
Abstract
Key message Major A 1 cytoplasm fertility restoration loci, Rf 2 and Rf 5 , were found in the West African sorghum. A potential causative mutation for Rf 2 was identified. KASP markers were validated on independent material.
Abstract
To accelerate the identification and development of hybrid parental lines in West African (WA) sorghum, this study aimed to understand the genetics underlying the fertility restoration (Rf) in WA A1 cytoplasmic male sterility system and to develop markers for a routine use in WA breeding programs. We genotyped by sequencing three F2 populations to map the Rf quantitative trait loci (QTL), validated the molecular KASP markers developed from those QTL in two F2:3 populations, and assessed the most promising markers on a set of 95 R- and B-lines from WA breeding programs. Seven QTL were found across the three F2 populations. On chromosome SBI-05, we found a major fertility restorer locus (Rf5) for two populations with the same male parent, explaining 19 and 14% of the phenotypic variation in either population. Minor QTL were detected in these two populations on chromosomes SBI-02, SBI-03, SBI-04 and SBI-10. In the third population, we identified one major fertility restorer locus on chromosome SBI-02, Rf2, explaining 31% of the phenotypic variation. Pentatricopeptide repeat genes in the Rf2 QTL region were sequenced, and we detected in Sobic.002G057050 a missense mutation in the first exon, explaining 81% of the phenotypic variation in a F2:3 population and clearly separating B- from R-lines. The KASP marker developed from this mutation stands as a promising tool for routine use in WA breeding programs.
Similar content being viewed by others

Introduction
Sorghum bicolor (L.) Moench is of vital importance for many smallholder farmers and their families in various regions of West Africa (WA). However, average grain yields for farmers in this zone are of 1 t/ha. Sorghum hybrids for WA were explored between the 1960s and 1990s (Andrews 1975; Toure and Scheuring 1982; Atokple 2003), but no hybrids were released or commercialized. A major challenge with the initial hybrids was their poor and unacceptable grain quality (Toure and Scheuring 1982). Recent work in WA to develop both male and female parents using the widely cultivated sorghum Guinea-race germplasm has resulted in hybrids with suitable grain quality combined with yield superiorities over farmer’s local varieties under diverse productivity conditions (Rattunde et al. 2013; Kante et al. 2017; Weltzien et al. 2018). The positive results with these hybrids under on-farm farmer-managed testing, and indications that farmers are starting to adopt these new hybrids (Smale et al. 2014) justify establishing a full-scale hybrid breeding pipeline to produce hybrids that meet farmers’ demands in this major sorghum producing zone.
Intensive hybrid breeding and seed production in many crop species were made possible by the identification and characterization of a stable and heritable cytoplasmic male sterility (CMS) mechanism. CMS is a maternally inherited defect where, as the result of specific nuclear and mitochondrial interactions, plants fail to produce functional pollen, or to ensure normal anther dehiscence, without affecting the female fertility (Duvick 1959; Laughnan and Gabay-Laughnan 1983; Hanson and Conde 1985; Levings and Brown 1989). It is attributed to abnormal transcripts usually coding for chimeric open reading frames (ORFs) (Schnable and Wise 1998; Tang et al. 1998; Hanson and Bentolila 2004). A CMS system depends therefore on a set of male-sterility-causing cytoplasms and dominant or recessive alleles in the nuclear genome, which either restore the fertility or maintain the sterility (Maunder and Pickett 1959; Rooney and Wayne Smith 2000). Several male-sterility-inducing cytoplasms were described in sorghum since the identification of a stable CMS system by Stephens and Holland (1954).
The A1 (milo) cytoplasm was first documented (Conner and Karper 1927) and is most widely used in sorghum hybrid development worldwide, followed by the A2 cytoplasm (Schertz 1977; Schertz and Ritchey 1978). Other types of CMS, namely A3 (Quinby 1980), A4 (Rao et al. 1984; Worstell et al. 1984), A5, A6, 9E (Webster and Singh 1964) and KS (Ross and Hackerott 1972), were also described. However, their use in commercial hybrid breeding programs is limited by the negative effects on grain yield of A3 cytoplasm (Moran and Rooney 2003), the low environmental stability of the restoration and, consequently, the lack of elite male restorer lines.
Fertility is restored in the progenies when a cytoplasmic male-sterile female is crossed with a male carrying the corresponding nuclear-encoded genes (restorer of fertility, Rf) that partially or completely restore the fertility. Recent quantitative trait loci (QTL) studies that used American, Australian and Indian sorghum material found, depending on the restorer line and the CMS type involved, one or several major loci controlling the fertility restoration in the A1 and A2 cytoplasms, complemented by genes with smaller effects (partial restorer) and modifier genes (Maunder and Pickett 1959; Murty and Gangadhar 1990; Qian 1990; Klein et al. 2001, 2005; Wen et al. 2002; Sanjana Reddy et al. 2010; Jordan et al. 2011; Yin et al. 2013). The so-far-suggested sorghum Rf genes for the sorghum A1 and A2 cytoplasm encode proteins that belong to the pentatricopeptide repeat (PPR) protein family (Klein et al. 2005; Jordan et al. 2010, 2011).
The A1-cytoplasm Rf1 locus was mapped on linkage group (LG) 8 (Klein et al. 2001), which corresponds to sorghum chromosome SBI-08 (Peng et al. 1999; Kim et al. 2005a, b). Two microsatellite markers flanking the Rf1 locus covered a 22.8-cM region with low recombination frequencies of 6.5 and 6.8% between the two markers and the gene. Further, sorghum PPR13 was cloned and reported as a candidate for the sorghum Rf1 gene (Klein et al. 2005). Rf2, a major A1-cytoplasm fertility restorer locus mapped on chromosome SBI-02, explained approximately 87% of the seed-set phenotypic variation (Jordan et al. 2010). The presence of modifier genes, revealed by a continuum of partially restored fertility, was observed in both used populations. Further, the locus on chromosome SBI-02 includes one PPR gene (Sb02g004810.1 with Sbi1.4 reference genome and Sobic.002G057050 with the Sbi3.4 reference genome) that co-segregates with the fertility restoration phenotype and could be a potential candidate gene. Recently, the Rf5 locus, located on chromosome SBI-05, has been associated with the fertility restoration in both A1 and A2 cytoplasms (Jordan et al. 2011). It explained 42 and 45% of the phenotypic variation in percent seed set on the A1 and A2 cytoplasm, respectively. A minor or partial restorer locus was also detected on chromosome SBI-04 explaining 10% of the variation of the percent seed set. Moreover, the Rf6 fertility restoration locus was mapped on chromosome SBI-04 and restores fertility in both A1 and A2 cytoplasms (Praveen et al. 2015). Informative markers based on these Rf genes could help in a cost-effective marker-assisted screening of breeding material for developing and identifying fertility restorer or maintainer lines.
A good understanding of the restoration and maintenance capacity of WA germplasm is essential for a long-term WA hybrid breeding program using the A1-type cytoplasm. To date, however, a detailed genetic study of fertility restoration in West African and Guinea-race germplasm has not been done, with prior studies focusing on Australian, Indian and US breeding materials. The Guinea-race of sorghum, predominantly cultivated across WA from Senegal to the western border of Nigeria, is known to be most genetically diverse and distinct from the other major sorghum races (Folkertsma et al. 2005; Deu et al. 2006). Most of the WA sorghum germplasm restores the A1 cytoplasm, and therefore, it is essential to have tools in hand which would ease the identification of potential maintainer and restorer lines.
