
Abstract
The Synaptotagmin (SYT) family of proteins play key roles in regulating membrane trafficking at neuronal synapses. Using both Ca2+-dependent and Ca2+-independent interactions, several SYT isoforms participate in synchronous and asynchronous fusion of synaptic vesicles (SVs) while preventing spontaneous release that occurs in the absence of stimulation. Changes in the function or abundance of the SYT1 and SYT7 isoforms alter the number and route by which SVs fuse at nerve terminals. Several SYT family members also regulate trafficking of other subcellular organelles at synapses, including dense core vesicles (DCV), exosomes, and postsynaptic vesicles. Although SYTs are linked to trafficking of multiple classes of synaptic membrane compartments, how and when they interact with lipids, the SNARE machinery and other release effectors are still being elucidated. Given mutations in the SYT family cause disorders in both the central and peripheral nervous system in humans, ongoing efforts are defining how these proteins regulate vesicle trafficking within distinct neuronal compartments. Here, we review the Drosophila SYT family and examine their role in synaptic communication. Studies in this invertebrate model have revealed key similarities and several differences with the predicted activity of their mammalian counterparts. In addition, we highlight the remaining areas of uncertainty in the field and describe outstanding questions on how the SYT family regulates membrane trafficking at nerve terminals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Synaptic communication and the synaptic vesicle release machinery
The nervous system relies on regulated secretion of neurotransmitters to meditate synaptic communication between neurons [1, 2]. Synaptic transmission typically occurs at specialized release sites in presynaptic terminals known as active zones (AZs). Highly conserved AZ scaffold proteins concentrate voltage-gated Ca2+ channels and synaptic vesicles (SVs) to position the release machinery near clustered neurotransmitter receptors in the postsynaptic membrane [3,4,5,6,7,8,9,10,11,12,13,14]. During action potential propagation along the axon, membrane depolarization triggers the opening of voltage-gated Ca2+ channels and influx of extracellular Ca2+ to trigger SV fusion at release sites [2, 15,16,17,18,19,20]. SV exocytosis is a highly stochastic process at individual AZs, with the probability of an individual fusion event varying over a wide range depending on the neuronal population [21,22,23]. The evoked response recorded postsynaptically represents the probability of SV fusion events that occur over a population of individual release sites.
The timing for single SV fusion events can occur over a relatively broad temporal window of ~ 1–200 ms at individual AZs following an action potential (Fig. 1a). Release kinetics have been loosely classified into two distinct phases termed synchronous and asynchronous release [24]. The synchronous phase accounts for the majority of neurotransmitter release at most synapses, with SV fusion events decaying within several milliseconds after presynaptic Ca2+ influx. The asynchronous phase (also known as the delayed response) is less pronounced at most synapses but becomes more robust during high frequency nerve stimulation. In this slower component of release, individual SV fusion events occur within several hundred milliseconds across the AZ population [19, 25, 26]. In addition to the two phases of evoked release, single SVs can fuse spontaneously (termed minis) in the absence of nerve stimulation [23, 26,27,28].

Regulation of synchronous and asynchronous SV fusion. a Model depicting phases of synchronous and asynchronous release after nerve stimulation (arrow). b Structure of the fusion machinery that bridges the SV and plasma membrane, with SYT1 (grey) and the SNARE complex (Syntaxin—red; SNAP25—green; Synaptobrevin—blue). c Model of synchronous and asynchronous release in relation to presynaptic Ca2+ entry (shaded). Following Ca2+ influx, SVs fuse during a synchronous phase at AZs that occurs within milliseconds. SYT1 acts as the Ca2+ sensor for synchronous release and resides on SVs. A slower asynchronous component can last for hundreds of milliseconds and include fusion of SVs farther from release sites. SYT7 has emerged as a candidate for the asynchronous Ca2+ sensor with higher affinity than SYT1 for Ca2+ binding. The role of SYT7 is still controversial, with studies in Drosophila suggesting it controls SV availability and fusogenicity
All three pathways for SV release (synchronous, asynchronous, spontaneous) require a highly conserved fusion machinery that controls the regulated assembly of a four stranded coiled-coil SNARE complex that bridges the SV and presynaptic membranes [29,30,31]. This complex includes the v-SNARE Synaptobrevin on the SV, and the t-SNAREs Syntaxin and SNAP-25 on the presynaptic plasma membrane (Fig. 1b). Multiple SNARE chaperones, including the UNC13 and UNC18 families, position and regulate the timing of the highly energetic assembly of the SNARE alpha-helices during the SV cycle [2, 32,33,34,35]. Current models suggest the coiled-coil SNARE bundle partially assembles at the SV-plasma membrane interface prior to fusion in a cis-conformation where the transmembrane domains of Synaptobrevin and Syntaxin remain separated in the SV and plasma membrane, respectively. The assembly and arrest of SNARE zippering at this intermediate state is regulated by the cytosolic SNARE-binding protein Complexin (CPX), which is hypothesized to provide a clamping brake on fusion until Ca2+ entry occurs [36,37,38,39,40,41,42,43]. The SYT1 family of SV Ca2+ sensors binds Ca2+ ions through the action of a cluster of negatively charged aspartate residues present in loop structures that emerge from its two C2 domains (Fig. 1b). The Ca2+-bound loops of SYT1 interact with negatively charged lipid headgroups in the presynaptic membrane. This interaction alters local lipid structure and helps trigger full zippering of the SNARE complex into a trans-SNARE state where the transmembrane domains of the SNARE proteins reside together in the fused membrane [2, 40, 44,45,46]. In addition to activating release, SYT1 also inhibits spontaneous fusion through a clamping mechanism similar to the role of CPX [47,48,49,50,51,52,53,54,55]. Together, these proteins act to drive full collapse of the SV into the plasma membrane during presynaptic Ca2+ influx to exocytose neurotransmitters through the fast synchronous pathway [24, 51, 56,57,58,59,60,61,62,63,64].
