
Abstract
Aims/hypothesis
The offspring of mothers with pre-gestational type 1 diabetes (PGDM) may be at increased risk of glucose intolerance and cardiovascular disease in childhood. The underlying causes of these observations, and whether they persist into adulthood, are unknown. The aim of the present study was to test the hypothesis that fetal chromosomal telomere oxidative DNA damage resulting from maternal PGDM programmes the offspring towards a senescent phenotype that is detectable in young adulthood.
Methods
We studied 21 young adult offspring (age 16–23 years) with a maternal history of PGDM and 23 age- and weight-matched controls with no maternal history of diabetes. All participants underwent anthropometric assessments, a standard 75 g OGTT, measurement of peripheral blood mononuclear cell and skin fibroblast telomere length, fibroblast senescence, cell DNA damage (by determination of 8-oxoguanine levels using flow cytometry), plasma lipoprotein profiles (determined by nuclear magnetic resonance) and plasma levels of soluble adhesion molecules and inflammatory markers.
Results
The groups did not differ significantly with respect to anthropometric measures, glucose tolerance, fasting and 2 h plasma insulin levels during OGTT, estimated peripheral insulin resistance, peripheral blood mononuclear cell or fibroblast telomere length, DNA damage or senescence in vitro, plasma NMR lipoprotein profiles or levels of high-sensitivity C-reactive protein. Plasma concentrations of soluble intercellular adhesion molecule 1 (sICAM-1; p < 0.05) and IL-6 (p = 0.08) were higher in the PGDM offspring.
Conclusions/interpretation
Young adult offspring of mothers with PGDM do not differ in terms of glucose tolerance, DNA damage or telomere length from controls of the same weight and BMI. This does not preclude such abnormalities at an earlier age, but there is no evidence of telomere damage as a pre-programming mechanism in the young adults enrolled in this study.
Similar content being viewed by others


Introduction
The view that an adverse intrauterine environment during pregnancy contributes to poor long-term health in the offspring is well established [1–3]. There is some evidence that the offspring of women with pre-gestational type 1 diabetes (PGDM), the archetypal adverse intrauterine environment, are at risk of developing glucose intolerance and cardiovascular disease in young childhood and adolescence [4–10]. The reason(s) for these observations are unknown, but the literature has emphasised nutritional or genetic models of pre-programming [1–3]. It is unknown whether these abnormalities persist into adulthood.
Telomeres are repeat DNA sequences that cap eukaryotic chromosomes and shorten at each cell division [11, 12]. Once telomeres are shortened to a critical length, cells are triggered into replicative senescence, resulting in cell cycle arrest and phenotypic and functional alterations [11, 12]. Previous studies have demonstrated telomere shortening in adults with type 1 diabetes [13, 14] and we and others have shown that patients with type 2 diabetes have shorter telomeres than age-matched controls [15, 16]. Rates of telomere shortening at cell division are highly dependent on oxidatively induced strand breaks in telomeric DNA and oxidative DNA damage [11, 12, 17, 18]. Feto-placental telomere attrition is an attractive hypothetical bridge between the adverse intrauterine environment and pre-programming of the offspring towards senescent phenotypes [19–21], as an adverse intrauterine environment increases oxidative damage to feto-placental DNA and fetal vascular endothelium [22–24]. We have previously suggested that this mechanism could account for a senescent phenotype in the adult offspring of women with type 1 diabetes [25].
In the present study we tested the hypothesis that young adult offspring of women with PGDM would demonstrate evidence of telomere attrition, glucose intolerance and increased vascular risk markers.
Methods
Recruitment strategy
Recruitment to this project began on 1 February 2006 and finished on 1 February 2008. We reviewed the cases of all women with PGDM who received their pregnancy care and delivered a live baby at our site (Norfolk and Norwich University Hospital NHS Trust), whose offspring would be 16–23 years old (inclusive) at the time of the study. We excluded any women who had a baby born with a major neurological or cardiac malformation. Between 1977 and 2007, all pregnant women with PGDM in our total population of 500,000 have been delivered at this site. Between 1984 and 1991 (excluding 1989 because of missing data), 117 women with PGDM gave birth at our site and 92 met the offspring age criteria. These 92 mothers were invited to participate in the study and asked to inform their 16–23-year-old offspring about the study and invite them to participate.