In order to accelerate the development of hybrid parental lines for WA and facilitate broadening the genetic pool of WA sorghum females, this study aims to understand the underlying genetics of pollen fertility restoration in key WA hybrid parents, to develop diagnostic and cost-efficient molecular markers for fertility restoration that can be used in applied WA hybrid breeding programs, and to assess the potential utility of those markers. We report here (1) the evaluation of several pollen fertility restoration traits and relationships among them, (2) the identification of QTL for fertility restoration in the A1 CMS system in WA germplasm and (3) molecular markers developed for further use in WA sorghum hybrid breeding programs.
Materials and methods
F2-based mapping study
Plant material
Three F2 populations derived from the hybridization of two A- and two R-lines were created for the segregation analysis. The two female inbred lines, “Combine Kafir-60” [CK60A] and FambeA, have the A1-type of CMS. FambeA and the two male inbred lines, 97-SB-F5DT-298 [hereafter called DT_298] and Lata, are widely used parents in hybrid breeding research in Mali. FambeA is derived from a Malian Guinea-race local variety, Lata was derived from a random-mating population based mostly on Guinea landrace germplasm but with somewhat more than 12% introduced genetic background, while DT_298 was the product of bi-parental pedigree breeding with inter-racial Guinea-Caudatum parentage. CK60A is an old A-line developed in Texas (USA), which is known to have fewer genes leading to partial fertility, and was therefore used as the source of sterile cytoplasm in creating FambeA.
The three F1 crosses, CK60AxDT_298 [POPCD, Caudatum × inter-racial intercross], FambeAxDT_298 [POPFD, Guinea race × inter-racial intercross] and FambeAxLata [POPFL, Guinea × Guinea intercross] were developed in the 2014 rainy season, at the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) research station, near Bamako, Mali. Randomly chosen F1 plants were selfed in the 2015 off-season, and F2 seeds of one F1 plant of each of the three crosses were harvested, hand-threshed and stored in a cold chamber.
Field trial and phenotyping
For each F2 family, 220 hills were sown on June 8, 2015, in two bands of 10 ridges each. The 3-m long ridges were separated by 75 cm with 11 hills per ridge. The parental male lines were sown in two rows and each A-line in three rows. Two weeks after sowing, each hill was thinned to one plant. The trial was weeded 3 weeks after sowing, and the operation was repeated frequently throughout plant development. Basic soil fertilization was applied with 100 kg ha−1 of diammonium phosphate while preparing the soil before sowing. Organic manure was hand-applied directly in the hill-holes prior to sowing, and 50 kg ha−1 of urea was applied after the first weeding. Due to low vigor and mortality in the field, only 168, 125 and 175 F2 plants were phenotyped in POPCD, POPFD and POPFL, respectively (Table 1). All available F2 panicles and three random panicles of each parental line were selfed at heading stage with paper bags. The bags were removed only for harvest to avoid cross-pollination and bird damage. Daily temperatures during the flowering period (from July 30 to October 11, 2015) were within the normal range of sorghum production temperatures in Mali (18 °C min. in the mornings and 45 °C max. in the midday–early afternoon). A direct impact of high temperatures on panicle fertility was not observed in any of our trials.
F2 panicles were individually harvested at maturity and visually evaluated for seed set using a fertility restoration score. The fertility score used a 0–10 scale with 0 for complete sterility with no seed set and 10 for fully fertile panicles with complete seed set. The classification of panicles for fertility phenotype considered panicles with scores of 0–2 to be sterile (i.e., maintainer reaction) and those with fertility scores of 8–10 to be fertile (i.e., fertility restorer reaction), with all remaining intermediate fertility scores as partially fertile. The Chi-square (χ2) testing segregation ratios for major fertility genes used fertility score ranges of 0–2 as sterile and 3–10 as fertile. Besides, panicle length, panicle dry weight, threshed grain weight and 100 seed weight were directly observed and grain number per panicle was estimated using grain weight and 100 seed weight. Due to the ambiguous fertility score distributions in POPCD and POPFD, we set up a validation study during the 2016 rainy season using 100 F2 plants from the remaining seeds of each of the F2 populations. Fertility score was recorded in these repeated trials as in the F2 mapping populations.
Genotyping
Leaf sampling and DNA extraction Three weeks after sowing, when plants were at stage 2 (Vanderlip and Reeves 1972), leaf samples were collected from each viable F2 plant. With a leaf puncher, 30 disks per plant were put in a labeled tea bag and directly dried on silica gel.
DNA extraction and purification followed the cetyltrimethylammonium bromide (CTAB) protocol with 20 disks of leaf sample per individual F2 plant. The extracted DNA was solubilized into 100 µL of TE buffer. Finally, the 482 samples were diluted to 30–100 ng/µL and shipped to Cornell University for genotyping by sequencing (GBS) (Elshire et al. 2011). These samples comprised the 168, 125 and 175 individuals from POPCD, POPFD and POPFL (Table 1), along with two samples of the female CK60A and four of each of the three remaining parents, FambeA, Lata and DT_298.
Sequence analyses and single nucleotide polymorphism (SNP) calling and filtering GBS libraries were constructed in 192- and 96-plex using the ApeKI restriction enzyme. The TASSEL 5 (Trait Analysis by Association, Evolution and Linkage) GBS v2 pipeline (Glaubitz et al. 2014) was used to extract informative SNPs from the raw sequencing data. Alignment of tags to the Sorghum bicolor reference genome version 3 (McCormick et al. 2018) was achieved using the Burrows–Wheeler alignment (BWA) tool (Li and Durbin 2009). Finally, we obtained with no specific SNP filtering 148,376 SNPs for all three F2 populations and parental lines.
With TASSEL 5, a cladogram was constructed with parental and F2 individuals to confirm the supposed crosses made in the field. Four F2 individuals that were detected as outliers were removed from the study (Table 1).
Using R (R Development Core Team 2011) and VCFtools (Danecek et al. 2011), individual populations were filtered by removing monomorphic sites between both parents, sites containing missing data for any of the parents, as well as sites with a coverage < 10 and/or a minor allele frequency (MAF) < 0.2. After filtering, 7821, 5701 and 5197 sites remained in POPCD, POPFD and POPFL, respectively. Missing data were then imputed with FSFHap in TASSEL 5. POPCD, POPFD and POPFL had 6.6, 2.8 and 11.9% of missing data before any imputation and 6.1, 2.5 and 11.4% of missing data after the TASSEL imputation. Due to undercalling of heterozygous loci using low coverage GBS data, we corrected the imputed data with ABHGenotypeR package (Reuscher and Furuta 2016). Undercalled heterozygous and short miscalled stretches were corrected based on flanking alleles with a window (maxHapLength) of four markers. This procedure reduced missing data to 2.0, 0.7 and 3.7% in POPCD, POPFD and POPFL, respectively.
Linkage map construction and QTL mapping
Prior to the map construction, χ2-tests were conducted for all SNPs of all populations to detect segregation-distorted sites. Distorted markers (p < 0.001; 44, 12 and 65% of markers for POPCD, POPFD and POPFL, respectively), as well as duplicated sites, were removed with R/qtl (Broman et al. 2003). After this filtering step, the remaining 3859, 4119 and 1574 SNPs for POPCD, POPFD and POPFL, respectively, were used for the linkage map construction. The R/qtl cross-file was converted into a BC0F2 format with the convert2bcsft function for a proper mapping input-data format for the package ASMap (Wu et al. 2008; Taylor and Butler 2017). The Kosambi mapping function, implemented with the MSTmap algorithm, was used for the linkage map construction, with a p value of 1 × 10−6, and for imputing the remaining missing marker data. Marker order was controlled based on the physical positions, and individual chromosome orientation was flipped when required. A composite interval mapping (CIM) analysis with the fertility score was executed using R/qtl, and the QTL allele probability was computed for every ten centimorgan with five background markers. The LOD significance was assessed following the method described in Van Ooijen (1999), and the Bayesian confidence interval, as well as the phenotypic variance explained by significant QTL, was calculated. A 10,000-permutation test was computed on all populations, separately, to estimate the genome-wide significance threshold. A 200 independently sampled fivefold cross-validation was computed using Plabmqtl (Utz 2012) with the dominance and two-loci additive × additive epistatic effects included into the model. These analyses provided information on the robustness on the detected QTL.