The asynchronous release pathway uses a similar fusion machinery with the major exception of not requiring SYT1 (Fig. 1c). Several mechanisms have been proposed that differentiate asynchronous and synchronous release [16, 24, 65,66,67,68,69,70,71,72,73,74], including distinct Ca2+ sensors, heterogeneity in SV protein content, SV distance from Ca2+ channels, distinct Ca2+ entry pathways, and regulation of Ca2+ extrusion and buffering. Asynchronous release is enhanced in Syt1 mutants [51, 57, 75], suggesting Ca2+ can activate this slower pathway through a distinct Ca2+ sensor(s). Manipulations of SYT7 change the amount of release occurring through the slower asynchronous SV fusion pathway, but whether the protein functions as a Ca2+ sensor for the fusion process itself or modifies Ca2+-dependent SV availability is unclear [58, 76,77,78,79,80,81,82,83,84]. Following fusion of SVs through either pathway, α-SNAP binding to trans-SNARE complexes in the plasma membrane recruits the AAA ATPase NSF to disassemble the complex and recharge individual SNAREs for additional rounds of release [29, 85,86,87,88,89,90]. NSF can also disassemble trans-SNARE complexes present on the SV that escape endocytosis control, releasing free v-SNAREs to form productive cis-SNARE complexes required for fusion [91]. Following endocytosis, SNARE chaperones and the priming machinery reposition fusogenic SVs at AZs for additional cycles of release.
In the current review, we discuss models for how the SYT family regulates membrane trafficking at synapses. In particular, we focus on studies performed in Drosophila that examine the function of the three most abundant SYT isoforms (SYT1, SYT4, and SYT7) that are found at most synapses. Similar to Drosophila, the homologs of these three SYT isoforms are among the most abundant members in mammals [84]. SYT1 and SYT7 have been shown to regulate SV trafficking in both systems, while SYT4 has been linked to presynaptic exosomes and postsynaptic retrograde signaling in Drosophila. Electrophysiology, imaging and structure–function studies have provided insights into how SYT1, SYT4, and SYT7 regulate synaptic communication in vivo. We review this data, as well as examine what is known about the remaining family members. Finally, we highlight recent studies identifying mutations in SYT family proteins in human neurological disorders and describe work on potential pathological mechanisms using Drosophila models.
Analyzing synaptic transmission at Drosophila neuromuscular junctions
Electrophysiological and imaging analysis at Drosophila 3rd instar larval neuromuscular junction (NMJ) synapses has proven highly effective for dissecting the roles of SYTs in synapse biology. At this connection, motoneurons form glutamatergic synapses onto muscles, with motor axons containing tens of en passant presynaptic boutons with hundreds of individual AZs highlighted by a T-bar filamentous structure at the center (Fig. 2a, b) [14]. SV release from motoneurons can be measured using electrophysiological recordings of synaptic currents from the muscle or by optical imaging of postsynaptic fusion events. Individual AZs at the NMJ are aligned to discrete postsynaptic glutamate receptor (GluR) fields. This arrangement facilitates use of modified GCaMPs expressed postsynaptically to image SV fusion events by visualizing spatially localized Ca2+ influx following GluR opening (Fig. 2c) [22, 23, 92,93,94]. This toolkit makes the Drosophila NMJ one of the only models where every SV fusion event can be imaged and assigned to single AZs, greatly increasing the spatial resolution of SV release compared to electrophysiology alone. These imaging approaches demonstrate single quanta are released at AZs following an action potential, as multi-vesicular release at individual sites is rare [22, 23, 92,93,94,95]. Similar to other systems, AZs formed by a single Drosophila motoneuron display heterogeneity in SV release probability (Pr), with a small population of strong AZs present amongst many weaker ones (Fig. 2c). Highlighting the stochastic nature of the release process, the average AZ Pr is ~ 0.07 at the NMJ, indicating most AZs release a SV less than 10% of the time in response to single action potentials [22]. This quantal imaging approach has been applied to Syt1 and Syt7 mutants to examine SV release at individual AZs.