Of this population, 18 (19.6%) did not respond to recruitment contact or were untraceable, and 13 (14.1%) declined to contact their offspring. In addition, a further 12 mother–offspring pairs (13.0%) were excluded for the following reasons : offspring major cardiac malformation (n = 1), cerebral palsy (n = 1), twin pregnancy (n = 1), classical type 1 diabetes diagnosed in childhood (n = 3), Addison’s disease and type 1 diabetes (n = 1), severe rheumatoid arthritis (n = 1), current offspring pregnancy (n = 2), missed appointments or outside age range (n = 2). Of the remaining 49 mother–offspring pairs, 28 (57.1%) of the offspring aged 16 to 23 years declined to participate, leaving a study population of 21 offspring of PGDM, or 22.8% of the total possible. Controls were selected by media advertisement inviting individuals who were born at the Norfolk and Norwich University Hospital NHS Trust between 1984 and 1991 whose mother did not have diabetes at the time of conception or during pregnancy. No selection criteria other than age and location of birth were applied to the recruitment of the control group; no attempt was made to weight-match controls (n = 23). All participants, except one control, were white. All individuals participated after providing written informed consent, and after review and approval of the study by the local research ethics committee.
Study protocol
All participants underwent a standard early morning OGTT, using 75 g of anhydrous glucose, after 10 h of fasting. Peripheral venous samples were collected from individuals in a recumbent position at −10, −5, 0, 60 and 120 min following the oral consumption of glucose. All plasma analyses were undertaken on fasting blood samples, other than plasma insulin, which was measured at time 0 and 120 min of the OGTT. Anthropometric measurements were made after the OGTT. Participants were also asked if they were prepared to undergo a transdermal skin biopsy for skin fibroblast studies.
Anthropometric and clinical measurements
We recorded height (cm; without shoes), weight (kg; unclothed), waist circumference (cm), waist:hip ratio, BMI (kg/m2), skinfold thickness (in mm; triceps, abdominal and subscapular). Body fat mass and fat-free mass were measured by bioimpedance (segmental Body Composition Analyser BC-418MA Tanita UK, West Drayton, UK). Peripheral vascular disease was defined by determination of the ankle–brachial pressure index using Doppler pressure measurement [26]. The blood pressure of the participants was measured by an Accoson sphygmomanometer (BS 2744; Accoson Works, Harlow, UK) after they had rested for 5 min, with three readings taken at 1 min intervals and the mean of the last two readings recorded. At the end of the visit each participant was given a food diary to complete and return. Participants were asked to record a 3 day dietary history over 1 week, including two weekdays and one weekend day. The diaries were analysed by the Department of Nutrition and Dietetics (Norfolk and Norwich University Hospital NHS Trust) using CompEat software [27]. Each adult participant was assigned a social deprivation score based on current address, derived by the UK government for all small areas in England from a range of economic, social and housing variables [28]. In addition, for each adult volunteer, an estimate of relative birthweight ratio, derived from actual recorded birthweight and normative data for gestational age- and sex-related 50th percentile for birthweight was calculated [29].
Insulin, glucose and the homeostatic assessment model of insulin resistance
Fasting and 2 hour plasma insulin levels were measured using a commercially available human insulin specific ELISA (Bio-stat Diagnostic Healthcare, Stockport, UK). Plasma glucose was analysed by the glucose oxidase method (Abbott Laboratories, Maidenhead, UK). Homeostasis model assessment of insulin resistance (HOMA-IR) was used to estimate peripheral insulin sensitivity [30]. Fasting glucose and insulin values are shown as the mean of samples collected at −10, −5 and 0 min.
Monoclonal antibodies
Mouse anti-human CD3:AF647, CD19:AF647, CD14:AF647 and CD45RA:PE antibodies were purchased from AbD Serotec (Oxford, UK), and mouse anti-human CD3:PE, CD19:PE-Cy7, CD14:PE-TR and CD45RA:PE-Cy5 antibodies were purchased from Caltag Medsystems (Buckingham, UK).
Isolation of peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMCs) were isolated using BD Citrate Vacutainer CPT tubes (Southern Syringe Services, Leicester, UK) according to the manufacturer’s instructions. Briefly, the cells were centrifuged at 1,500 g for 20 min, washed twice in PBS and then stored in liquid nitrogen until analysis. On average, the total number of viable cells was 95% when counted under a microscope with a haemocytometer using trypan blue staining.