KASP marker development
The SNP markers detected within and flanking the QTL regions on chromosome SBI-05 and SBI-02 were converted into kompetitive allele-specific polymerase chain reaction (KASP) markers. DNA sequences flanking the respective SNP, primer sequences and polymerase chain reaction (PCR) conditions are shown in Supplemental Table S1. All KASP assays were established and run on the Roche LightCycler480II using 10 µL PCR volumes and the KASP master mix with low ROX provided by LGC Genomics (www.lgcgroup.com).
Candidate gene sequencing
Pentatricopeptide repeat (PPR) genes are known to be involved in fertility reactions; hence, we Sanger-sequenced a set of selected PPR genes in the QTL regions on chromosome SBI-05 for POPCD and POPFD. Due to the narrow confidence interval on chromosome SBI-02 (1.2 cM) and no known PPR genes within the confidence interval, we selected some of the closest PPR genes upstream of the QTL interval on chromosome SBI-02 and Sanger-sequenced them. The reference sequences and the functional annotations of the selected genes were retrieved from Phytozome (Goodstein et al. 2012, www.phytozome.com, Table 2). The primer sequences and the used PCR conditions are shown in Supplemental Table S2. We first sequenced the four parents. If we detected any potential mutations (missense, frameshifts, etc.), which discriminated the female and male parents, we used a set of other diverse B- and R-lines from ICRISAT breeding program (Supplemental Table S3) and sequenced them for the region of interest. Only in Sobic.002G057050, we detected a missense mutation, which could be validated to differentiate well B- and R-lines. This SNP was finally converted into a KASP marker named S002G057050_1090.
All of the 11 KASP markers shown in Supplemental Table S1 were used in the validation study for different purposes. First, with one of the KASPs, namely S2_6843380 on chromosome SBI-02, we re-genotyped the entire POPFL F2 family to validate the accuracy of our GBS derived SNPs and our imputation steps. A very high match (93%) between raw GBS data and genotyped F2 individuals, and a 99% agreement with the imputed data indicated few genotyping errors in our markers. Secondly, we used the KASPs on chromosome SBI-05 and the KASPs on chromosome SBI-02 to be validated in F2:3 plants of POPCD and POPFL, respectively.
QTL validation in F3 families
A single F2 panicle from both POPCD and POPFL was selected based on the high number of seeds and high proportion of heterozygous SNPs. The 92 POPCD and 93 POPFL F2:3 seeds from these panicles were used for validation of detected QTL. In the 2016 rainy season, all seeds were sown at the ICRISAT station, Samanko, with the same agronomic treatments as in the F2 populations in 2015.
Panicles were covered with selfing bags at heading stage. Individual bags were temporarily removed 5–7 days later to score the pollen quantity, anther color and the presence or absence of an anther pore. Pollen quantity was scored before 10 a.m. using a visual rating from one (no pollen) to five (high pollen quantity) by gently shaking each panicle and observing the pollen quantity shed. Further, the color of the anthers was rated following a visual scoring of 1 (very bright yellow to white) to 5 (dark yellow). Panicles with a pore on the anthers were scored 1 and those without pore 0. Cross-pollination from neighboring plants was prevented by having all neighboring plants bagged during individual panicle scoring and washing the hands with ethanol if necessary before opening a new bag. At maturity, all bags were removed and panicles harvested and dried. Apart from panicle length, all data recorded in the F2 populations (used in the mapping study) were also evaluated in the F3 validation populations. Leaf samples were taken from all F2:3 plants, dried in silica gel and brought to the University of Hohenheim for DNA extraction and KASP marker analyses. The F2:3 plants from POPCD (hereafter called POPCD_F3) were analyzed with the KASP markers on chromosome SBI-05, whereas the F2:3 plants from POPFL (hereafter called POPFL_F3) were analyzed with the KASPs on chromosome SBI-02. The phenotypic variances explained by each marker were tested in a linear regression model with fertility score as depending variable.
QTL validation in R- and B-lines
Additionally to the QTL validation in the F3 families, a validation of the most promising markers was conducted using a diverse set of 95 lines of interest for the hybrid breeding programs of ICRISAT-Mali and the Institut d’Economie Rurale (IER-Mali) (Supplemental Table S3). These lines were classified as fertility restorers (R-lines, N = 50) or maintainers (B-Lines, N = 45) based on prior classification or by testcrossing those lines onto the male-sterile female CK60A during the 2017 rainy season and evaluating the fertility of the resulting hybrids. KASPs either being the QTL peak or spanning the QTL region, namely S2_6045380, S002G057050_1090, S2_6843380, S5_1180493 and S5_2174322, were evaluated for these 95 lines.
Results
Descriptive analysis of fertility phenotypes in three F2 populations
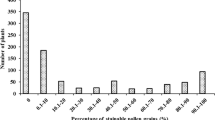
The fertility scores of the parents corresponded to expectations; all female parents were completely sterile, and male parents were completely fertile. The fertility scores of POPFL F2 plants showed high frequency of fertile panicles (104 of 174 panicles) and low frequency of sterile panicles (n = 33), whereas POPCD and POPFD showed low frequency of fertile panicles (19 of 166, and 27 of 124 panicles, respectively) and a high frequency of sterile panicles (n = 123 and 87, respectively) (Fig. 1). Partially fertile panicles with intermediate fertility scores were observed in all populations, but their total numbers were less than that of the combined classes of fertile and sterile panicles (Fig. 1). The χ2 test revealed no statistically significant deviation from the 3:1 segregation ratio at α = 5% in POPFL (χ2 = 3.52 with one degree of freedom). POPCD and POPFD deviated significantly from the 3:1 segregating ratio (χ2 = 208.29 for POPCD and 137.41 for POPFD, with one degree of freedom).

Frequency distribution of- and correlations between- phenotypic traits recorded on POPCD (a), POPFD (b) and POPFL (c) sorghum F2 mapping populations, with 166, 124 and 174 individuals, respectively. Pa_Length, Pa_Weight and Gr_Weight are the panicle length, the panicle dry weight and the individual panicle grain weight and seed number, respectively
No strong correlations were found between fertility score with measures of phenology (heading date) or panicle length (Fig. 1). The fertility score was strongly correlated with measures of panicle weight, threshed grain weight and seed number.
Genetic maps
The linkage maps of the three F2 populations consisted of 10 LG (chromosome SBI-01 to SBI-10), which spanned a cumulative distance of 1602.4 cM for POPCD, 1736.7 cM for POPFD and 1518.3 cM for POPFL (Supplemental Fig. S1). The number of SNP markers in each LG varied from 66 markers on chromosome SBI-06 of POPFL to 700 markers on chromosome SBI-01 of POPFD, with an average of 386, 412 and 157 SNPs per linkage group for POPCD, POPFD and POPFL, respectively. The average distance between markers across the 10 linkage groups was 0.4 cM for both POPCD and POPFD, and 1.0 cM for POPFL.