The Drosophila larval NMJ as a model for synaptic function. a Immunolabeling of the nerve terminal showing AZs (anti-BRP, magenta) and post-synaptic densities (anti-Glutamate Receptor III staining, green) at a larval NMJ. b EM of a single synaptic bouton with an AZ T-bar denoted (arrow). c The left panel shows a model of synaptic boutons with multiple individual AZs (left). The middle panel shows an NMJ expressing the presynaptic Ca2+ channel (Cac-GFP, green) and evoked SV release events (red) visualized with a modified jRGECO Ca2+ indicator expressed postsynaptically. The right panel shows quantal imaging of evoked release probability at the same NMJ, revealing heterogeneity in AZ strength as noted on the color-coded heat map. a Modified from [105], b modified from [82], and c modified from [22]
The Synaptotagmin superfamily
SYTs are conserved family of membrane-trafficking proteins containing a single transmembrane domain and two paired cytosolic C2 domains (Figs. 1b, 3). Their expression is largely restricted to the nervous system, though a few isoforms participate in a smaller subset of trafficking pathways in non-neuronal cells. Genes encoding SYT proteins are not found in bacterial or yeast genomes, indicating intracellular membrane fusion events like ER to Golgi trafficking do not require the function of this protein family. The first members of the SYT family emerged during evolution in the placozoans, a primitive branch of multi-cellular metazoans that lack neurons. Homologs of SYT1 and SYT7 are encoded in the Trichoplax adhaerens genome, suggesting a non-neuronal origin of SYTs during a period when multicellular communication was emerging [96, 97]. The SYT family expanded during invertebrate evolution, and the Drosophila genome encodes seven distinct SYT proteins. With genome duplications in vertebrates, greater diversification occurred and 17 SYT family members are encoded in the human genome [96]. Many SYT proteins can be grouped into functional orthologs based on sequence similarity across evolution, while other isoforms are more divergent without clear orthologs between invertebrate and vertebrate proteins (Fig. 3a).

Conservation, abundance and localization of Drosophila SYTs. a Phylogenetic tree of SYT homologs in Drosophila melanogaster (d), Mus musculus (m), and Homo sapiens (h). The SYT1, SYT4, SYT7, SYT12, and SYT14 subfamilies are highlighted. The sequences of each SYT were extracted from NCBI and the tree was generated using neighbor clustering algorithm. b Expression level of Drosophila Syt genes in larval and adult brain using RNAseq. c Expression of endogenously CRISPR-tagged SYT7-GFP compared to the AZ protein BRP in a single larval NMJ bouton. SYT7 surrounds AZs and localizes to an interconnected tubular membrane compartment within the peri-AZ. d Localization of SYT1 to SVs and SYT4 to postsynaptic puncta at the larval NMJ using immunocytochemistry. The motor axon is stained with anti-HRP (green). e Model of the subcellular localization of Drosophila SYTs. SYT1 is attached to SVs and triggers synchronous SV fusion. SYT7 localizes to an internal peri-AZ compartment and negatively regulates SV re-entry into the readily releasable pool and SV fusogenicity. SYT4 localizes to presynaptic exosomes that are released from multi-vesicular bodies (MVBs) to transfer the protein to the postsynaptic compartment where it mediates retrograde signaling. The graph in panel b was generated by plotting gene expression levels in the CNS of 7-day-old males reported in [368], panel c was modified from [100], and panel d was modified from [82]
SYTs can be subdivided into whether or not they are likely to bind Ca2+ based on conservation of negatively charged aspartate residues within their C2 domains that mediate this interaction [98]. For the seven Drosophila SYT homologs, five are predicted to bind Ca2+ (SYT1, SYT4, SYT7, SYTα, SYTβ), while two lack conserved Ca2+ binding aspartate residues (SYT12, SYT14) [99, 100]. Three Drosophila SYT isoforms are highly expressed in neurons (SYT1, SYT4, SYT7) based on in situ mRNA expression and RNAseq analysis (Fig. 3b) [100]. SYT1 is homologous to mammalian SYT1, SYT2, and SYT9 proteins, all of which reside on SVs (Fig. 3c) and function as Ca2+ sensors driving fast synchronous SV fusion in specific neuronal populations [57, 60, 61, 63, 101]. Drosophila Syt1 null mutants generally die as embryos or during early larval development [63, 102], though some can survive to adulthood at low frequency when cultured directly on food where minimal movement is required [103]. These surviving adults are completely ataxic and die within several days. SYT7 is encoded by single gene in both Drosophila and mammals and has been linked to asynchronous SV release [80, 82, 84]. The Drosophila homolog resides on a tubular membrane compartment present within the peri-AZ, a synaptic domain implicated in endocytosis and protein sorting (Fig. 3d). Drosophila SYT4 is homologous to mammalian SYT4 and SYT11 and is transferred to the postsynaptic compartment from exosomes (Fig. 3b), where it functions in retrograde signaling [97, 100, 104,105,106,107]. Mutations in Drosophila Syt4 and Syt7 are viable and fertile as adults [82, 97, 104, 105], though no studies to date have examined potential roles in synaptic plasticity and learning in adult animals. SYT12 and SYT14 are more divergent in sequence, but appear orthologous to mammalian SYT12 and SYT14 [99]. These isoforms are expressed at low levels in Drosophila and little is known about their function. SYTα and SYTβ are present on dense core vesicles (DCVs) in neuroendocrine neurons and are presumed to function as DCV Ca2+ sensors in Drosophila [100, 108,109,110]. Their precise relationship to specific mammalian SYT isoforms is unclear. No mutants in these final four SYT isoforms have been described. Beyond these seven SYT family members in Drosophila, there are multiple genes encoding proteins that contain C2 domains that are orthologs of other synaptic and non-synaptic proteins that will not be discussed. These include the ER residents extended SYT (ESYT) and multiple C2 domain protein (MCTP) family members [111,112,113,114], homologs of the synaptic proteins Rabphilin, Rim, and Munc13 [99, 115, 116], as well as homologs of mammalian Otoferlin (Misfire) [117] and Granulophilin (Bitesize) [118]. In contrast to mammals, the Drosophila genome does not encode a homolog of the DOC2 family, a group of SYT-like proteins with two cytosolic C2 domains lacking a transmembrane domain. The DOC2 family has been implicated in SV trafficking [119,120,121,122], but they appear to represent a novel adaptation to the process found only in vertebrates.