Isolation and culture of skin fibroblasts
Human skin fibroblasts were derived from skin biopsy samples taken from the inside upper arm of 15 individuals, as previously described [31]. The biopsy samples were transferred to cell culture dishes (60 mm diameter) and incubated at 37°C, in an atmosphere of 5% CO2 and 95% air, in Eagle’s minimum essential medium (MEM; Sigma-Aldrich, Gillingham, UK) supplemented with 10% (vol./vol.) fetal bovine serum to allow fibroblast outgrowth. After two passages the cells were grown to confluence, washed and either used in the senescence assay or stored in liquid nitrogen until needed for telomere analysis.
Telomere length analysis
Telomere length in individual PBMCs was measured using a Telomere Peptide Nucleic Acid (PNA) kit/fluorescein isothiocyanate (FITC) for flow cytometry (Bio-stat Diagnostic Healthcare, Stockport, UK). Telomere length was measured in different leucocyte subsets as previously described [18], with minor modifications. Briefly, 5 × 105 cells were incubated for 30 min at 4°C with either CD3:AF647, CD19:AF647 or CD14:AF647. The labelled cells were resuspended in either hybridisation solution containing the telomere probe or hybridisation solution alone as a negative control. The cells were then incubated for 10 min at 80°C in a water bath, followed by overnight hybridisation in the dark at room temperature. The cells were incubated twice for 10 min at 40°C with wash buffer, centrifuged and then resuspended in 50 μl of PBS. The cells labelled with CD3:AF647 and CD19:AF647 were further incubated with CD45RA:PE for 30 min at 4°C, washed once and then resuspended in DNA staining solution to identify cells in G0/G1 of the cell cycle. After 2–3 h 20,000 events were acquired on a Cytomics FC500MPL flow cytometer (Beckman Coulter, High Wycombe, UK) and CXP software (Beckman Coulter) was used for analysis. The different leucocyte subsets were identified by their forward and side scatter properties and staining with the appropriate antibody. Naive T cells were identified as CD3+/CD45RA+, memory T cells as CD3+/CD45RA−, B cells as CD19+ and monocytes as CD14+. The mean telomere fluorescence of each cell type was calculated by subtracting the mean background fluorescence of unlabelled control cells from the mean fluorescence of cells hybridised in the presence of the FITC-PNA probe. Telomere fluorescence was converted into molecular equivalents of soluble fluorochrome (MESF) using Quantum FITC premixed low-level MESF beads (Bangs Laboratories, Fishers, IN, USA), which were run in each experiment. Telomere length in cultured fibroblasts was measured using the Telomere PNA kit/FITC, as described above. In addition, one tube of cells per sample was incubated with mouse anti-human CD90/Thy 1 (FITC), a fibroblast marker, for 30 min at 4°C, fixed with 1% (vol./vol.) paraformaldehyde and stored at 4°C until analysed by flow cytometry.
Oxidative DNA damage
An OxyDNA kit (Biotrin, Dublin, Republic of Ireland) was used to evaluate oxidative DNA damage in different leucocyte subsets as previously described [18], with some modifications. Briefly, 5 × 105 cells were washed in PBS and incubated for 15 min at room temperature with the antibodies CD3:PE, CD14:PE-TR, CD19:PE-Cy7 and CD45RA:PE-Cy5 (Caltag Medsystems, Buckingham, UK) to identify the different cell types. After washing, the cells were fixed and permeabilised with a Fix and Perm cell permeabilisation kit (Invitrogen, Paisley, UK), followed by a further wash. The cells were then incubated for 1 h at 37°C with 50 μl of Biotrin blocking solution, washed and incubated for 1 h in the dark at room temperature with 100 μl of FITC-conjugate probe specific for 8-oxoguanine. Following incubation, cells were washed twice, analysed by flow cytometry with the acquisition of 20,000 events and the mean fluorescence intensity recorded. A control sample was run in each experiment to account for inter-assay variation.
Fibroblast senescence assay
Fibroblast senescence was measured by senescence-associated β-galactosidase activity using the fluorogenic substrate 5-dodecanoylamino fluorescein di-β-d-galactopyranoside (C12FDG; Invitrogen, Paisley, UK), as previously described [32] with some minor modifications. Briefly, cells were incubated with either C12FDG (33 μmol/l), or DMSO as a control, for 2.5 hours. Following a wash with PBS the cells were trypsinised, analysed by flow cytometry; 10,000 events were acquired and the mean fluorescent intensity recorded.
HbA1c, lipid and lipoprotein profiles
HbA1c was measured by HPLC on a Biomen HA-8160 analyser (Menarini Diagnostics, Wokingham, UK). Plasma lipoprotein analysis was performed by Lipomed Biosciences (Liposcience, Raleigh, NC, USA) using nuclear magnetic resonance (NMR) spectroscopy [33].