QTL analyses
Seven significant QTL (LOD score > 4) for fertility restoration were identified on five of the ten sorghum chromosomes based on associations with fertility scores across the three F2 populations (Fig. 2, Table 3). The QTL that explained the greatest phenotypic variation for fertility score were identified on SBI-02 in POPFL (Table 3), accounting for over 31% and having 97% match in cross-validation runs. The next QTL, in order of percent of phenotypic variation explained, were identified on SBI-05 in POPCD and POPFD and on SBI-10 in POPFD. These QTL had LOD scores not only superior to four (computed LOD significance threshold) but also exceeding the population-specific 10,000-permutation LOD thresholds (Fig. 2, Table 3). In addition, QTL were identified on chromosome SBI-05 of POPCD and POPFD with 76 and 62% of matches from the cross-validation runs, respectively.

QTL scan from composite interval mapping of the fertility (A1 cytoplasm) score for three sorghum F2 populations (POPCD = a; POPFD = b; POPFL = c)
Candidate gene sequences
Three candidate PPR genes on chromosome SBI-02, close to the narrow confidence interval of the detected QTL in POPFL mapping population, were sequenced (Table 2, Fig. 3). Several mutations for Sobic.002G054100 were detected, but only one missense SNP (262 bp) caused an amino acid change. However, sequencing R- and B-lines for Sobic002g054100, we could not see any consistency and discriminating ability between R- and B-lines at this position. Direct evidence of effect of this gene is therefore lacking. No SNP was detected in Sobic.002G059700 between the parents of POPFL. Several mutations in Sobic.002G057050 were detected, including one missense SNP (1090 bp) and one 2 bp InDel (2687–2688 bp) causing a frameshift mutation. These two mutations were also detected in multiple R- and B-lines, differentiating the groups very well, except for one R-line (76R) that contained the deletion (Fig. 4, see Supplemental Table S3). The SNP at position 1090 bp was therefore chosen and converted into a KASP marker for further validation.

Scan of QTL for male fertility reaction (A1 cytoplasm) in sorghum POPFL for chromosome SBI-02, and a high-resolution genome map showing the selected and sequenced PPR genes and their positions (indicated with arrows) among the SNPs within this region. The heatmap shows the R2 among the different SNPs, delimiting three major linkage blocks, whereas the QTL peak falls within the middle one

Aligned sequences of Sobic.002G057050 around the two mutations at 1090 bp and 2686 bp, putatively related to male fertility reaction in sorghum. The two first rows show the sorghum reference genome, followed by the parents of POPFL mapping population, Fambe (A-line) and Lata (R-line), and followed by five B- and five R-lines sampled from ICRISAT breeding material (Supplemental Table S3)
Sequencing candidate genes on chromosome SBI-05 (Table 2) did not result in the detection of any mutations among the parental lines for Sobic.005G011000. Several SNPs in Sobic.005G017100 were detected in the first exon, but none of these mutations clearly separated R- and B-lines. Several SNPs in the 3′-UTR were detected in Sobic.005G020600 and Sobic.005G026400, but none of them could be validated by effectively discriminating among R- and B-lines. We cannot rule out that Sobic.005G017100, Sobic.005G020600 and Sobic.005G026400 are somehow involved in the fertility reaction caused by the QTL on chromosome SBI-05 since we only sequenced a limited number of R- and B-lines.
Validation of QTL
The female and male parents in the validation study showed complete sterility (score = 0) and fertility (scored 10), respectively (Fig. 5). In POPCD_F3, 34% of the 92 scored panicles were sterile, and 48% were fertile. In POPFL_F3, 26% of the 93 scored panicles were sterile, and 63% were fertile. Both F3 populations showed plants with partial fertility representing 18% and 11% of the total plants in POPCD_F3 and POPFL_F3, respectively. The χ2 test revealed a statistically significant deviation from the 3:1 segregation ratio at α = 5% for POPCD_F3 population (χ2 = 83.71 with one degree of freedom). POPFL_F3 approximated the 3:1 fertile to sterile segregation ratio (χ2 = 0.03 with one degree of freedom) (Fig. 5). Panicle weight, grain weight and grain number had the same trend as the fertility-related scores.

Frequency distribution of fertility (A1 cytoplasm) scores of 92 F3 plants of POPCD_F3 (a) and 93 F3 plants of POPFL_F3 (b) validation populations and parental means
The fertility scores of the F3 validation plants showed no correlation with heading date in either POPCD_F3 or POPFL_F3, but were positively correlated with pollen quantity (r = 0.57 and r = 0.79, respectively) and negatively correlated with anther color (r = − 0.60) for POPFL. The fertility scores of both F3 populations were strongly correlated with panicle weight, grain weight and seed number (ranging from 0.64 to 0.68 for POPCD_F3 and 0.70 to 0.78 for POPFL_F3; α < 0.001) similarly to the F2 populations.
Markers on chromosome SBI-05 explained between 0.97 and 7.83% of the phenotypic variation of POPCD_F3, thus showing a very low consistency between the mapping and validation study (Table 4). Contrarily, the markers on chromosome SBI-02 explained between 54 and 81% of the variation in POPFL_F3. KASP S002G057050_1090, developed from the missense SNP in Sobic.002G057050 (1090 bp), explained 81% of the variation in POPFL_F3 (Table 4, Fig. 6). However, it explained only 25% of the phenotypic variation in the F2 mapping population (data not shown). The KASP S2_6843380, co-located with our peak SNP in the linkage map, explained 62% and 30% of the phenotypic variation in the POPFL_F3 validation set and in the F2 mapping population, respectively.

Boxplot showing the fertility scores of different F3 individuals as related to their genotypic values for the KASP marker Sobic.002G057050_1090. A is the recessive (female/maintainer) allele, and T is the dominant (restorer) allele. Crosses indicate the mean and the horizontal line the median of each group
Moreover, with the screening of a set of 95 R- and B-lines (Supplemental Table 4) for the most promising KASPs on chromosomes SBI-02 and SBI-05, the marker Sobic.002G057050_1090 discriminated R- and B-lines most accurately. This marker had a false positive rate of 0.0% (45 out of 45 correct) for B-lines and 26.0% (37 out of 50 correct) for R-lines. All the other KASPs had much higher false positive rates (Supplemental Table S3).
Discussion
This study is the first known on the genetics of fertility restoration in the A1 cytoplasm for West African breeding materials. In addition, this is the first known use of Guinea-race derived materials/germplasm for such study. CK60A, one of the two female parents, is a well-known male-sterile female line used in breeding programs in WA and worldwide. FambeA, DT_298 and Lata are derived from local germplasm and used in local breeding programs, thus allowing a very suitable basis for mapping fertility restorer genes in the WA sorghum germplasm. Markers developed from this study would be a valuable tool for direct use in WA sorghum breeding programs.
Phenotyping and genetics of fertility restoration in WA germplasm
The patterns of fertility score distributions showed consistency that suggests that the scoring was effective for phenotypic evaluation of fertility restoration. Parents showed no variation from the expected phenotypes. POPFL followed the anticipated distribution having more fertile than sterile plants, but also a continuum of partially restored fertility, as in the two other F2 populations. A continuum of partially restored fertility, showing the presence of partial restorer genes, was likewise found in sorghum fertility restoration studies (Jordan et al. 2010, 2011). The distribution of fertility score in POPCD and POPFD was contrary to what is generally reported in sorghum (Klein et al. 2001; Jordan et al. 2011; Praveen et al. 2015, 2018), with most of plants being sterile (Fig. 1). The fertility score distributions of the “repeated” F2 populations were similar to the corresponding F2 set used for the mapping study (data not shown). Extreme temperatures (mostly low night temperatures) around heading date could reduce the pollen quantity and viability, and consequently the seed set, in sorghum (Downes and Marshall 1971; Brooking 1976; Prasad et al. 2006; Hatfield and Prueger 2015). Therefore, we looked at the temperatures around heading time during our experiments and could not find any influence of temperature on fertility score; neither had we found significant correlations between heading date and the fertility score in the F2 and F3 populations (Fig. 1, Supplemental Table S4). Hence, we concluded that no specific year effects, e.g., extreme weather events, confounded our fertility reaction phenotyping.