The domain structure of the SYT family is highly conserved across all isoforms, with a single-pass transmembrane domain, a variable linker and two tandem cytosolic C2 domains, termed C2A and C2B [98,99,100, 123, 124]. C2 domains are found in a wide array of proteins and often function as Ca2+-dependent lipid binding modules. They represent one of several Ca2+ binding domains found among proteins, with EF hands representing another prominent motif that translates intracellular Ca2+ rises into downstream effector responses [125,126,127,128]. Within the protein kinase C (PKC) family, the C2 domain serves to bring the kinase to the plasma membrane in response to internal Ca2+ elevation through its lipid-binding properties. Such an evolutionarily conserved role can be postulated for some SYT family members, which are tethered to intracellular membrane organelles like SVs via their transmembrane domains. This would allow the C2 domains of SYTs to bridge distinct membrane compartments and bring two lipid bilayers in close proximity for potential fusion or lipid mixing. Although C2 domains are largely considered Ca2+-dependent lipid binding modules, they also mediate a host of Ca2+-independent interactions. As noted above, multiple SYT family members lack the required aspartate residues that coordinate Ca2+ binding, suggesting these isoforms use the C2 domain as a protein–protein interaction module instead. This is also the case for SYTs that display Ca2+-dependent lipid binding, as other regions of the C2 domain interact with multiple effector proteins, including the SNARE complex [38, 129]. In addition, a polybasic motif on the surface of the C2 domains of some SYT proteins can interact with lipids in a Ca2+-independent manner to facilitate vesicle docking [56, 98, 130,131,132].
Beyond the role of specific isoforms in SV fusion, SYT family members also function as Ca2+ sensors for DCV fusion that mediates release of neuropeptides and neuromodulators [108, 133,134,135,136,137,138,139,140,141,142]. Though Ca2+ regulation of SV and DCV release by SYTs in presynaptic terminals represent the most-well characterized role, there is also evidence they function in postsynaptic vesicle trafficking [97, 104, 105, 143,144,145,146,147,148]. Postsynaptic Ca2+ influx is required to regulate membrane trafficking to support retrograde signaling and postsynaptic neurotransmitter receptor cycling. Family members of the SYT4/SYT11 and SYT3 subgroups, along with SYT1 and SYT7, have been implicated in these postsynaptic processes. Finally, the group of SYT proteins with degenerate Ca2+ binding sites within their C2 domains are likely to participate in distinct Ca2+-independent membrane trafficking steps [98,99,100]. In general, little is known about these more obscure SYT isoforms.
Synaptotagmin 1 functions as the major Ca2+ sensor for triggering synchronous SV fusion
SYT1 is the best characterized member of the SYT family. It is found on SVs and functions as the primary Ca2+ sensor for activating fast synchronous fusion in all species examined to date. In mammals, SYT1 is the most highly expressed member of the SYT subgroup found on SVs and serves as the sole Ca2+ sensor for synchronous fusion for most neurons in the CNS. SYT2 and SYT9 share largely redundant roles with SYT1 in regulating SV fusion for a smaller population of CNS neurons and most PNS neurons [60, 101]. In Drosophila, a single gene encodes SYT1 and its loss disrupts evoked release as well [49, 51, 57, 63, 75, 149,150,151,152,153,154,155,156]. Although the role of SYT1 as a Ca2+ sensor for fusion is widely accepted, the protein also functions in additional steps of the SV cycle, including docking, priming, and endocytosis [57, 150, 155, 157]. We focus our discussion primarily on SYT1 studies performed in Drosophila, as multiple reviews describing mammalian SYT1 are available [40, 44, 70, 86, 158,159,160,161,162,163].