Assays
Commercially available ELISA kits were used to analyse plasma levels of TNFα, IL-6, soluble cell adhesion molecules (soluble intercellular adhesion molecule 1 [sICAM-1] and soluble vascular cell adhesion molecule 1 [sVCAM-1]) (all R&D Systems Europe, Abingdon, UK) high-sensitivity C-reactive protein (hs-CRP) (Kalon Biological, Guildford, UK).
Data analysis
Data are expressed as mean (SD) or as median (interquartile range) as appropriate. All data were normally distributed, except plasma IL-6 and hs-CRP concentrations. Differences between groups were analysed by the unpaired Student’s t test or Mann–Whitney U test as appropriate. A p value of <0.05 was considered significant. In our recent studies [18], monocyte telomere length was 5. 74 (0.88) MESF in controls, which suggested an initial sample size of 30 in each group was necessary to provide 89% power at the 5% level to detect a 0.75 SD difference between groups. In the present study, based on actual mean and SD data for telomere length in PBMCs in both groups (Table 4), the study had 90.1% power at the 5% level to detect a 0.75 SD in mean telomere length compared with the control group without a maternal history of diabetes.
Results
Baseline clinical characteristics
Baseline clinical data for the mothers with PGDM and the mothers in the control group are shown in Table 1. Mothers in the control group were slightly older (p < 0.05) and had a longer duration of pregnancy (p < 0.0001). The offspring born to mothers with PGDM had a higher birthweight ratio but did not differ from the control offspring with respect to any other clinical or anthropometric measurement (Table 1). In particular, the two groups of offspring were almost identical in terms of waist circumference, waist:hip ratio and BMI (Table 1). Eleven of the 21 PGDM offspring and 14 of the 23 controls completed the 3 day dietary history. No significant differences in dietary intake were observed between the two groups (data not shown).
Baseline metabolic characteristics of the PGDM and control offspring
The PGDM offspring did not differ significantly from the control offspring in terms of mean plasma glucose or insulin concentrations during the OGTT, HOMA-IR assessments of insulin sensitivity, or fasting lipid profiles (Table 2). NMR lipoprotein profiles did not differ significantly between groups in terms of particle size or the concentration of any particle type or subtype (Table 3; remaining NMR lipoprotein data not shown). Plasma sICAM-1 concentrations were significantly higher in the group of PGDM offspring (p < 0.05), and plasma IL-6 concentrations were also higher, but not significantly, in this group (p = 0.08).
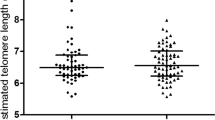
Telomere length and oxidative DNA damage in nucleated blood cells
There were no significant differences in telomere length or DNA damage in the PBMCs overall or any subset of PBMCs (Table 4).
Telomere length and senescence in cultured skin fibroblasts
There were no significant differences in fibroblast telomere length or in the percentage of skin fibroblasts with a senescence phenotype in cells obtained from skin biopsies from PGDM offspring compared with those from control offspring (Table 4).
Relationships between variables
There were no significant relationships between telomere length (in either group) and birthweight, deprivation index, birthweight ratio, inflammatory marker concentrations or estimates of insulin resistance (data not shown; all p > 0.1). In addition, there was no significant relationship (all p > 0.1) between pre-conception levels of HbA1c in the PGDM mothers and any variable in the young adult offspring, including plasma glucose during an OGTT, NMR LDL particle size, HOMA–IR, telomere length or DNA damage.
Discussion
We found that the young adult offspring (age 16–23 years) of women with PGDM were indistinguishable from controls without this maternal history in terms of clinical characteristics, NMR plasma lipoprotein profiles, insulin sensitivity and glucose tolerance. Although these results do not exclude such abnormalities in a younger age group, or the development of such derangements in middle age, they are in contrast with the commonly held view that these young offspring are at substantial risk of glucose intolerance and at increased cardiovascular risk. We also found no evidence of telomere length abnormalities, oxidative DNA damage or in vitro senescence in several cell types in the PGDM offspring. These observations do not exclude such abnormalities in younger offspring, but do not provide evidence for a persistent detectable senescent phenotype characterised by telomere shortening in this group.