Consequently, distributions in the mapping populations, as well as those of the validation sets and the χ2 tests, indicate that, as previously described in sorghum (Maunder and Pickett 1959; Miller and Pickett 1964; Wen et al. 2002; Jordan et al. 2010, 2011), one or multiple dominant loci plus modifier and/or partial restorer genes may control the fertility restoration in WA sorghum. One single gene seems to control the fertility restoration/sterility maintenance in POPFL, whereas the genetics of fertility restoration/sterility maintenance in POPCD and POPFD are not well understood with more sterile than fertile plants; hence, further studies would be needed for clarification. However, the low phenotypic variance explained by our markers on chromosome SBI-05 in the POPCD_F3 validation population, as well as the phenotypic distributions of POPCD and POPFD, points to a highly quantitative trait with multiple potential partial restorer genes.
Fertility restoration loci and partial restorers
The removal of distorted sites prior to the genetic map construction provided us with a less dense but more accurate linkage map, by avoiding bias in marker order and in the distances between distorted markers (Lorieux et al. 1995; Liu et al. 2010). Distorted markers were reported in various mapping studies (Mace et al. 2009; Kong et al. 2018; Boyles et al. 2017) and were associated with the population type, the specific cross and the type of molecular marker. In our study, the difference in amount of distorted markers between populations suggests that the difference between parental lines may have influenced the segregation distortion (Paterson et al. 2009). Further, most of distorted markers (19, 15 and 23% in, respectively, POPCD, POPFD and POPFL) were located and evenly distributed on chromosome SBI-01. This chromosome was reported to contain most of the distorted makers in various sorghum mapping studies (Menz et al. 2002; Mace et al. 2009; Kong et al. 2018). However, for a higher coverage, inclusion of these markers in the linkage map construction should be considered with more fitting statistical models (Lorieux et al. 1995; Xu 2008). A deeper characterization and insight of the excluded distorted sites would be worthwhile for breeding purposes, given that the population with 100% Guinea-race background presented a higher rate of distorted markers and distortion favored generally the male parent’s alleles.
The QTL region on chromosome SBI-05 found in POPCD and POPFD is located in the same 2.6-Mbp region detected by Jordan et al. (2011) as a major locus for fertility restoration and described as Rf5 locus, using Australian material. This major locus restored fertility in both A1 and A2 cytoplasms and was associated with a modifier or partial fertility restorer locus on chromosome SBI-04. We found a small effect QTL on the same chromosome SBI-04 in POPCD that was different from the one found in the above-cited study. It is possible that major loci are common across Australian and West African germplasm, and, depending on environmental conditions, the partial restorer loci will be expressed or their effects masked in a particular genotype. With more phenotypic variance explained, QTL on chromosomes SBI-05 in POPCD and POPFD had relatively larger effects than those on chromosomes SBI-02, SBI-03 and SBI-04. The effect of the minor QTL on chromosome SBI-10 in POPFD was large and rather similar to the effect of the major QTL on chromosome SBI-05 (Fig. 2, Table 3). The small sample size of POPFD could have led to the likely overestimation of its effect (Utz and Melchinger 1994), whereas QTL with larger effect on chromosome SBI-05 could still be detected (Vales et al. 2005). Further, the fact that we found the same QTL region on SBI-05 for POPCD and POPFD (same male parent) gives more confidence in this major QTL than on the population-specific minor QTL. Small effect QTL need therefore more focus with larger and repeatable populations in order to more accurately map them, estimate their true effects, and understand how they affect fertility restoration in WA germplasm in different environments.
The effect of the only QTL found on chromosome SBI-02 in POPFL was largest across populations with 31% of the variance explained (Table 2). This indicates that despite the relative low marker density in that region (Fig. 3) and the relatively limited population size, this QTL stands as a strong candidate for fertility restoration. However, the power and the accuracy of detected QTL could be decreased as compared to larger populations (Schön et al. 2004). Further, that same region had been mapped and described as Rf2 locus, restoring male fertility in the A1-type CMS in Australian germplasm (Jordan et al. 2010), and recently in Indian germplasm (Praveen et al. 2018). This locus seems therefore to control fertility restoration in some WA lines, as well as in the Australian and Indian germplasm.
Further, Caudatum, Kafir, Durra and inter-racial lines (Additional Table 1 in Mace et al. (2008)) were used in the studies of both Jordan et al. (2010) and Praveen et al. (2018), while our POPFL has a 100% Guinea-race background. Thus, there is no evidence that the fertility restoration loci are race specific. Identified QTL or developed markers should therefore be useful either for Guinea-race-oriented breeding programs or for the development of non-Guinea-race hybrid parents.
PPR-Rf genes and a potential causative mutation for Rf 2
The capacity of Rf genes to encode mitochondria-targeted PPR proteins was first discovered by the cloning of Rf-PPR592 gene from petunia (Bentolila et al. 2002). Further, except the Rf2 gene in maize, which is a member of the aldehyde dehydrogenase gene family (Cui et al. 1996; Liu et al. 2001), all Rf genes identified so far in other crop species, including rice (Kazama and Toriyama 2003; Komori et al. 2004; Inagaki et al. 2004) and sorghum (Klein et al. 2005), are members of the PPR gene family. Rf-like PPR genes evolve rapidly (Dahan and Mireau 2013) through a “birth and death” process (Geddy et al. 2007), and their local duplication occurs rather frequently, for them to ensure corrective functions (Dahan and Mireau 2013). In our study, clusters of PPR genes were found in the QTL regions. Since we could only use gene model data of the currently available reference genome, it is possible that in our tested parental lines further PPR gene duplicates are present. In POPCD, POPFD and POPFL, we found 12, 13 and 7 PPR genes, respectively, spanning 2.01, 1.30 and 1.87 Mbp genomic segments (with a respective average distance of 179, 257 and 152 kbp between PPR genes), on the QTL regions on chromosome SBI-05 (POPCD and POPFD) and chromosome SBI-02 (POPFL). We could not sequence all PPR genes in these regions and therefore only selected the ones located in—or linked to—the confidence intervals for validation and marker development.
In the region spanning the Rf5 locus, Jordan et al. (2011) identified a cluster of six PPR genes exhibiting a strong homology with the rice Rf1 gene. The four shared PPR genes to POPCD and POPFD were located in a region of sorghum chromosome SBI-05 from 0.98 to 2.37 Mbp (Table 2), whereas in the above-cited study, the six candidate PPR genes for the Rf5 locus were located in the region from 2.45 to 2.78 Mbp of the chromosome SBI-05. Therefore, there was no overlap of PPR genes between these two studies. In the present study, we could not detect any plausible mutations in any of the sequenced PPR genes on chromosome SBI-05. However, due to our reduced set of sequenced genotypes and sample size, we cannot rule out any direct impact of the sequenced genes, and hence, further research is needed here.
