPKβ1 (1:1000), p-Ser428 liver kinase B1 (LKB1; 1:1000), p-Ser9 glycogen synthase kinase 3β (GSK3β; 1:1000), GSK3β (1:1000), p-Ser307 IRS1 (1:1000), p-Ser133 cyclic AMP response element-binding protein (CREB; 1:1000), CREB (1:1000), p-Ser473 Akt (1:1000) and Akt (1:1000) antibodies were obtained from Cell Signaling Technology (Boston, MA, USA). Other antibodies used in the study included anti-GLUT2 (1:1000) and anti-GLUT4 (1:1000) (Abcam, Cambridge, UK); anti-LKB1 (1:1000, Merck Millipore); anti-CRTC2 (1:2000, Calbiochem, Merck Millipore); anti-GRK2 (1:200, Santa Cruz); anti-AMPKγ1 (1:1000, Thermo Scientific, Waltham, MA, USA); anti-FSHR (1:500) and anti-glucose-6-phosphatase (G6Pase) (1:1000) (OriGene, Rockville, MD, USA); anti-PEPCK (1:2000), PGC1α (1:1000), GFP (1:5000), LMB1 (1:5000) and GAPDH (1:7500) (Proteintech, Wuhan, China), and anti-p-Ser670 GRK2 (1:200, Bioss, Beijing, China). All the antibodies used in the article were validated by companies from which they were bought respectively, using methods such as, siRNA knock-down, phosphatase and activator treatment, positive/negative cell line and lot-to-lot testing. The appropriate secondary antibodies conjugated to horseradish peroxidase (HRP; ZSGB-Bio, Beijing, China) were used at 1:5000. Immune complexes were detected using the chemiluminescence (see ESM Methods).
Co-immunoprecipitation
Co-immunoprecipitation was carried out to test the binding of GRK2 with AMPK. Protein was extracted from cells and incubated with rabbit anti-AMPKα or rabbit anti-AMPKβ1 antibodies. The pre-immune sample was generated by combining protein extracts with rabbit IgG (Cell Signaling Technology). The following day, Protein A/G plus agarose slurry beads (Santa Cruz) were added to samples for 2 h. The eluted proteins were separated on 10% (wt/vol.) SDS-PAGE and transferred to a PVDF membrane. The membrane was probed with an anti-GRK2 antibody (Santa Cruz) and then with a secondary HRP-conjugated antibody (1:5000). Immune complexes were detected using chemiluminescence (see ESM Methods for details).
Statistical analysis
The data are expressed as the means ± SEMs. Statistical analyses were performed using either two-tailed unpaired Student’s t tests (two groups) or one-way ANOVA tests (more than two groups). A p < 0.05 is considered to be statistically significant.
Results
FSH upregulates hepatic gluconeogenesis in ovariectomised mice
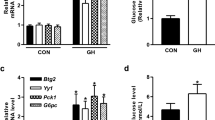
OVX mice receiving an E2-supplemented diet to maintain normal levels of serum E2 were injected with L-FSH or H-FSH (Fig. 1a–c). No significant changes in body weight, alanine aminotransferase (ALT), aspartate aminotransferase (AST) and hepatic histology were observed among the four groups (ESM Fig. 1a–d). FSH increased fasting glucose levels in a dose-dependent manner (Fig. 1d). An OGTT showed that FSH affected whole-body glucose metabolism (Fig. 1e) [21], whereas it did not obviously alter insulin levels (ESM Fig. 1e). ITTs were also performed to evaluate insulin sensitivity (Fig. 1f). Unchanged protein levels of phospho-IRS1 (Ser306), phospho-Akt (Ser473) and GLUT4 in skeletal muscles after FSH injection indicated that FSH did not influence insulin sensitivity (ESM Fig. 1f) [22]. A PTT was used to determine glucose production by the liver because pyruvate is a major substrate for glucose production [13]. The PTTs showed that hepatic gluconeogenesis was elevated in a dose-dependent manner in mice receiving FSH (Fig. 1g). Even in a refed state, serum glucose increased after FSH injection, demonstrating the critical role of FSH in gluconeogenesis regulation (Fig. 1h). HepG2 cells, which exhibit a wide variety of liver-specific metabolic responses to different types of stimuli [23, 24], were used to explore the influence of FSH in vitro. FSH increased glucose production, which is widely used to evaluate gluconeogenesis [13], and insulin, as a positive control, reduced glucose production in HepG2 cells (Fig. 1i). These results confirm that FSH increases hepatic gluconeogenesis.