The view that PGDM offspring are at increased risk of glucose intolerance in childhood and adolescence has been held for more than a decade, since Silverman et al. [6] described a heterogeneous group of 88 offspring (age 10–16 years old) born to women with diabetes during pregnancy (72% had PGDM). According to the results of a standard 1.75 g/kg OGTT, the overall prevalence of impaired glucose tolerance (IGT) was 19.5% in the offspring of women with diabetes compared with 2.5% in the control offspring group without this maternal history (p < 0.005). IGT was uncommon under the age of 10 years old. However, the offspring of mothers with diabetes were significantly more obese (mean BMI 2.8 kg/m 2 higher; p = 0.001), and there were differences in ethnicity between the two groups (p = 0.073). Furthermore, there was little analysis of the different maternal diabetic groups. Similar observations have been made in more homogeneous groups of offspring of mothers with type 1 diabetes [7] studied at a younger age (2–5 years), particularly in those who were obese or macrosomic at birth. Other uncontrolled observational studies have also described a high prevalence (9.4 %) of IGT in the very young offspring of PGDM mothers (age 1–4 years), which was even higher (17.4%) in those aged 5–9 years [9]. In contrast, others have found no difference in insulin sensitivity, fasting plasma insulin or fasting plasma glucose in young PGDM offspring (age 5–11 years) with the same BMI as controls [4]. As far as we are aware, only one substantial study [5] has examined older (age 18–27 years) PGDM offspring (n = 160), and this reported that 17 offspring (11%) had type 2 diabetes, IGT or impaired fasting glucose compared with 5 out of 128 (4%) controls at a mean age of 22.5 years. Although the PGDM offspring were significantly more obese (BMI 24.4 vs 22.4 kg/m2) and from a lower social class (p < 0.001), this increase in prevalence remained significant after some adjustment for maternal and offspring weight and BMI, with a reported OR of 4.0 (95% CI 1.31–12.33). In the present study the two groups were identical in terms of age, BMI, waist:hip ratio and social class, and we found no differences in glucose tolerance, NMR lipoprotein profiles or insulin sensitivity. This may suggest that obesity or increased BMI is the primary abnormality in the PGDM offspring that drives glucose intolerance. IGT is uncommon in children, and the high prevalence of IGT in the young children of PGDM mothers is a striking observation [6, 7, 9]. It should be stressed, however, that the prevalence of IGT and glucose intolerance is higher in obese and overweight children and adolescents even without such a maternal diabetes history [34, 35], and that it is unclear whether glucose intolerance in these children is independent of increased BMI. There are also other possible explanations for childhood glucose intolerance and increased BMI in the offspring of PGDM mothers. Breast milk from women with type 1 diabetes has a higher glucose and energy content than breast milk from women without diabetes [36]. In some studies of the offspring of mothers with type 1 diabetes, the only variable that independently predicted offspring relative body weight at 2 years old was the volume of breast milk from the diabetic mother ingested during the first week of life, and the risk of childhood IGT was significantly lower in offspring fed banked breast milk from mothers without diabetes [37]. Other data have demonstrated a direct relationship between the duration of breast feeding and the 2 h plasma glucose level measured during a standard OGTT (r = 0.46; p = 0.002) in these offspring [7]. In the UK, recently published national guidance on diabetes and pregnancy has recommended that women with type 1, type 2 or gestational diabetes should be advised that their offspring are at increased risk of diabetes and obesity in later life [38]. Our data suggest that this advice may be premature for women with type 1 diabetes and that there is a need for larger longitudinal cohort studies following glucose tolerance and cardiovascular risk markers in these offspring from birth to young adulthood.
Type 1 diabetes offers a very hostile intrauterine environment, and is associated with high rates of fetal morbidity, including increased risks of congenital malformation, spontaneous abortion and perinatal death [39]. The fetal morbidity associated with maternal diabetes (and other embryopathies) reflects increased intrauterine oxidative stress and fetal oxidative DNA damage [11, 12, 17–24]. The increased fetal DNA damage described in diabetes pregnancy must lead to increased telomeric DNA damage, as the GGG sequence in telomeric DNA is particularly susceptible to DNA damage [17]. It therefore follows that elevated levels of telomere attrition at cell division during fetal development should lead to offspring born programmed towards senescent angiogenic and endothelial precursors [19–21], with shorter telomeres than their peers born to mothers without diabetes. We hypothesised that this would extend into early adulthood [30], as others have shown in animal models that adverse fetal environments are associated with shortened telomere lengths and adult morbidity [40–42]. Telomere attrition has attracted attention in the last 2 years as a possible predeterminant of later risk of cardiovascular disease [19–21]. However, we found no evidence of residual telomere shortening in multiple blood cell lines or in fibroblasts, and no evidence of an increased risk of senescence in vitro in fibroblasts. This does not exclude the presence of these abnormalities in neonates or younger children, but we found no evidence of the continuation of any assumed telomeric damage into young adulthood in these offspring.