On chromosome SBI-02, we sequenced several PPR genes including Sobic.002G057050 (Sb02g004810 in the previous version of sorghum reference genome) which was proposed by Jordan et al. (2010) as a candidate gene for fertility restoration in the A1 CMS system. Jordan et al. (2010) suggested that additional fine mapping, along with additional experimental observations, would be necessary to confirm the identity of the Rf2 gene. Very recently, the 236 kb Rf2 locus was fine mapped to 10.32 kb and Sobic.002G057050 was the only PPR gene located in this interval (Praveen et al. 2018). The same study found that Sobic.002G057050 was more expressed in the sorghum inflorescence as compared to the stem or the leaves, and 12 times more expressed in inflorescence tissues of male-fertile (restorer) lines as compared to the male-sterile (female) ones. Further, Praveen et al. (2018) found 44 and 43 SNPs between the parental female line (296A) and the two restorer lines (RS29 and DSV1, respectively) used in their study, and a 3 bp deletion in the coding DNA sequence of 296A. In our study, we found the same deletion in all the sequenced B-lines and in one of our R-lines as well (Fig. 4). Further, we detected a missense mutation (1090 bp) leading to an amino acid exchange and a clear separation of B- and R-lines. This same SNP was also found by Praveen et al. (2018) separating their A- and R-lines (Supplemental Fig. S2). The developed marker from this mutation, namely Sobic.002G057050_1090, explained around 81% of the phenotypic variation in our POPFL_F3 validation population (Table 4). All genotyped B-lines were carrying the recessive (female/maintainer) allele of Sobic.002G057050_1090, and 37 out of the 50 genotyped R-lines had the male parent’s allele (Supplemental Table S3). The few R-lines that did not carry the restorer allele of Sobic.002G057050_1090 (excluding the heterozygous status) had the restorer allele for marker S5_1180493 on chromosome SBI-05. This points to the two major loci Rf2 and Rf5 as being complementary; hence, either of them must be present to restore fertility in the tested material. We must point out though that most of the characterized B-lines had the same or similar genetic background. For a better characterization of WA B-lines, genotypes with more diverse background have to be assessed with the developed markers. Putting the results of Jordan et al. (2010), Praveen et al. (2018) and ours together point very strongly to Sobic.002G057050 as the underlying gene of the Rf2 locus and to the SNP at 1090 bp as the causative mutation. Awaiting the validation of Sobic.002G057050, Sobic.002G057050_1090 can be a useful tool for an initial molecular identification and selection of maintainer and restorer lines in WA breeding programs; nonetheless, caution must be paid to the partial restorers and the QTL on chromosome SBI-05.
Markers and options for increasing effectiveness of selection for hybrid parents in WA
Maintaining the sterility of A-lines while duplicating their seeds is necessary for commercial hybrid seed production. Maintainers should not have restorer alleles to prevent male fertility restoration of female lines while maintaining A/B pairs. Apart from the already-discussed locus on chromosome SBI-10 of POPFD, QTL with smaller effects poorly explained the phenotypic variation in the respective mapping populations (Fig. 2, Table 3). In any case, these QTL regions may contain partial restorer genes and understanding how they affect the fertility restoration is essential for breeding programs. A-lines must possess the highest level of male sterility to ensure 100% hybrid seed production. Partially fertile A-lines lead to a degradation of hybrid seed quality and can reduce hybrid purity and yield in field crops. Markers for fertility restoration/sterility maintenance, if available, would allow a marker-assisted assessment of fertility restoration or sterility maintenance of WA breeding lines, and could be more efficient, labor- and cost-effective than direct field evaluation. Such markers would help excluding the (partial) restorer alleles from the A- and B-lines, as partial fertility restorer genes can stay unobserved for generations and be expressed under particular environmental conditions (Jordan et al. 2010, 2011). Alternatively, the combination of major restorer genes/loci and partial restorers can lead to a recovery of complete fertility in the F1 hybrids. Therefore, understanding and introgressing several Rf loci, either full or partial restorer, in restorer lines used in the breeding programs could lead to complete restoration of fertility in hybrids and therefore ensure a more complete panicle filling for more grain yield. However, until molecular tools will be at hand, including for minor QTL found in this study, breeders would still need to rely on extensive multi-location phenotypic evaluations to identify stable R- and B-lines in the advanced generations, specifically in environment corresponding to seed production.
Conclusion
The final markers created in this study are the first set of markers for fertility restoration in WA sorghum germplasm. QTL on chromosome SBI-05 and partial restorers need to be better understood. The marker developed from the mutation in 1090 bp of the PPR gene Sobic.002G057050 clearly separated B- from R-lines. This, along with the high phenotypic variance the marker Sobic.002G057050_1090 explained in the F3 population and previous researches confirm that this gene is a strong candidate for fertility restoration in the WA A1 CMS, and that the mutation in 1090bp may be the causative mutation underlying Rf2. QTL on chromosomes SBI-02 and SBI-05 seem to work separately. Therefore, either combining both QTL in- or excluding them from- one genotype to ensure more stable R- or A/B-lines could be beneficial for WA breeding programs. For a certain set of genotypes, WA breeders can directly use Sobic.002G057050_1090 to preselect potential B- and R-lines. Nevertheless, and awaiting for the validation of PPR Sobic.002G057050, this marker and its stability would need to be assessed on genetically more diverse B-lines and more environments.
Author Contribution statement
HFR, WLL, EW and BIGH conceived the study. MK, BN and WLL collected the data; MK performed the analyses; MK, WLL, BIGH, and HFR wrote the paper. All authors approved the final manuscript.
Abbreviations
- CMS:
-
Cytoplasmic male sterility
- CTAB:
-
Cetyltrimethylammonium bromide
- DNA:
-
Deoxyribonucleic acid
- GBS:
-
Genotyping by sequencing
- ICRISAT:
-
International Crops Research Institute for the Semi-arid Tropics
- IER:
-
Institut d’Economie Rurale
- KASP:
-
Kompetitive allele-specific polymerase chain reaction
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
- PPR:
-
Pentatricopeptide repeat
- QTL:
-
Quantitative trait loci
- Rf:
-
Fertility restoration locus/gene
- SNP:
-
Single nucleotide polymorphism
- WA:
-
West Africa
References
Andrews DJ (1975) Sorghum grain hybrids in Nigeria. Exp Agric 11:119–127. https://doi.org/10.1017/S0014479700006566
Atokple ID (2003) Sorghum and millet breeding in West Africa in practice. In: Belton P, Taylor JR (eds) Workshop on the proteins of sorghums and millets: enhancing nutritional and functional properties for Africa. Pretoria, South Africa
Bentolila S, Alfonso AA, Hanson MR (2002) A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proc Natl Acad Sci U S A 99:10887–10892. https://doi.org/10.1073/pnas.102301599
Boyles RE, Pfeiffer BK, Cooper EA et al (2017) Genetic dissection of sorghum grain quality traits using diverse and segregating populations. Theor Appl Genet 130:697–716. https://doi.org/10.1007/s00122-016-2844-6