FSH upregulates hepatic gluconeogenesis in ovariectomised mice. (a) Schematic of C57BL/6 mice receiving different treatments. Fasting levels for (b) serum E2, (c) serum FSH and (d) glucose. *p < 0.05, ***p < 0.001 vs sham; §p < 0.05, §§p < 0.01, §§§p < 0.001 vs OVX+E2. (e) OGTT together with its AUC. (f) ITT and Kitt. (g) PTT together with its AUC. Sham (n = 7), black circles with solid line; OVX+E2 (n = 5), white squares with dashed line; OVX+E2+L-FSH (n = 5), black triangles with solid line; OVX+E2+H-FSH (n = 5), white circles with dotted line. (h) Glucose levels of mice that were fasted or refed (fasted for 8 h and then received food ad libitum for 6 h). OVX+E2 (n = 5), light grey bars; OVX+E2+H-FSH (n = 3), black bars. *p < 0.05, **p < 0.01 OVX+E2+H-FSH vs sham; §p < 0.05, §§p < 0.01, §§§p < 0.001 OVX+E2+H-FSH vs OVX+E2; †p < 0.05 OVX+E2+L-FSH vs sham. (i) Glucose production in HepG2 cells treated with FSH (50 ng/ml, 24 h) or insulin (100 nmol/l, 4 h). *p < 0.05 vs control, §§p < 0.01 vs FSH. Each figure part represents at least three independent experiments. Data are means ± SEM
FSH enhances the expression of key enzymes of gluconeogenesis in the liver
Hepatic glucose metabolism is associated with glucose uptake, gluconeogenesis, glycolysis and glycogen metabolism [14]. To elucidate the mechanisms underlying the influence of FSH, we examined the expression of genes involved in glucose metabolism (Fig. 2a). FSH did not alter the glycogen content in the liver, as assessed by PAS staining (ESM Fig. 2a). In addition, phospho-GSK3β, a critical regulator of glycogen synthesis [14], was unaltered by FSH administration in vivo or stimulation in vitro. FSH affected neither mRNA nor protein levels of GLUT2 and GLUT4 in the liver (ESM Fig. 2b–e), indicating that FSH has no obvious effect on glucose uptake [25]. The expression of PEPCK and G6Pase, key enzymes of hepatic gluconeogenesis [26], increased in the livers of fasted mice receiving FSH (Fig. 2a, b), and their mRNA levels were also significantly elevated after FSH injection in a refed state (Fig. 2c, d). In vertebrates, gluconeogenesis occurs mainly in the liver and to a lesser extent in the kidneys [27]. However, the mRNA levels of Pepck and G6pase in the kidney remained at nearly the same level among the groups (ESM Fig. 2f). As shown in Fig. 2e, FSH increased PEPCK and G6Pase mRNA levels in a time-dependent manner in HepG2 cells. Furthermore, PEPCK and G6Pase promoter luciferase activities increased after FSH treatment (Fig. 2f). These results suggest that FSH enhances the transcription of these two genes through the promoter regions.

FSH enhances the expression of key enzymes of gluconeogenesis in the liver. (a) Real-time PCR of genes related to liver glucose metabolism. The mRNA levels were calculated relative to the sham levels. (b) PEPCK and G6Pase protein levels in the liver as analysed by western blotting. (c, d) Relative Pepck and G6pase mRNA levels measured by real-time PCR in the livers of fasted or refed mice. Sham (n = 10), white bars; OVX+E2 (n = 6), light grey bars; OVX+E2+L-FSH (n = 6), dark grey bars; OVX+E2+H-FSH (n = 6), black bars. *p < 0.05 vs sham; §p < 0.05, §§p < 0.01, §§§p < 0.001 vs OVX+E2. (e) Real-time PCR for the relative PEPCK and G6Pase mRNA levels in HepG2 cells treated with FSH (50 ng/ml) for 6 h or 24 h. Control, white bars; FSH 6 h, grey bars; FSH 24 h, black bars. *p < 0.05, **p < 0.01 vs control; §§p < 0.01, §§§p < 0.001 vs FSH 6 h. (f) The relative luciferase activities of the PEPCK-luc and G6Pase-luc reporter constructs in HepG2 cells treated with FSH (50 ng/ml) or insulin (100 nmol/l) for 6 h. Control, white bars; FSH, grey bars; insulin, black bars. *p < 0.05 vs control; §§p < 0.01, §§§p < 0.001 vs FSH. Each figure part represents at least three independent experiments. Data are means ± SEM
CRTC2 is activated by FSH and mediates the regulatory role of FSH in hepatic gluconeogenesis
CRTC2 is one of the major coactivators that stimulate the transcription of PEPCK and G6Pase, which subsequently increase hepatic gluconeogenesis [15]. As shown in Fig. 3a, no obvious changes in Crtc2 and Pgc1α (another transcriptional coactivator regulating gluconeogenesis [28]) mRNA levels were detected after the administration of L-FSH or H-FSH. Notably, prominent dose-dependent increases in cytoplasmic and nuclear fractions of non-phosphorylated CRTC2 protein were observed in the livers of mice injected with FSH (Fig. 3b). Correspondingly, FSH caused concentration- and time-dependent increases in non-phosphorylated CRTC2, but not PGC1α, levels in cytoplasmic and nuclear fractions of HepG2 cells (Fig. 3c). In addition, even with refeeding or glucagon treatment, FSH regulated CRTC2 activity (Fig. 3d, e).