We found that the PGDM offspring had a significantly higher plasma sICAM-1 concentration than the control offspring. Nelson et al. [43] have recently reported a similar finding in umbilical cord blood plasma from PGDM offspring, and other groups have described higher concentrations of other soluble adhesion molecules, such as sVCAM-1 and E-selectin [44] in younger PGDM offspring. Increased plasma concentrations of sICAM-1 in populations with or without diabetes are an independent marker of increased vascular risk [44]. Elevated sICAM-1 concentrations are felt to be a marker of endothelial dysfunction and enhanced ICAM-1 shedding from endothelial cells [45]. This observation is the only distinguishing feature of the PGDM offspring compared with the control offspring, and may imply some degree of endothelial dysfunction, but other endothelial markers were not measured in this study, and we cannot confirm whether this abnormality has persisted since birth throughout childhood. However, results from the Framingham Offspring Study indicate that there is significant heritability of sICAM-1, suggesting a genetic component to systemic inflammation [46]. Telomere length is also strongly genetically determined [47]. It has recently been shown that telomere length is shorter in healthy offspring with an increased familial risk of coronary artery disease, and a significant positive correlation in mean telomere length was observed between offspring and their parents [48], but the possibility of postnatal factors influencing telomere length could not be excluded.
The present study is subject to certain limitations. First, the PGDM mothers had excellent glycaemic control during pregnancy and before conception. It is possible that the offspring of PGDM mothers with poorer glycaemic control during pregnancy would show abnormalities in glucose tolerance or telomere attrition. However, it should be stressed that PGDM mothers with good glycaemic control still suffer a high rate of fetal morbidity [49], and diabetic populations with good glycaemic control still demonstrate evidence of DNA damage and telomere attrition [18, 50]. Second, we cannot exclude the possibility that the offspring of PGDM mothers who declined to participate were different from those who consented with respect to diet, lifestyle, social class and anthropometric measurements. However, the PGDM offspring were identical to the non-selected control group enrolled from the general population with respect to diet and deprivation indices, which makes this bias less likely. Finally, the study was powered to detect differences in telomere length in the offspring groups, not glucose tolerance in the offspring. However, the study had 91% power at the 5% level to detect a 0.5 SD difference between the two groups in terms of fasting plasma glucose or 2 h plasma glucose during the OGTT (0.2 and 0.65 mmol/l, respectively, based on control data).
In summary, we found that young adults with a maternal history of PGDM did not differ from controls with respect to glucose tolerance, NMR lipoprotein profiles, insulin sensitivity, body fat mass, BMI or anthropometric measurements. This is in contrast with other published data, and may reflect the lack of difference in BMI and body fat mass between the two groups in the present study. We found no evidence that hypothesised differences in telomere length at birth are present in this age group, but we did find some minor differences in soluble adhesion molecule concentrations, which may imply some degree of endothelial dysfunction even in this young adult population. There is a need for a large prospective cohort study of PGDM offspring from birth to young adulthood, as it is possible that the substantial abnormalities in glucose intolerance described in very young offspring of PGDM mothers may not persist.