Broman KW, Wu H, Sen S, Churchill GA (2003) R/QTL: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Brooking I (1976) Male sterility in Sorghum bicolor (L.) Moench induced by low night temperature. I. Timing of the stage of sensitivity. Aust J Plant Physiol 3:589–596. https://doi.org/10.1071/PP9760589
Conner AB, Karper RE (1927) Hybrid vigor in sorghum. Agricultural and Mechanical College in Texas, College Station
Cui X, Wise RP, Schnable PS (1996) The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 272:1334–1336. https://doi.org/10.1126/SCIENCE.272.5266.1334
Dahan J, Mireau H (2013) The Rf and Rf-like PPR in higher plants, a fast-evolving subclass of PPR genes. RNA Biol 10:1469–1476. https://doi.org/10.4161/rna.25568
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Deu M, Rattunde F, Chantereau J (2006) A global view of genetic diversity in cultivated sorghums using a core collection. Genome 49:168–180
Downes R, Marshall D (1971) Low temperature induced male sterility in Sorghum bicolor. Aust J Exp Agric Anim Husb 11:352–356
Duvick DN (1959) The use of cytoplasmic male-sterility in hybrid seed production. Econ Bot 13:167–195. https://doi.org/10.1007/BF02860581
Elshire RJ, Glaubitz JC, Sun Q et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Folkertsma RT, Rattunde HFW, Chandra S et al (2005) The pattern of genetic diversity of Guinea-race Sorghum bicolor (L.) Moench landraces as revealed with SSR markers. Theor Appl Genet 111:399–409. https://doi.org/10.1007/s00122-005-1949-0
Geddy R, Brown GG, Small I et al (2007) Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genom 8:130. https://doi.org/10.1186/1471-2164-8-130
Glaubitz JC, Casstevens TM, Lu F et al (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9:e90346. https://doi.org/10.1371/journal.pone.0090346
Goodstein DM, Shu S, Howson R et al (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:D1178–D1186. https://doi.org/10.1093/nar/gkr944
Hanson MR, Bentolila S (2004) Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 16:S154–S169
Hanson MR, Conde MF (1985) Functioning and variation of cytoplasmic genomes: lessons from cytoplasmic-nuclear interactions affecting male fertility in plants. Int Rev Cytol 94:213–267. https://doi.org/10.1016/S0074-7696(08)60398-8
Hatfield JL, Prueger JH (2015) Temperature extremes: effect on plant growth and development. Weather Clim Extrem 10:4–10. https://doi.org/10.1016/J.WACE.2015.08.001
Inagaki A, Akagi H, Nakamura A et al (2004) Positional cloning of the rice Rf-1 gene, a restorer of BT-type cytoplasmic male sterility that encodes a mitochondria-targeting PPR protein. TAG Theor Appl Genet 108:1449–1457. https://doi.org/10.1007/s00122-004-1591-2
Jordan DR, Klein RR, Sakrewski KG et al (2011) Mapping and characterization of Rf5: a new gene conditioning pollen fertility restoration in A1 and A2 cytoplasm in sorghum (Sorghum bicolor (L.) Moench). Theor Appl Genet 123:383–396. https://doi.org/10.1007/s00122-011-1591-y
Jordan DR, Mace ES, Henzell RG et al (2010) Molecular mapping and candidate gene identification of the Rf2 gene for pollen fertility restoration in sorghum [Sorghum bicolor (L.) Moench]. Theor Appl Genet 120:1279–1287. https://doi.org/10.1007/s00122-009-1255-3
Kante M, Rattunde HFW, Leiser WL et al (2017) Can tall guinea-race sorghum hybrids deliver yield advantage to smallholder farmers in west and central Africa? Crop Sci 57:833–842. https://doi.org/10.2135/cropsci2016.09.0765
Kazama T, Toriyama K (2003) A pentatricopeptide repeat-containing gene that promotes the processing of aberrant atp6 RNA of cytoplasmic male-sterile rice. FEBS Lett 544:99–102. https://doi.org/10.1016/S0014-5793(03)00480-0
Kim J-S, Klein PE, Klein RR et al (2005a) Chromosome identification and nomenclature of Sorghum bicolor. Genetics 169:1169–1173. https://doi.org/10.1534/genetics.104.035980
Kim J-S, Klein PE, Klein RR et al (2005b) Molecular cytogenetic maps of sorghum linkage groups 2 and 8. Genetics 169:955–965. https://doi.org/10.1534/genetics.104.026765
Klein RR, Klein PE, Chhabra AK et al (2001) Molecular mapping of the rf1 gene for pollen fertility restoration in sorghum (Sorghum bicolor L.). Theor Appl Genet 102:1206–1212. https://doi.org/10.1007/s001220100575
Klein RR, Klein PE, Mullet JE et al (2005) Fertility restorer locus Rf1 [corrected] of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the colinear region of rice chromosome 12. Theor Appl Genet 111:994–1012. https://doi.org/10.1007/s00122-005-2011-y
Komori T, Ohta S, Murai N et al (2004) Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryza sativa L.). Plant J 37:315–325. https://doi.org/10.1046/j.1365-313X.2003.01961.x
Kong W, Kim C, Zhang D et al (2018) Genotyping by sequencing of 393 Sorghum bicolor BTx623 × IS3620C recombinant inbred lines improves sensitivity and resolution of QTL detection. G3 (Bethesda). https://doi.org/10.1534/g3.118.200173
Laughnan JR, Gabay-Laughnan S (1983) Cytoplasmic male sterility in maize. Annu Rev Genet 17:27–48. https://doi.org/10.1146/annurev.ge.17.120183.000331
Levings CS, Brown GG (1989) Molecular biology of plant mitochondria. Cell 56:171–179. https://doi.org/10.1016/0092-8674(89)90890-8
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Liu F, Cui X, Horner HT et al (2001) Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 13:1063–1078
Liu X, Guo L, You J et al (2010) Progress of segregation distortion in genetic mapping of plants. Res J Agron 4:78–83. https://doi.org/10.3923/rjagr.2010.78.83
Lorieux M, Perrier X, Goffinet B et al (1995) Maximum-likelihood models for mapping genetic markers showing segregation distortion. 2. F2 populations. Theor Appl Genet 90:81–89. https://doi.org/10.1007/BF00220999
Mace ES, Xia L, Jordan DR et al (2008) DArT markers: diversity analyses and mapping in Sorghum bicolor. BMC Genomics 9:26. https://doi.org/10.1186/1471-2164-9-26
Mace ES, Rami J-F, Bouchet S et al (2009) A consensus genetic map of sorghum that integrates multiple component maps and high-throughput Diversity Array Technology (DArT) markers. BMC Plant Biol 9:13. https://doi.org/10.1186/1471-2229-9-13
Maunder AB, Pickett RC (1959) The genetic inheritance of cytoplasmic-genetic male sterility in grain sorghum1. Agron J 51:47. https://doi.org/10.2134/agronj1959.00021962005100010016x
McCormick RF, Truong SK, Sreedasyam A et al (2018) The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J 93:338–354. https://doi.org/10.1111/tpj.13781
Menz MA, Klein RR, Mullet JE et al (2002) A high-density genetic map of Sorghum bicolor (L.) Moench based on 2926 AFLP®, RFLP and SSR markers. Plant Mol Biol 48:483–499. https://doi.org/10.1023/A:1014831302392
Miller D, Pickett R (1964) Inheritance of partial male-fertility in Sorghum vulgare Pers. Crop Sci 4:1–4
Moran JL, Rooney WL (2003) Effect of cytoplasm on the agronomic performance of grain sorghum hybrids. Crop Sci 43:777–781. https://doi.org/10.2135/cropsci2003.7770
Murty U, Gangadhar G (1990) Milo and non-milo sources of cytoplasms in Sorghum bicolor (L.) Moench. III. Genetics of fertility restoration. Cereal Res. Commun. 18:111–116
Paterson AH, Bowers JE, Bruggmann R et al (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556. https://doi.org/10.1038/nature07723
Peng Y, Schertz KF, Cartinhour S, Hart GE (1999) Comparative genome mapping of Sorghum bicolor (L.) Moench using an RFLP map constructed in a population of recombinant inbred lines. Plant Breed 118:225–235. https://doi.org/10.1046/j.1439-0523.1999.118003225.x
Prasad PVV, Boote KJ, Allen LH (2006) Adverse high temperature effects on pollen viability, seed-set, seed yield and harvest index of grain-sorghum [Sorghum bicolor (L.) Moench] are more severe at elevated carbon dioxide due to higher tissue temperatures. Agric For Meteorol 139:237–251. https://doi.org/10.1016/J.AGRFORMET.2006.07.003