CRTC2 is activated by FSH and mediates the regulatory role of FSH in hepatic gluconeogenesis. (a) Real-time PCR for relative Crtc2 and Pgc1α mRNA levels in the liver calculated relative to sham levels. Sham (n = 8), white bars; OVX+E2 (n = 6), light grey bars; OVX+E2+L-FSH (n = 6), dark grey bars; OVX+E2+H-FSH (n = 6), black bars. (b) Western blotting indicating CRTC2 protein levels in cytoplasmic and nuclear fractions of the liver and (c) HepG2 cells treated with 0, 10, 50 and 100 ng/ml FSH for 24 h. (d) HepG2 cells were treated with or without glucagon (100 nmol/l, 2 h) and then FSH (50 ng/ml, 24 h) was added or not. (e) CRTC2 protein in the livers of fasted or refed mice. GAPDH and LaminB1 were used as internal markers for cytoplasmic and nuclear fractions, respectively. (f) PCR analysis demonstrating CRTC2 fragments generated from the tails of Crtc2+/+, Crtc2+/− and Crtc2−/− mice. (g) Immunoblot of CRTC2 protein in hepatic extracts from Crtc2+/+ and Crtc2−/− mice. (h) Schematic of Crtc2+/+ and Crtc2−/− mice subjected to different treatments. (i) PTT and the AUC. Crtc2+/+ + vehicle (n = 5), white squares with dashed line; Crtc2+/+ + FSH (n = 8), black circles with solid line; Crtc2−/− + vehicle (n = 5), white circles with dotted line; Crtc2−/− + FSH (n = 5), black triangles with solid line. **p < 0.01 Crtc2+/+ + FSH vs Crtc2+/+ + vehicle; §p < 0.05 Crtc2−/− + vehicle vs Crtc2+/+ + vehicle; ††p < 0.01, †††p < 0.001 Crtc2−/− + FSH vs Crtc2+/+ + FSH (j) Pepck and G6pase mRNA levels in the liver. Crtc2+/+ + vehicle, white bars; Crtc2+/+ + FSH, light grey bars; Crtc2−/− + vehicle, dark grey bars; Crtc2−/− + FSH, black bars. *p < 0.05, ***p < 0.001 vs Crtc2+/+ + vehicle; §§§p < 0.001 vs Crtc2+/+ + FSH. (k) Relative luciferase activities of the PEPCK-luc-WT and PEPCK-luc-CRE-mutant reporter constructs in HepG2 cells treated with or without FSH (50 ng/ml, 6 h). Control, white bars; FSH, black bars. *p < 0.05, ***p < 0.001 vs PEPCK-luc-WT control, §§§p < 0.001 vs PEPCK-luc-WT + FSH. (l) Protein levels (measured by western blotting) in HepG2 cells treated with or without PKI (10 μmol/l, 2 h) and then FSH (50 ng/ml, 24 h) was added or not. Each figure part represents at least three independent experiments. Data are means ± SEM
To determine the involvement of CRTC2 in FSH-induced hepatic gluconeogenesis, we used Crtc2 knockout mice. The results of PCR genotyping and the efficiency of Crtc2 gene deletion by western blotting are presented in Fig. 3f, g. All mice were ovariectomised, supplemented with E2 to maintain the normal E2 levels and then injected with H-FSH (shown in Fig. 3h). Crtc2−/− mice exhibited reduced PTT results compared with Crtc2+/+ littermates; however, Crtc2−/− mice receiving H-FSH exhibited approximately the same PTT results as the Crtc2−/− mice receiving vehicle (Fig. 3i). Changes in hepatic Pepck and G6pase mRNA levels were consistent with the PTT results (Fig. 3j). Moreover, when the CRTC2-CREB complex-binding site (CRE) [15] in the PEPCK promoter was mutated, the effect of FSH was significantly attenuated (Fig. 3k). To explore the involvement of CREB, we conducted experiments with PKI, a specific pharmacological PKA inhibitor. The results showed that FSH did not alter CREB phosphorylation; however, inhibition of CREB activity by PKI partly blocked FSH-related increases in PEPCK and G6Pase (Fig. 3l). Considered together, these findings suggest that CRTC2 plays an indispensable role in FSH-induced hepatic gluconeogenesis.
AMPK is essential for the FSH-mediated regulation of gluconeogenesis
AMPK is important in energy metabolism and its activity can be regulated by autophosphorylation [17, 18]. We found that the phosphorylation of AMPK Ser485 was increased in a dose-dependent manner in the liver after injection of FSH and that phosphorylation at Thr172 caused the opposite change (Fig. 4a). The results in HepG2 cells were consistent with those in the liver (Fig. 4b). Pre-treatment with a specific AMPK activator, AICAR, resulted in increased phosphorylation of AMPK at Thr172 and the inhibition of PEPCK and G6Pase production. Furthermore, AICAR abolished the effect of FSH on PEPCK and G6Pase levels (Fig. 4c). To confirm the specific contribution of AMPK Ser485 phosphorylation, we overexpressed a GFP-tagged non-phosphorylatable mutant of AMPK Ser485, AMPK S485A, in HepG2 cells. Overexpressed cell lines demonstrated comparable GFP-AMPK fusion protein (~90 kDa) levels (cells transfected with vector plasmids only expressed GFP at ~26 kDa) (Fig. 4d) and remarkably increased AMPK mRNA levels (Fig. 4e). The overexpression of AMPK S485A completely blocked FSH-regulated AMPK Ser485 and Thr172 phosphorylation and PEPCK and G6Pase enhancement (Fig. 4f). We also observed that neither mutant AMPK nor AICAR affected CREB (ESM Fig. 3a,b), which is consistent with previous research [29].