The multi-functional nature of SYT1 has made it challenging to separate its role for specific steps in SV trafficking, as null mutations disrupt all its properties. This has been partially addressed in Drosophila using point mutants altering a single amino acid, or by rescuing null mutants with SYT1 transgenes containing mutations in one or several residues [49, 51, 57, 62, 75, 131, 150, 152, 156, 164,165,166,167,168]. Although these approaches have improved resolution, several functions of SYT1 are likely to require similar interactions and are difficult to separate. A second issue is that some point mutants have dominant-negative activity and disrupt release more than the complete absence of SYT1, confounding interpretations for these alleles. This has been particularly problematic for C2B domain Ca2+ binding mutants that act in a dominant-negative manner [49, 57, 62, 75, 156]. A final issue in interpreting defects in Syt1 mutants has been the predominant use of the 3rd instar NMJ preparation for analysis. Although this synaptic connection is excellent for physiological studies, it requires animals to progress through earlier larval stages that last for days where compensation through homeostatic mechanisms could mask some phenotypes. In addition, reduced activity in Syt1 mutants may have consequences on synaptic development. A few studies have used the more difficult embryonic NMJ preparation where developmental defects and compensation are less likely to occur [57, 75, 151]. Acute inactivation approaches have also been used to avoid these issues, though their specificity is somewhat unclear [157, 169].
The initial analysis of Drosophila Syt1 mutants revealed multiple defects in SV release at larval NMJs. The first key observation was that evoked release was dramatically reduced while spontaneous mini frequency was elevated (Fig. 4a) [63, 154]. This result indicated SYT1 played distinct roles in SV release by promoting evoked fusion and clamping Ca2+-independent spontaneous release. Although reductions in evoked release can be secondary to many potential defects in the SV cycle, the finding that the Ca2+ dependence of release was altered in some Syt1 hypomorphic mutants provided support for the protein acting as a Ca2+ trigger for fusion [51]. Subsequent work in mice identified specific point mutants in SYT1 that shifted the Ca2+ sensitivity of release, further pointing towards a release defect tied to Ca2+ sensing [170]. Following the initial characterization, additional studies of Drosophila Syt1 mutants indicated its role in evoked release was twofold. Loss of SYT1 caused a dramatic reduction in fast synchronous release with enhanced release through the slower asynchronous fusion pathway at both embryonic and larval NMJs [57, 75, 151]. This work confirmed SYT1 was not the sole Ca2+ sensor for SV fusion, consistent with earlier studies on null mutants [102]. A two Ca2+ sensor model for SV exocytosis emerged in the field (Fig. 1c), with SYT1 driving the majority of rapid synchronous release and an unknown Ca2+ sensor activating asynchronous fusion. Candidates for the asynchronous Ca2+ sensor included other SYT family members, with SYT4 and SYT7 being the only attractive isoforms in Drosophila given their broad expression throughout the nervous system [100]. However, electrophysiological studies of Syt1/Syt4 and Syt1/Syt7 double mutants found these animals retain asynchronous release [153, 171], indicating Ca2+-sensitive mechanisms outside of the SYT family contribute to the slower release pathway at Drosophila synapses.

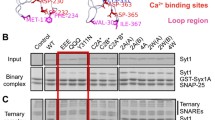
Function of SYT1 in SV fusion. a Syt1 null mutants reduce synchronous fusion and enhance asynchronous release and mini frequency. Rescue with a Syt1 transgene with defective Ca2+ binding to C2A(*) and C2B(*) fails to support synchronous fusion and causes higher rates of spontaneous release. b Overexpression of a C2B Ca2+ binding mutant (D1,2N) suppresses release compared to overexpression of wildtype SYT1. Twenty essential residues mapping to C2A (blue) or C2B (magenta) were identified in an intragenic suppressor screen that blocked the dominant-negative effects. c. Structure of the primary and tripartite interface of the SYT1/SNARE/CPX complex. d Location of mutations disrupting the primary SNARE interface on the SYT1 C2B domain. The polybasic stretch is shown in grey and localizes to the opposite C2B surface. SNARE-binding mutations fail to rescue release defects in Syt1 null mutants (right panel). e Location of the R250H mutation at the SYT1 dimer interface that disrupts oligomerization. This mutation reduces SV release (right panel), though not as severely as SNARE-binding mutants. a Modified from [49], b, d, and e modified from [62], and c modified from [38]
Initial models for SYT1 function in autaptic neuronal cultures from Syt1 mutant mice suggested loss of the protein resulted in shifts in the time course of fusion without altering the actual number of SVs released. This view was not consistent with the dramatic reduction in the total number of SVs released in Drosophila Syt1 mutants. Subsequent studies in mice confirmed observations from Drosophila that the total number of SVs released was dramatically reduced in the absence of SYT1 [53, 60, 64, 84, 101]. Quantal imaging of SV release at individual AZs in Drosophila Syt1 mutants further demonstrated a profound reduction in release probability [22], confirming loss of SYT1 reduces synchronous SV fusion and dramatically decreases the total number of SVs released overall. The dual roles of SYT1 in activating synchronous release and suppressing asynchronous fusion were genetically separated and mapped to distinct C2 domains [75]. These observations supported a model whereby SYT1 actively inhibited the asynchronous pathway, versus the alternative where SVs normally destined for synchronous release were simply being released asynchronously. Together, these data suggested some similarities between SYT1 and rapidly inactivating ion channels that open to allow ion flow and undergo a second conformational change that inactivates the channel. For SYT1, Ca2+ binding triggers a conformational state or set of interactions that increase the likelihood of a SV undergoing fusion within several milliseconds. Shortly after, SYT1 undergoes another conformational change, potentially triggered by Ca2+ unbinding, that actively inhibits SV release and reduces the probability of any slower asynchronous fusion events. This dual activity of SYT1 helps ensure SVs are released within precise temporal windows that are needed for rapid neuronal computations.