Praveen M, Anurag Uttam G, Suneetha N et al (2015) Inheritance and molecular mapping of Rf6 locus with pollen fertility restoration ability on A1 and A2 cytoplasms in sorghum. Plant Sci 238:73–80. https://doi.org/10.1016/j.plantsci.2015.05.020
Praveen M, Uttam AG, Tonapi VA, Ragimasalawada M (2018) Fine mapping of Rf2, a major locus controlling pollen fertility restoration in sorghum A 1 cytoplasm, encodes a PPR gene and its validation through expression analysis. Plant Breed 137:148–161. https://doi.org/10.1111/pbr.12569
Qian ZQ (1990) Discussions on the inheritance of A1 cytoplasmic male sterility and the establishment of differentiating line for restoring genotype in sorghum. Hered Beijing 12:11–12
Quinby J (1980) Interaction of genes and cytoplasms in male sterility in sorghum. In: 35th Corn and sorghum research conference. American Seed Trade Association, Chicago, IL, USA
R Development Core Team (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rao NGP, Tripathi DP, Rana BS (1984) Genetic analysis of cytoplasmic systems in sorghum. Indian J Genet Plant Breed 44:480–496
Rattunde HFW, Weltzien E, Diallo B et al (2013) Yield of photoperiod-sensitive sorghum hybrids based on guinea-race germplasm under farmers’ field conditions in Mali. Crop Sci 53:2454–2461. https://doi.org/10.2135/cropsci2013.03.0182
Reuscher S, Furuta T (2016) ABHgenotypeR: easy visualization of ABH genotypes (R package ABHgenotype, R version 1.0.1). Bioscience and Biotechnology Center, Nagoya University, 464-8601, Japan
Rooney W, Wayne Smith C (2000) Techniques for developing new cultivars. In: Wayne Smith C, Frederiksen RA (eds) Sorghum: origin, history, technology, and production. Wiley, New York, pp 329–348
Ross WM, Hackerott HL (1972) Registration of seven isocytoplasmic sorghum germplasm lines1 (Reg. Nos. GP 9 to GP 15). Crop Sci 12:720. https://doi.org/10.2135/cropsci1972.0011183X001200050083x
Sanjana Reddy P, Rao D, Reddy BV, Ashok Kumar A (2010) Inheritance of male-fertility restoration in A1, A2, A3 and A4 (M) cytoplasmic male-sterility systems of sorghum [Sorghum bicolor (L.) Moench]. Indian J Genet Plant Breed 70:240–246
Schertz KF (1977) Registration of A2 Tx2753 and B Tx2753 sorghum germplasms1 (Reg. No. GP 30 and 31). Crop Sci 17:983. https://doi.org/10.2135/cropsci1977.0011183X001700060056x
Schertz KF, Ritchey JM (1978) Cytoplasmic-genic male-sterility systems in sorghum1. Crop Sci 18:890. https://doi.org/10.2135/cropsci1978.0011183X001800050055x
Schnable PS, Wise RP (1998) The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci 3:175–180. https://doi.org/10.1016/S1360-1385(98)01235-7
Schön CC, Utz HF, Groh S et al (2004) Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics 167:485–498. https://doi.org/10.1534/GENETICS.167.1.485
Smale M, Assima A, Weltzien E, Rattunde F (2014) An overview and economic assessment of sorghum improvement in Mali. International development working paper 137. Michigan State University, East Lansing, MI
Stephens JC, Holland RF (1954) Cytoplasmic male-sterility for hybrid sorghum seed production. Agron J 46:20–23. https://doi.org/10.2134/agronj1954.00021962004600010006x
Tang HV, Chang R, Pring DR (1998) Cosegregation of single genes associated with fertility restoration and transcript processing of sorghum mitochondrial orf107 and urf209. Genetics 150:383–391
Taylor J, Butler D (2017) R package ASMap: efficient genetic linkage map construction and diagnosis. J Stat Softw 79:1–29. https://doi.org/10.18637/jss.v079.i06
Toure AB, Scheuring JF (1982) Presence de genes mainteneurs de l’androsterilité cytoplasmique parmi les variétés locales de Sorgho au Mali. L’agronomie Trop 37:362–365
Utz HF (2012) PLABMQTL, A Computer Programme to Map QTL, Version 0.5.s. Institute of Plant Breeding, Seed Science, and Population Genetics, University of Hohenheim. Stuttgart, Germany
Utz HF, Melchinger EA (1994) Comparison of different approaches to interval mapping of quantitative trait loci. In: Van Ooijen JW, Jansen J (eds) Biometrics in plant breeding: applications of molecular markers. Proceedings of the ninth meeting of the EUCARPIA Section Biometrics in Plant Breeding. Wageningen, The Netherlands, pp 195–204
Vales MI, Schön CC, Capettini F et al (2005) Effect of population size on the estimation of QTL: a test using resistance to barley stripe rust. Theor Appl Genet 111:1260–1270. https://doi.org/10.1007/s00122-005-0043-y
Van Ooijen JW (1999) LOD significance thresholds for QTL analysis in experimental populations of diploid species. Heredity (Edinb) 83:613–624. https://doi.org/10.1038/sj.hdy.6886230
Vanderlip RL, Reeves HE (1972) Growth Stages of Sorghum [Sorghum bicolor, (L.) Moench.]. Agron J 64:13–16. https://doi.org/10.2134/agronj1972.00021962006400010005x
Webster OJ, Singh SP (1964) Breeding behavior and histological structure of a nondehiscent anther character in Sorghum vulgare Pers. 1. Crop Sci 4:656–658. https://doi.org/10.2135/cropsci1964.0011183X000400060032x
Weltzien E, Rattunde HFW, van Mourik TA, Ajeigbe HA (2018) Sorghum cultivation and improvement in West and Central Africa. In: Rooney W (ed) Achieving sustainable cultivation of sorghum: sorghum utilization around the world. Burleigh Dodds Science Publishing, Cambridge
Wen L, Tang HV, Chen W et al (2002) Development and mapping of AFLP markers linked to the sorghum fertility restorer gene rf4. TAG Theor Appl Genet 104:577–585. https://doi.org/10.1007/s00122-001-0774-3
Worstell JV, Kidd HJ, Schertz KF (1984) Relationships among male-sterility inducing cytoplasms of sorghum1. Crop Sci 24:186–189. https://doi.org/10.2135/cropsci1984.0011183X002400010044x
Wu Y, Bhat PR, Close TJ, Lonardi S (2008) Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet 4:e1000212. https://doi.org/10.1371/journal.pgen.1000212
Xu S (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208. https://doi.org/10.1534/genetics.108.090688
Yin R, Liu R, Mei Y et al (2013) Characterization of bio-oil and bio-char obtained from sweet sorghum bagasse fast pyrolysis with fractional condensers. Fuel 112:96–104. https://doi.org/10.1016/j.fuel.2013.04.090
Acknowledgements
The authors thank the technical staff of the sorghum breeding program in ICRISAT-Mali and specifically Abdoul Karim Touré and Marthe Dembélé for their contribution in phenotyping. We also acknowledge the support of the lab personnel at the State Plant Breeding Institute of the University of Hohenheim, particularly Stefanie Laucher, for candidate gene sequencing and marker creation. We would like to acknowledge the funding by the German Ministry for Economic Cooperation and Development (BMZ) via the BMZ-Heterosis Project (ICRISAT Project 13.1432.7-001.00), and also the supplemental financial support from the McKnight Foundation Collaborative Crop Research Program via discretionary research funds provided to B.I.G. Haussmann. This work was undertaken as a part of the CGIAR Research Program on Dryland Cereals.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The authors declare that all experiments conform to the current laws in Mali and in Germany.
Additional information
Communicated by Hai-Chun Jing.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kante, M., Rattunde, H.F.W., Nébié, B. et al. QTL mapping and validation of fertility restoration in West African sorghum A1 cytoplasm and identification of a potential causative mutation for Rf2. Theor Appl Genet 131, 2397–2412 (2018). https://doi.org/10.1007/s00122-018-3161-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-018-3161-z