AMPK is essential for the FSH-mediated regulation of gluconeogenesis. (a) Western blot showing p-AMPK (Ser485), p-AMPK (Thr172) and total AMPK protein levels in the liver. (n = 6 for each group). (b) Western blot showing p-LKB1 (Ser428) and total LKB1, p-AMPK (Ser485), p-AMPK (Thr172) and total AMPK protein levels in HepG2 cells treated with 0, 10, 50, 100 ng/ml FSH for 24 h. (c) HepG2 cells were treated with or without AICAR (1 mmol/l, 2 h) and then FSH (50 ng/ml, 24 h) was added or not. (d) GFP and GFP-AMPK fusion protein levels determined by western blotting and (e) relative mRNA levels of AMPK determined by RT-PCR in HepG2 cells transfected with GFP-tagged AMPK S485A or vector plasmids for 48 h. *p < 0.05. (f) 48 h after the addition of GFP-tagged AMPK S485A, HepG2 cells were treated with 50 ng/ml FSH or not for another 24 h and protein levels were determined by western blotting. Each figure part represents at least three independent experiments. Data are means ± SEM
FSH regulates AMPK-dependent hepatic gluconeogenesis via GRK2
The protein levels of GRK2 in the liver increased after OVX+E2 mice received FSH in a dose-dependent manner (Fig. 5a). Moreover, FSH induced the translocation of GRK2 from the cytosol to the plasma membrane, as demonstrated through confocal laser-scanning microscopy, revealing that FSH can activate GRK2 (Fig. 5b). To explore the role of GRK2, a commonly used GRK2-specific inhibitor, methyl ([5-nitro-2-furyl]vinyl)-2-furoate, was used. This inhibitor blocks GRK2 kinase activity and prevents GRK2 from interacting with some molecules [30]. Hyperphosphorylation at Ser670 indicates GRK2 inactivity [31]. GRK2 activity was decreased by the GRK2 inhibitor, as shown by the reciprocal of ratio for phospho-GRK2/total GRK2 (ESM Fig. 4a). The ability of FSH to increase hepatic mRNA levels of Pepck and G6pase in OVX+E2 mice was abolished with administration of the GRK2 inhibitor (Fig. 5c). Notably, the GRK2 inhibitor also prevented the effects of FSH on phospho-AMPK and CRTC2 levels (Fig. 5d). In vitro, GRK2 inhibition decreased AMPK Ser485 and induced Thr172 phosphorylation (Fig. 5e). Overexpression of GRK2 produced the opposite result (Fig. 5f); however, overexpression of AMPK S485A blocked these effects (Fig. 5g).

FSH regulates AMPK-dependent hepatic gluconeogenesis via GRK2. (a) Western blotting and (b) laser-scanning confocal microscope images demonstrating GRK2 protein levels in the liver. Scale bars, 10 μm. (c) Pepck and G6pase mRNA levels, and (d) p-AMPK (Ser485), p-AMPK (Thr172), total AMPK and CRTC2 protein levels in the liver. In (c) and (d), the OVX+E2 mice underwent different treatments with a GRK2 inhibitor (inh) or FSH. Control (n = 7), white bars; FSH (n = 7), light grey bars; GRK2 inh (n = 5), dark grey bars; GRK2 inh + FSH (n = 7), black bars. *p < 0.05, **p < 0.01 vs control, §p < 0.05, §§p < 0.01 vs FSH. (e, f) The protein levels were analysed by western blotting in HepG2 cells transfected with Grk2 siRNA for 72 h (e) or GRK2 WT or vector plasmids for 48 h (f). (g) HepG2 cells were transfected with AMPK S485A 24 h before GRK2 WT. (h) Co-immunoprecipitation of GRK2 and AMPK in HepG2 cells treated with FSH (50 ng/ml, 24 h) or not. (i) Co-immunoprecipitation of GRK2 and AMPK in HepG2 cells treated with FSH (50 ng/ml, 24 h) and then a GRK2 inhibitor (100 μmol/l, 24 h) was added or not. (j–k) Co-immunoprecipitation of GRK2 and AMPK in HepG2 cells transfected with AMPKα1, AMPKα2, AMPKβ1 or AMPKγ1 siRNAs for 72 h (added with 50 ng/ml FSH for the final 24 h). (l) Western blotting after HepG2 cells were transfected with AMPKβ1 or AMPKγ1 siRNAs for 72 h (added with 50 ng/ml FSH for the final 24 h). N.C., negative control. Each figure part represents at least three independent experiments. Data are means ± SEM
To thoroughly explore the mode of action of GRK2, we performed co-immunoprecipitation assays. Intriguingly, AMPK and GRK2 co-immunoprecipitated, and this was more prominent after FSH stimulation (Fig. 5h). Moreover, the GRK2 inhibitor attenuated the binding of AMPK to GRK2 (Fig. 5i). AMPK consists of three subunits: α, β and γ. Two or three isoforms of each subunit (α1, α2, β1, β2, γ1, γ2 and γ3) are encoded by different genes [32]. The α1- and α2-containing complexes each account for approximately half of the total AMPK activity in liver extracts, β1- and γ1-containing complexes account for over 90%. To identify which AMPK subunit contributes to GRK2 binding, we knocked down AMPK α1, α2, β1 and γ1 by siRNA (ESM Fig. 4b–d). The co-immunoprecipitation results demonstrated that both AMPKα1 and α2, but not β1 or γ1, contributed to the binding (Fig. 5j, k). Knockdown of neither β1 nor γ1 had an effect on AMPK Ser485 and Thr172 phosphorylation, which is consistent with the co-immunoprecipitation results (Fig. 5l).