Abbreviations
- HOMA-IR:
-
homeostasis model assessment of insulin resistance
- hs-CRP:
-
high-sensitivity C-reactive protein
- IGT:
-
impaired glucose tolerance
- MESF:
-
molecular equivalents of soluble fluorochrome
- NMR:
-
nuclear magnetic resonance
- PBMCs:
-
peripheral blood mononuclear cells
- PGDM:
-
pre-gestational type 1 diabetes
- sICAM-1:
-
soluble intercellular adhesion molecule 1
- sVCAM-1:
-
soluble vascular cell adhesion molecule 1
References
Godfrey KM, Barker DJ (2000) Fetal nutrition and adult disease. Am J Clin Nutrition 71:1344S–1522S
Sattar N (2004) Do pregnancy complications and CVD share common antecedents. Atheroscler Suppl 5:3–7
Hattersley AT, Tooke JE (1999) The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet 353:1789–1792
Manderson JG, Mullan B, Patterson CC, Hadden DR, Traub AI, McCance D (2002) Cardiovascular and metabolic abnormalities in the offspring of diabetic pregnancy. Diabetologia 45:991–996
Clausen TD, Mathiesen ER, Hansen T et al (2008) High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes—the role of intrauterine hyperglycaemia. Diabetes Care 31:340–346
Silverman BL, Cho N, Rizzo TA, Metzger BE (1998) Long term effects of the intrauterine environment. Diabetes Care 21:B142–B149
Buinauskiene J, Baliutaviciene D, Zalinkevicius R et al (2004) Glucose tolerance of 2-5 year old offspring of diabetic children. Pediatr Diabetes 5:143–146
Weiss P, Scholz HS, Haas J et al (2000) Long term follow up of infants of mothers with type 1 diabetes: evidence for hereditary and nonhereditary transmission of diabetes and precursors. Diabetes Care 23:905–911
Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G (1997) Glucose tolerance and insulin secretion in children of mothers with pregestational IDDM or gestational diabetes. Diabetologia 40:1094–1100
Persson B, Gentz J, Möller E (1984) Follow-up of children of insulin-dependent (type I) and gestational diabetic mothers. Growth pattern, glucose tolerance, insulin response, and HLA types. Acta Paediatr Scand 73:778–784
von Zglinicki T (2002) Oxidative stress shortens telomeres. Trends Biochem Sci 27:339–344
von Zglinicki T (2000) Role of oxidative stress in telomere length regulation and replicative senescence. Ann N Y Acad Sci 908:99–110
Jeanclos E, Krolewski A, Skurnick J et al (1998) Shortened telomere length in white blood cells of patients with IDDM. Diabetes 47:482–486
Uziel O, Singer JA, Danicek V et al (2007) Telomere dynamics in arteries and mononuclear cells of diabetic patients: effect of diabetes and of glycemic control. Exp Gerontol 42:971–978
Fitzpatrick AL, Kronmal RA, Gardner JP et al (2007) Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol 165:14–21
Adaikalakoteswari A, Balasubramanyam M, Ravikumar R, Deepa R, Mohan V (2007) Association of telomere shortening with impaired glucose tolerance and diabetic macroangiopathy. Atherosclerosis 195:83–89
Petersen S, Saretzki G, von Zglinicki T (1998) Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp Cell Res 239:152–160
Sampson MJ, Winterbone M, Hughes JC (2006) Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care 29:283–289
Stefanec T (2004) How the endothelium and its bone marrow derived progenitors influence development of disease. Med Hypotheses 62:247–251
Demerath EW, Cameron N, Gillman MW (2004) Telomeres and telomerase in the fetal origins of cardiovascular disease: a review. Hum Biol 76:127–134
Edo MD, Andres V (2005) Aging, telomeres and atherosclerosis. Cardiovasc Res 66:213–221
Wiktor H, Kankofer M, Schmerold I et al (2004) Oxidative DNA damage in placentas from normal and pre-eclamptic pregnancies. Virchows Arch 445:74–78
Bekaert S, Derradji H, Baatout S (2004) Telomere biology in mammalian germ cells and during development. Dev Biol 274:15–30
Kudo T, Izutsu T, Sato T (2000) Telomerase activity and apoptosis as indicators of ageing in placenta with and without intrauterine growth retardation. Placenta 21:493–500
Sampson MJ, Hughes DA (2006) Chromosomal telomere attrition as a mechanism for the increased risk of epithelial cancers and senescent phenotypes in type 2 diabetes. Diabetologia 23:1008–1015
Ankle Brachial Index Collaboration (2008) Ankle brachial index combined with Framingham risk score to predict cardiovascular events and mortality: a meta-analysis. JAMA 300:197–208
Bingham SA, Welch AA, McTaggart A et al (2001) Nutritional methods in the European Prospective Investigation of Cancer in Norfolk. Public Health Nutr 4:847–858
The English indices of deprivation 2007. Available from http://www.communities.gov.uk/publications/communities/indiciesdeprivation07, accessed October 2008