Beyond a role in regulating fusion, a number of studies also identified defects in SV docking, priming and endocytosis, indicating SYT1 acts at multiple steps to regulate SV cycling in Drosophila [57, 150, 155, 157]. Although Syt1 mutants have defects in docking and endocytosis, the protein is not essential for either pathway as these processes continue with reduced efficacy in the absence of the protein. Interactions between SYT1 and clathrin adapter proteins provide candidate mechanisms for its role in facilitating endocytosis [150, 172,173,174,175,176,177,178,179]. In contrast, how SYT1 supports SV docking is less clear. Ca2+-independent membrane interactions mediated by a polybasic amino acid stretch in the two C2 domains, together with its SNARE-binding properties, represent candidate mechanisms. Overall, initial studies of SYT1 provided models for how it regulates SV cycling. Loss of SYT1 did not eliminate spontaneous or asynchronous release and had no obvious effects on spontaneous SV fusion kinetics. As such, SYT1 does not appear to be essential for fusion or dynamics of the SV fusion pore itself, but acts as a Ca2+-dependent trigger to increase the likelihood SNARE-dependent fusion occurs and is limited to a short temporal window. Although the kinetics of single SV fusion events do not appear to be controlled by SYT1, there is evidence the protein can regulate fusion pore kinetics of DCVs and in reconstituted in vitro fusion systems [138, 139, 180,181,182,183,184,185,186,187,188]. These early studies in Drosophila also highlighted differences between the mechanisms for fast synchronous SV release that require SYT1 versus asynchronous and spontaneous fusion that are negatively regulated by the protein.
Role of C2 domain Ca2+ binding and SNARE interactions for Synaptotagmin 1 function
To directly characterize the role of the C2 domain Ca2+ binding loops in Drosophila, a subset of the key aspartate residues in each loop (numbered D1–D5) were mutated to neutralize the negative charge and prevent Ca2+ binding. The most common approach was to generate D to N substitutions to create D1,2N and D3,4N mutants in either or both C2 domains and use these transgenes to rescue Syt1 null mutants. This approach indicated the C2B domain had a critical role in evoked release, with a near complete loss of synchronous SV fusion in C2B D1,2N or C2B D3,4N mutants [49, 75, 156]. Similar transgenes disrupting the C2A domain did not block evoked release, but rather failed to clamp asynchronous fusion [49, 75]. Together with studies in mice, these findings pointed towards Ca2+ binding to the C2B domain as the critical trigger for exocytosis, while C2A Ca2+ binding appeared less important. However, these results have become more difficult to interpret with the observation that mutations in the C2B Ca2+ binding pocket cause dominant-negative phenotypes in Drosophila, mice, and humans [49, 57, 75, 152, 156, 164, 189,190,191]. Indeed, release in Syt1 null mutants rescued with C2B D1,2N or C2B D3,4N display less SV fusion that the null mutant itself [75, 156]. This has led to re-evaluation of how these substitutions are altering C2 domain function. One hypothesis is neutralization of the charged residues makes SYT1 more likely to undergo Ca2+-independent membrane interactions via the loops that potentially block fusion sites. As such, the preferential role of Ca2+ binding to C2B as the sole trigger for SV fusion requires re-evaluation. More recent mutagenesis work using different amino acid substitutions within the C2 domain loops that alter hydrophobicity of residues rather than charge indicate C2A Ca2+ binding also has an important role in SYT1 [165, 192,193,194,195]. These observations suggest Ca2+ binding to both C2 domains is important for the full activity of SYT1 in triggering synchronous SV fusion. Mutations disrupting Ca2+ binding to both C2 domains of SYT1 still support SV docking and endocytosis, indicating these functions of the protein are largely Ca2+-independent [49], though Ca2+ binding may enhance endocytosis rates [176]. Interestingly, C2A–C2B Ca2+ binding-defective SYT1 animals have a far greater increase in spontaneous release compared to null mutants (Fig. 4a), indicating restoring docking and endocytosis magnifies the role of SYT1 in clamping spontaneous fusion that is less pronounced in nulls due to reduced SV number [49].
One candidate mechanism for Ca2+-dependent membrane binding by SYT1 is based on structural similarity of the C2 domain loops to fusion peptides in viral fusion proteins like hemagglutinin [196]. For viral fusion proteins, conformational changes in the acidic environment of endosomes expose the loops, allowing them to drive fusion between viral and endosomal membranes and release viral content. The fusion loops in SYT1 contain negatively charged aspartate residues nested within them, unlike the loops of viral fusion proteins. In the absence of Ca2+, these residues repel C2 domains from interacting with negatively charged phospholipid containing membranes. In the presence of Ca2+, the negative charge on these residues are neutralized and the C2 domains engage the lipid bilayer, potentially allowing the loops to function similarly to those of viral fusion proteins.