Fshr knockout blocks the effects of FSH
To determine the contribution of FSHR to hepatic gluconeogenesis in vivo, we used a Fshr−/− mouse model. The genotypes were confirmed by PCR (Fig. 6a) and the gene deletion efficiency was assessed by western blotting (Fig. 6b). To exclude the potential influence of E2 deficiency and to amplify the effect of FSH, OVX+E2+H-FSH mice were used (Fig. 6c). Thus, Fshr−/− mice and their Fshr+/+ littermates exhibited similar E2 and FSH serum levels (Fig. 6d, e).

FSHR knockout blocks the effect of FSH. (a) PCR analysis revealing FSHR fragments generated from the tails of Fshr+/+, Fshr+/− and Fshr−/− mice. (b) Immunoblot of FSHR protein in primary hepatocyte extracts from Fshr+/+ and Fshr−/− mice. (c) Schematic of Fshr+/+ and Fshr−/− mice subjected to ovariectomy and treated with E2 and H-FSH daily for 2 weeks. (d, e) Fasting serum E2 and FSH levels. (f) Fasting glucose levels. (g) PTT together with its AUC. Fshr+/+, black circles; Fshr−/−, white circles. (h) Pepck and G6pase mRNA levels in the liver. Fshr+/+ (n = 6), grey bars; Fshr−/− (n = 8), black bars. *p < 0.05, **p < 0.01 vs Fshr+/+. (i) Western blot showing GRK2, p-AMPK (Ser485), p-AMPK (Thr172) and total AMPK, CRTC2, PEPCK and G6Pase protein levels in the liver. (j) Pepck and G6pase mRNA levels in the livers of OVX+E2 mice receiving the Fshr siRNA or scrambled siRNA followed by FSH or vehicle. *p < 0.05, ***p < 0.001 vs scrambled siRNA + vehicle, §§p < 0.01, §§§p < 0.001 vs scrambled siRNA + FSH. (n = 6, 3, 6, 8 for each group, respectively). Each figure part represents at least three independent experiments. Data are means ± SEM. (k) Schematic representation of the role of FSH in hepatic gluconeogenesis. FSH induces AMPKα hyperphosphorylation at Ser485 and subsequent AMPKα Thr172 dephosphorylation by activating GRK2. These changes result in enhanced nucleus translocation of CRTC2 and elevated gluconeogenesis in the liver
Compared with their Fshr+/+ littermates, fasting serum glucose levels were decreased (Fig. 6f) and PTT results were significantly reduced in Fshr−/− mice (Fig. 6g). Consistently, the mRNA and protein levels of PEPCK and G6Pase were also decreased in Fshr−/− mice (Fig. 6h, i). Moreover, GRK2, phospho-AMPK (Ser485) and CRTC2 protein levels were attenuated, and phospho-AMPK (Thr172) levels were increased in Fshr−/− mice (Fig. 6i). FSHR deficiency, induced by injecting siRNA targeting Fshr via the tail vein, prevented the FSH-mediated stimulation of Pepck and G6pase transcription (Fig. 6j). These results demonstrate that the effects of FSH on hepatic gluconeogenesis depend on FSHR in the liver.
Discussion
We have discovered a novel extragonadal role for FSH in regulating hepatic gluconeogenesis via FSHR in the liver. FSH suppresses AMPK activity by stimulating GRK2-mediated AMPK Ser485 phosphorylation, thereby enhancing PEPCK and G6Pase transcription via CRTC2 (Fig. 6k).
Hepatic gluconeogenesis and glycogenolysis are the two main sources of fasting glucose. However, within 2 h of fasting, the glycogenolysis rate returns to the basal level, whereas gluconeogenesis remains active [12], suggesting the vital importance of gluconeogenesis, particularly in prolonged fasting. In our study, hepatic glycogen and several molecules involved in glycogen metabolism, including GSK3β, glycogen synthase 2 and liver glycogen phosphorylase, were unaltered after FSH injection. Additionally, the transcription of Pfkfb1 and Gck, important genes involved in glycolysis, was unchanged by FSH. These results indicate that FSH does not significantly affect glycogen metabolism and glycolysis in the liver. Approximately 80% of endogenous glucose is produced in the liver, and the remaining 20% in the kidneys [27]. We excluded the effects of FSH on kidney gluconeogenesis. Insulin sensitivity was evaluated using ITTs, OGTTs and by measuring insulin levels during the OGTT. Insulin tolerance was analysed through the insulin sensitivity index (Kitt) which can exclude the interference of changes in basal blood glucose level [33]. After a pyruvate load during a PTT, changes in glucose clearance influence glucose levels. Therefore, we measured p-IRS1, p-Akt and GLUT4 to evaluate glucose degradation and insulin sensitivity in skeletal muscles, and no significant changes were found. The results indicate that FSH does not significantly affect insulin levels and insulin sensitivity. Hence, we focused on the role of FSH in hepatic gluconeogenesis and the underlying mechanism.