Gairdner D, Pearson (1971) A growth chart for premature and other infants. Arch Dis Child 46:783–787
Wallace TM, Matthews DR (2002) The assessment of insulin resistance in man. Diabetic Med 19:527–534
Wall SJ, Sampson MJ, Levell N, Murphy G (2003) Elevated matrix metalloproteinase-2 and -3 production from human diabetic dermal fibroblasts. Br J Dermatol 149:13–16
Kurz DJ, Decary S, Hong Y, Erusalimsky JD (2000) Senescence-associated β-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113:3613–3622
Otvos JD (1997) Measurement of lipoprotein subclass profiles by nuclear magnetic resonance spectroscopy. In: Rifai N, Warnick G, Dominiczak MH (eds) Handbook of lipoprotein testing. AACC Press, Washington, pp 497–508
Mazur A, Grzywa M, Malecka-Tendera E, Telega G (2007) Prevalence of glucose intolerance in school age children. Population based cross-sectional study. Acta Paediatr 96:1799–1802
Sinha R, Fisch G, Teague B (2002) Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N Engl J Med 346:802–810
Jovanovic-Petersen L, Fuhrmann K, Hedden K, Walker L, Peterson CM (1989) Maternal milk and plasma glucose and insulin levels: studies in normal and diabetic subjects. J Am Coll Nutr 8:125–131
Plageman A, Harder T, Franke K, Kohlhoff R (2002) Long-term impact of neonatal breast feeding on body weight and glucose tolerance in children of diabetic mothers. Diabetes Care 25:16–22
National Institute for Health and Clinical Excellence (2008) Diabetes in pregnancy: management of diabetes and its complications from pre-conception to the postnatal period. Available from http://www.nice.org.uk/guidance/index.jsp?action=byId&o=11626, accessed October 2008
Macintosh MC, Fleming KM, Bailey JA et al (2006) Perinatal mortality and congenital abnormalities in babies of women with type 1 or type 2 diabetes in England, Wales and Northern Ireland: population-based study. BMJ 333:177–181
Ozanne SE, Fernandez-Twinn D, Hales CN (2004) Fetal growth and adult disease. Semin Perinatol 28:81–87
Hales CN (2001) Suicide of the nephron. Lancet 357:136–137
Jennings BJ, Ozanne SE, Hales CN (2000) Nutrition, oxidative damage, telomere shortening and cellular senescence: individual or connected agents of ageing. Mol Genet Metab 71:32–42
Nelson SM, Sattar N, Freeman DJ, Walker JD, Lindsay RS (2007) Inflammation and endothelial activation is evident at birth in offspring of mothers with type 1 diabetes. Diabetes 56:2697–2704
Witte DR, Broekmans WMR, Kardinaal AFM et al (2003) Soluble intercellular adhesion molecule 1 and flow-mediated dilatation are related to the estimated risk of coronary heart disease independently from each other. Atherosclerosis 170:147–153
Szekanecz Z, Shah MR, Pearce WH, Koch AE (1994) Intercellular adhesion molecule-1 (ICAM-1) expression and soluble ICAM-1 (sICAM-1) production by cytokine-activated human aortic endothelial cells: a possible role for ICAM-1 and sICAM-1 in atherosclerotic aortic aneurysms. Clin Exp Immunol 98:337–343
Keaney JF Jr, Massaro JM, Larson MG et al (2004) Heritability and correlates of intercellular adhesion molecule-1 in the Framingham Offspring Study. J Am Coll Cardiol 44:168–173
Slagboom PE, Droog S, Boomsma DI (1994) Genetic determination of telomere size in humans: a twin study of three age groups. Am J Hum Genet 55:876–882
Brouilette SW, Whittaker A, Stevens SE, van der Harst P, Goodall AH, Samani NJ (2008) Telomere length is shorter in healthy offspring of subjects with coronary artery disease: support for the telomere hypothesis. Heart 94:422–425
Temple RC, Aldridge VJ, Murphy HR (2006) Prepregnancy care and pregnancy outcomes in women with type 1 diabetes. Diabetes Care 29:1744–1749
Hannon-Fletcher MP, O’Kane MJ, Moles KW, Weatherup C, Barnett CR, Barnett YA (2000) Levels of peripheral blood cell DNA damage in insulin dependent diabetes mellitus human subjects. Mutat Res 460:53–60
Acknowledgements
This study was funded by a project grant from Diabetes UK (BDA:RD06/0003397). We would like to thank R. Bongaerts and K. Ivory for their expert advice on flow cytometric techniques. We are grateful to the UK Biotechnology and Biological Sciences Research Council for additional support; J. Thorp (Norfolk and Norwich University Hospital), B. Johal and S. Kelly (Ipswich Hospital NHS Trust); and to G. Zerbini (Istituto Scientifico San Raffaele, Milan, Italy) for advice on fibroblast culture.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cross, J.A., Brennan, C., Gray, T. et al. Absence of telomere shortening and oxidative DNA damage in the young adult offspring of women with pre-gestational type 1 diabetes. Diabetologia 52, 226–234 (2009). https://doi.org/10.1007/s00125-008-1207-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1207-5