Beyond the role of Ca2+-dependent lipid binding by the C2 domains, several other SYT1 interactions have been implicated in SV release in Drosophila. Syt1 transgenes containing mutations in the C2B Ca2+ binding pocket dominantly disrupt release (Fig. 4b) and cause lethality even in the presence of endogenous SYT1. Taking advantage of this observation, chemical mutagenesis screens have been performed to identify intragenic SYT1 suppressors where randomly generated second-site mutations in the dominant-negative transgene reduce the toxic effect [62]. This approach uncovered 20 essential residues within SYT1 that mapped to distinct areas of the protein (Fig. 4b). The screen identified the five essential C2B domain residues (S332 (S279 in mammalian SYT1), R334 (R281), Y391 (Y338), E348 (E295) and A455 (A402) that form the primary surface interaction site that docks one side of the C2B domain onto the SNARE complex based on the elucidated structure (Fig. 4c) [129]. Generation of several of these mutations into otherwise wildtype Syt1 transgenes demonstrated they failed to rescue evoked release defects in Syt1 nulls (Fig. 4d). Disruption of the SYT1–SNARE complex interaction caused a loss of synchronous release, enhanced asynchronous fusion, a reduction in the Ca2+ cooperativity curve, and elevated spontaneous release. As such, these SYT1 functions require SNARE complex binding. SV docking and endocytosis defects were not observed, suggesting these roles are independent of SNARE binding or redundant with other SYT1 binding partners.
Beyond SNARE interactions, mutations that disrupt the C2B polybasic stretch on the opposite surface of the C2B SNARE-binding domain were also identified in the screen. In the absence of Ca2+, SYT1 binds to PIP2 present in the plasma membrane through a polybasic stretch of amino acids lining this surface of the C2B domain. This interaction is predicted to occur before fusion as part of the docking/priming process, helping to increase membrane penetration of the C2 loops during Ca2+ entry by prepositioning SYT1 at fusion sites [44, 56, 130, 132, 165, 197,198,199,200,201,202]. Similar to prior structure function-studies, disruption of this Ca2+-independent lipid binding surface reduced evoked release, but caused far milder phenotypes than loss of Ca2+-dependent lipid binding or SNARE interactions [62, 131]. Together, these data suggest a model where SYT1 is prepositioned at the SV-plasma membrane interface in part through its C2B polybasic stretch and more fully by its C2B SNARE-binding interaction. Upon Ca2+ entry, the C2A and C2B Ca2+ binding loops pivot into the plasma membrane to drive lipid instability and facilitate complete SNARE zippering to activate fusion [44, 56, 203,204,205,206]. An alternative model includes two separate pools of SYT1 on the SV, one bound to SNARE complexes to support docking/priming and another pool not associated with SNAREs that mediate Ca2+-dependent lipid binding and membrane insertion of the protein.
The role of SYT1 tethering to SVs has also been examined by generating transgenes lacking the transmembrane domain or replacing the transmembrane domain with a myristoylation motif [152]. These transgenes could not support fast synchronous fusion in Syt1 null mutants, though cytosolic C2 domains enhanced asynchronous release even more than that observed in Syt1 nulls. These observations suggest transmembrane tethering of SYT1 positions the protein near release sites so its Ca2+ binding properties can rapidly drive fusion. An additional study examined the relevance of the individual SYT1 C2 domains for release by generating otherwise wildtype SYT1 proteins with only C2A or C2B alone, double C2A–C2A or C2B–C2B modules or reversed C2B–C2A order [49]. None of these manipulations rescued release defects in Syt1 null mutants, indicating both C2 domains are uniquely required for release and must be present in the correct sequence. Similar observations have been made for C2 domain swaps between mammalian SYT1 and SYT7, where chimeric proteins failed to rescue Syt1 mutant phenotypes in mice [207]. Although C2 domains may have a generalized function, they have uniquely evolved within each SYT family member, with the specific C2A and C2B domains serving distinct roles.
Open questions on the role of Synaptotagmin 1 in SV trafficking and fusion
It is widely accepted that the central role of SYT1 is centered around Ca2+-dependent lipid binding, but questions about additional interactions and their role in fusion are still being debated. For example, the precise role of SNARE binding is less clear. Studies in Drosophila provide strong in vivo evidence this interaction is required for SYT1 to promote synchronous fusion and suppress asynchronous release, but it is unknown when the interaction occurs during the SV cycle. Part of this confusion is tied to the lack of a precise understanding of the structure of the primed SNARE complex before fusion in vivo. Are SNARE complexes partially zippered before fusion as most models suggest, and can SYT1 bind to partially assembled SNAREs in this state? If so, this would place the interaction at a pre-fusion point to position SYT1 for future membrane insertion and/or regulate SNARE zippering. The interaction could also contribute to multimerization of SNARE complexes through the oligomerization properties of SYT1. Alternatively, the interaction might require fully assembled trans-SNARE complexes that are predicted to only form during fusion. This would place the interaction during the fusion process itself or after fusion has been completed. Although early biochemical work suggested SYT1 binding to individual t-SNAREs and the t-SNARE complex was Ca2+-dependent, current data suggest the primary binding mode is Ca2+-independent. As such, SNARE binding could act before, during or after Ca2+ entry.