GRK2 belongs to the GRK family, GRK1–7 [34], of which GRK2 is expressed most ubiquitously. In this study, GRK2 upregulated hepatic gluconeogenesis, exhibiting an identical function to FSH. Thus, the results demonstrated that the dominant role of GRK2 is as a signal molecule that helps to mediate FSH, rather than as a GPCR kinase desensitising FSHR and blocking FSHR signalling. Previous research has indicated that both the expression and the translocation of GRK2 can be regulated [11, 35]. Chronic or even acute administration of GPCR agonists can increase GRK2 levels, and the best-known case is the upregulation of GRK2 in heart failure [36]. GRK2 can be regulated by protein–protein interactions, as with many kinases including Akt [11]. When the GPCR is activated, Gβγ subunits bind to GRK2 and recruit it to the membrane, dependent on the presence of anionic phospholipids. GRK2 is also phosphorylated by C-terminal Src (c-Src) and the extracellular signal regulated kinase (ERK), this phosphorylation not only reduces the activity of GRK2 but also promotes its degradation. The phosphorylation of GRK2 (not only by c-Src and ERK) can affect its interaction with Gβγ and membrane recruitment. We hypothesised that after FSH activates FSHR, the Gβγ subunits of the G-protein bind to GRK2 and recruit it to the membrane. A reduction in GRK2 degradation may also occur after chronic FSH stimulation. However, knowledge of the regulation of GRK2 expression and translocation remains incomplete and further exploration is required. These new findings suggest a novel role for GRK2 in hepatic gluconeogenesis regulation and provide promising new insight into the effects of FSH on liver function.
GRK2 can directly interact with proteins [37], such as Akt [38] and p38 mitogen-activated protein kinases (p38 MAPK) [39]. AMPK, Akt and p38 MAPK all belong to the Ser/Thr kinases family, indicating that they potentially exhibit a similar phosphorylation pattern. Moreover, GRK2 is necessary for insulin signalling [40], which implies its involvement in energy regulation, and AMPK is crucial to energy metabolism [41]. Thus, we explored the relationship between GRK2 and AMPK. Based on our data, we hypothesise a possible mechanism in which GRK2 binds to the AMPK α1 and α2 catalytic subunits and phosphorylates AMPK at Ser485, leading to the inhibition of AMPK Thr172 phosphorylation and its inactivation.
Following demonstration that FSH stimulates AMPK Ser485 phosphorylation both in vivo and in vitro, we prepared an AMPK mutant in which Ser485 was altered to determine its role in affecting FSH signalling and biological actions. The overexpression of AMPK S485A blocked FSH-regulated AMPK Thr172 dephosphorylation and PEPCK and G6Pase production. Studies have found that phosphorylation of AMPK at Ser485 reduces the phosphorylation at the activating site Thr172 and decreases AMPK activity [17, 42], which our results confirmed. Although several kinases have been identified, LKB1 is thought to be the predominant upstream kinase responsible for AMPK activation in the liver. LKB1 is activated when phosphorylated at Ser428 [43]. Surprisingly, we found that FSH did not alter LKB1 Ser428 phosphorylation. Although AMPK phosphorylation results partly from the activation of calcium/calmodulin-dependent protein kinase kinase (CaMKK), only trace levels of CaMKK were observed in the liver [44]. Thus, it is unlikely that AMPK activation by CaMKK occurs in hepatocytes [45].
CRTC2, as a molecule downstream from AMPK, is a key regulator of glucose homeostasis during fasting [46]. In our study, Crtc2−/− mice exhibited reduced PTT results and decreased Pepck and G6pase mRNA levels, consistent with previous research [47]. Additionally, the CRE region mutation in the PEPCK promoter attenuated the enhanced effect of FSH on PEPCK luciferase activity. These results demonstrate the indispensability of CRTC2 in FSH functioning. However, knocking out Crtc2 could not rule out a FSH-mediated CREB activation to stimulate PEPCK and G6Pase production. We found that FSH did not alter CREB phosphorylation; however, inhibiting CREB activity by PKI partly blocked FSH-related increases in PEPCK and G6Pase levels. To explain, when CRTC2 is activated by FSH it must bind to CREB to increase its target gene transcription [47]; thus, CREB is indispensable in the FSH gluconeogenesis pathway. Therefore, both CRTC2 and CREB were found to participate in FSH signalling in our experiment, and FSH activates CRTC2 but not CREB. Thus, our main focus was CRTC2. Law et al reported that FSH/GPCR/PKA could activate insulin pathway in other cells [48]; a finding which is of great interest. Whether and how FSH affects the Akt pathway in the liver requires further exploration. While cyclic AMP/PKA could interact with the insulin pathway in the liver, it is widely reported that the cyclic AMP/PKA pathway inactivates insulin signalling, thereby enhancing gluconeogenesis and glycogenolysis [14]. Especially in a fasted state, insulin signalling is inhibited to a large extent. In addition, entirely different biological actions would occur in different cells, although the same proteins are expressed. Landomiel et al reported that activated FSHRs can couple to multiple transduction mechanisms, not just in the cyclic AMP/PKA pathway, including Gαq and the epidermal growth factor receptor [49].
In conclusion, we present evidence supporting a direct role for FSH in the pathogenesis of unrestrained gluconeogenesis, a surprising example of a previously undescribed metabolic function of FSH. However, the entire mechanism involved in glucose metabolism appears to be highly complex [50], and additional efforts are necessary to explore the potential involvement of FSH in glycometabolism. In our study, through FSHR and targeting GRK2, FSH suppressed AMPK activation by increasing AMPK Ser485 phosphorylation, then increased hepatic Pepck and G6pase transcription via CRTC2, and consequently enhanced hepatic gluconeogenesis independent of E2. Our findings highlight a novel pathophysiological role for FSH in regulating glucose metabolism in the liver and may provide an additional strategy for treating fasting hyperglycaemia.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AICAR:
-
5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- ALT:
-
Alanine aminotransferase
- AMPK:
-
AMP-activated protein kinase
- AST:
-
Aspartate aminotransferase
- CaMKK:
-
Calcium/calmodulin-dependent protein kinase
- CRE:
-
Cyclic AMP response element
- CREB:
-
Cyclic AMP response element-binding protein
- CRTC2:
-
Cyclic AMP-regulated transcriptional coactivator 2
- E2:
-
Oestradiol
- FSH:
-
Follicle-stimulating hormone
- FSHR:
-
Follicle-stimulating hormone receptor
- G6Pase:
-
Glucose-6-phosphatase
- GPCR:
-
G-protein-coupled receptor
- GRK2:
-
GPCR kinase 2
- GSK3β:
-
Glycogen synthase kinase 3β
- H&E:
-
Haematoxylin and eosin
- H-FSH:
-
High-dose FSH
- ITT:
-
Insulin tolerance test
- Kitt:
-
Insulin sensitivity index
- L-FSH:
-
Low-dose FSH
- LKB1:
-
Liver kinase B1
- MAPK:
-
Mitogen-activated protein kinase
- OVX:
-
Ovariectomised
- PAS:
-
Periodic acid–Schiff
- PKA:
-
Protein kinase A
- PTT:
-
Pyruvate tolerance test
References
Heianza Y, Arase Y, Kodama S et al (2013) Effect of postmenopausal status and age at menopause on type 2 diabetes and prediabetes in Japanese individuals: Toranomon Hospital Health Management Center Study 17 (TOPICS 17). Diabetes Care 36:4007–4014
Mauvais-Jarvis F, Clegg DJ, Hevener AL (2013) The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 34:309–338
Kulaksizoglu M, Ipekci SH, Kebapcilar L et al (2013) Risk factors for diabetes mellitus in women with primary ovarian insufficiency. Biol Trace Elem Res 154:313–320
Banerjee AA, Mahale SD (2015) Role of the extracellular and intracellular loops of follicle-stimulating hormone receptor in its function. Front Endocrinol 6:110
Liu XM, Chan HC, Ding GL et al (2015) FSH regulates fat accumulation and redistribution in aging through the Galphai/Ca(2+)/CREB pathway. Aging Cell 14:409–420
Onori P, Mancinelli R, Franchitto A et al (2013) Role of follicle-stimulating hormone on biliary cyst growth in autosomal dominant polycystic kidney disease. Liver Int 33:914–925
Song Y, Wang ES, Xing LL et al (2016) Follicle-stimulating hormone induces postmenopausal dyslipidemia through inhibiting hepatic cholesterol metabolism. J Clin Endocrinol Metab 101:254–263
Sun L, Peng Y, Sharrow AC et al (2006) FSH directly regulates bone mass. Cell 125:247–260
Vila-Bedmar R, Cruces-Sande M, Lucas E et al (2015) Reversal of diet-induced obesity and insulin resistance by inducible genetic ablation of GRK2. Sci Signal 8:ra73
Taguchi K (2015) The role of GRK2 and its potential as a new therapeutic target in diabetic vascular complications. J Pharm Soc Jpn 135:961–967
Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther 133:40–69
Sharabi K, Tavares CD, Rines AK, Puigserver P (2015) Molecular pathophysiology of hepatic glucose production. Mol Asp Med 46:21–33
Jing Y, Liu W, Cao H et al (2015) Hepatic p38alpha regulates gluconeogenesis by suppressing AMPK. J Hepatol 62:1319–1327
Rui L (2014) Energy metabolism in the liver. Compr Physiol 4:177–197
Jitrapakdee S (2012) Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int J Biochem Cell Biol 44:33–45
Koo SH, Flechner L, Qi L et al (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437:1109–1111
Ning J (2011) Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology 152:3143–3154
Cao J, Meng S, Chang E et al (2014) Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J Biol Chem 289:20,435–20,446
Chen Z, Gaudreau R, Le Gouill C, Rola-Pleszczynski M, Stankova J (2004) Agonist-induced internalization of leukotriene B(4) receptor 1 requires G-protein-coupled receptor kinase 2 but not arrestins. Mol Pharmacol 66:377–386
Kim YD, Park KG, Lee YS et al (2008) Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 57:306–314
Miyake K, Ogawa W, Matsumoto M, Nakamura T, Sakaue H, Kasuga M (2002) Hyperinsulinemia, glucose intolerance, and dyslipidemia induced by acute inhibition of phosphoinositide 3-kinase signaling in the liver. J Clin Invest 110:1483–1491
Tao W, Wu J, Xie BX et al (2015) Lipid-induced muscle insulin resistance is mediated by GGPPS via modulation of the RhoA/Rho kinase signaling pathway. J Biol Chem 290:20,086–20,097
Liu NC, Lin WJ, Kim E et al (2007) Loss of TR4 orphan nuclear receptor reduces phosphoenolpyruvate carboxykinase-mediated gluconeogenesis. Diabetes 56:2901–2909
Gao D, Nong S, Huang X et al (2010) The effects of palmitate on hepatic insulin resistance are mediated by NADPH oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J Biol Chem 285:29,965–29,973
Malla R, Wang Y, Chan WK, Tiwari AK, Faridi JS (2015) Genetic ablation of PRAS40 improves glucose homeostasis via linking the AKT and mTOR pathways. Biochem Pharmacol 96:65
Zhang X, Xie X, Heckmann BL, Saarinen AM, Czyzyk TA, Liu J (2014) Targeted disruption of G0/G1 switch gene 2 enhances adipose lipolysis, alters hepatic energy balance, and alleviates high-fat diet-induced liver steatosis. Diabetes 63:934–946
Gerich JE (2010) Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabetic Med 27:136–142
Lin HV, Accili D (2011) Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 14:9–19
Rice S, Elia A, Jawad Z, Pellatt L, Mason HD (2013) Metformin inhibits follicle-stimulating hormone (FSH) action in human granulosa cells: relevance to polycystic ovary syndrome. J Clin Endocrinol Metab 98:E1491–E1500
Chen W, Sang JY, Liu DJ et al (2013) Desensitization of G-protein-coupled receptors induces vascular hypocontractility in response to norepinephrine in the mesenteric arteries of cirrhotic patients and rats. Hepatobiliary Pancreat Dis Int 12:295–304
Taguchi K, Matsumoto T, Kamata K, Kobayashi T (2012) Inhibitor of G protein-coupled receptor kinase 2 normalizes vascular endothelial function in type 2 diabetic mice by improving beta-arrestin 2 translocation and ameliorating Akt/eNOS signal dysfunction. Endocrinology 153:2985–2996
Viollet B, Foretz M, Guigas B et al (2006) Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol 574:41–53
Park JS, Cho MH, Ahn CW, Kim KR, Huh KB (2012) The association of insulin resistance and carotid atherosclerosis with thigh and calf circumference in patients with type 2 diabetes. Cardiovasc Diabetol 11:62
Penela P, Murga C, Ribas C, Lafarga V, Mayor F Jr (2010) The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol 160:821–832
Penela P, Ribas C, Mayor F Jr (2003) Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal 15:973–981
Jurado-Pueyo M, Campos PM, Mayor F, Murga C (2008) GRK2-dependent desensitization downstream of G proteins. J Recept Signal Transduct Res 28:59–70
Evron T, Daigle TL, Caron MG (2012) GRK2: multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol Sci 33:154–164
Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC (2005) A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med 11:952–958
Peregrin S, Jurado-Pueyo M, Campos PM et al (2006) Phosphorylation of p38 by GRK2 at the docking groove unveils a novel mechanism for inactivating p38MAPK. Curr Biol 16:2042–2047
Copps KD, White MF (2012) Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55:2565–2582
Viollet B, Guigas B, Sanz GN, Leclerc J, Foretz M, Andreelli F (2012) Cellular and molecular mechanisms of metformin: an overview. Clin Sci 122:253–270
Horman S, Vertommen D, Heath R et al (2006) Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 281:5335–5340
Woods A, Johnstone SR, Dickerson K et al (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13:2004–2008
Anderson KA, Means RL, Huang QH et al (1998) Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J Biol Chem 273:31,880–31,889
Hasenour CM, Berglund ED, Wasserman DH (2013) Emerging role of AMP-activated protein kinase in endocrine control of metabolism in the liver. Mol Cell Endocrinol 366:152–162
Altarejos JY, Montminy M (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 12:141–151
Wang Y, Inoue H, Ravnskjaer K et al (2010) Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci U S A 107:3087–3092
Law NC, White MF, Hunzicker-Dunn ME (2016) G protein-coupled receptors (GPCRs) that signal via protein kinase A (PKA) cross-talk at insulin receptor substrate 1 (IRS1) to activate the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. J Biol Chem 291:27,160–27,169
Landomiel F, Gallay N, Jegot G et al (2014) Biased signalling in follicle stimulating hormone action. Mol Cell Endocrinol 382:452–459
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Acknowledgements
We thank Y. Wang (School of Life Sciences, Tsinghua University, China) for providing mice; H. Choi (School of Biological Sciences and Technology, Chonnam National University, Republic of Korea), J. Staňková (Department of Paediatrics, University of Sherbrooke, Canada), F. Mayor (Department of Molecular Biology, Universidad Autónoma de Madrid, Spain) and J. L. Benovic (Department of Biochemistry and Molecular Biology, Thomas Jefferson University, USA) for providing plasmids.
Funding
This work was supported by the National Natural Science Foundation (81670796) and the National Key R&D Programme of China (2017YFC1309800 and 0909600).
Author information
Authors and Affiliations
Contributions
JZ and LG designed and supervised the project. JZ, LG, CY, YS, YG and XQ designed the experiments. XQ, YG and DK performed the experiments. XQ analysed the data and wrote the manuscript. CY, DK, LF and LZ participated in analysis and interpretation of data. All authors revised the manuscript critically and approved the final version for publication. LG is the guarantor of this work.
Corresponding authors
Ethics declarations
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material
ESM
(PDF 626 kb)
Rights and permissions
About this article

Cite this article
Qi, X., Guo, Y., Song, Y. et al. Follicle-stimulating hormone enhances hepatic gluconeogenesis by GRK2-mediated AMPK hyperphosphorylation at Ser485 in mice. Diabetologia 61, 1180–1192 (2018). https://doi.org/10.1007/s00125-018-4562-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-018-4562-x