Several other binding modes between SYT1 and the SNARE complex have also been described beyond the primary interface on the C2B surface that has genetic support in Drosophila. One model based on X-ray crystal structure suggests formation of an alpha helix partially contributed by a piece of the SYT1 C2B domain and part of the SNARE-binding protein CPX (Fig. 4c) [38]. This “tripartite” interaction mode brings in another key component for fusion regulation in CPX, but no genetic data has implicated this region of SYT1 in Drosophila SV trafficking. However, studies in Drosophila indicate the activity of SYT1 and CPX are indeed intimately tied together in controlling SV release dynamics [37]. Cpx null mutants share similar phenotypes with milder Syt1 loss-of-function alleles. Both mutants have decreased synchronous release, enhanced asynchronous fusion and elevated rates of spontaneous release [37, 41, 208,209,210]. Although SYT1 is the primary candidate for a fusion clamp to prevent spontaneous release in mammals, that role is mediated more prominently by CPX at invertebrate synapses. Syt1 null mutants and animals with mutated C2A/C2B Ca2+ binding sites display a two–tenfold increase in mini frequency compared to controls [37, 49, 51], while Cpx null mutants have a far greater increase that can exceed 100-fold [36, 37, 41, 95, 208,209,210,211,212,213]. As such, both proteins negatively regulate spontaneous release by clamping fusion, but the balance of their effects have shifted during evolution. SYT1 and CPX also display genetic interactions in double mutants or following co-overexpression, suggesting CPX is likely to exert many of its effects by regulating the timing and activity of SYT1 during SV priming and fusion in Drosophila [37]. An attractive hypothesis involves CPX binding to the partially assembled SNARE complex to regulate when and how SYT1 interacts, allowing additional spatial and temporal control of SYT1-SNARE interactions during priming and fusion.
Another question remaining in the field is how many SNARE complexes and SYT proteins are required for fusion in vivo. Most estimates suggest 3–5 SNARE complexes drive evoked fusion, while the assembly of only a single SNARE complex might be sufficient to trigger spontaneous release [214,215,216,217]. Increasing the number of assembling SNARE complexes has been shown to increase release probability for SV fusion in vivo [31]. In terms of the number of SYTs required, estimates suggest ~ 15 SYT proteins are found on a single SV [218]. Do SYTs have to bind every SNARE complex at the fusion site or is binding to only one sufficient? Beyond the number of SYTs that bind SNAREs, how many SYTs are required overall to trigger an evoked fusion event and inhibit asynchronous and spontaneous release? If more than one SYT is required, do they need to form an oligomeric complex, or can they act independently? Do the number of SYT proteins activated during Ca2+ entry contribute to the steep Ca2+ cooperativity curve for SV fusion, or is that effect mediated through other mechanisms? Do individual SYTs bind SNAREs, while others drive Ca2+-dependent membrane insertion? What actual changes in the lipid structure of the presynaptic membrane occur following penetration of the SYT1 C2 domains that activates fusion? How specifically do SYT1 proteins prevent asynchronous and spontaneous release; are these shared or distinct mechanisms from its positive role in activating synchronous release? Defining the timing and precise role for each of SYT1’s interactions in the various routes of SV trafficking and fusion will require future studies with additional temporal and spatial resolution.
Additional questions in the field center on the role of oligomerization in SYT1 function. Although SYT1 forms oligomers in vitro [49,50,51, 219,220,221,222,223,224], the importance, stability, and timing of SYT1 oligomerization for SV release in vivo is unclear. Distinct SYT1 oligomerization states may also exist, as multimerization can occur via Ca2+-dependent and Ca2+-independent mechanisms and through interactions mediated by the linker domain, residues on the C2A surface or via the C2B polybasic region [49, 82, 177, 219, 221,222,223,224,225,226,227]. In the intragenic suppressor screen described above, an oligomerization-defective C2A mutant (R250H) was identified [62]. This mutant reduced evoked release but did cause disruptions to the synchronicity of fusion (Fig. 4e). As such, the R250H mutant suggests a potential role for oligomerization in enhancing the number of SVs that fuse, with release timing and suppression of asynchronous fusion independent of oligomerization. Although oligomerization is defective in R250H, this mutation also disrupts C2A–C2B intradomain interactions within individual SYT1 monomers, suggesting either interaction could contribute to defects observed in this mutant. It is also possible that other oligomerization states function independently of the C2A R250 residue to drive SYT1 activity. Defining the role of SYT1 multimers in SV trafficking requires future studies in the field.
Is Synaptotagmin 7 the Ca2+ sensor for asynchronous release?
The discovery that SYT1 mediates synchronous release but does not abolish asynchronous fusion raised the possibility that other members of the SYT family might function as the asynchronous Ca2+ sensor. SYT7 emerged as a promising candidate due to its higher affinity for Ca2+, strong lipid binding properties, slower dissociation from membranes, and widespread expression within the nervous system [228,229,230,231,232,233]. These properties could potentially allow SYT7 to activate SV fusion farther away from AZs and after the initial rise in Ca2+ influx (Fig. 1c). This would provide an attractive solution to the Ca2+ sensing problem for SV fusion: SYT1 functioning as the synchronous sensor and its homolog SYT7 acting as the asynchronous sensor. In contrast to the work on SYT1 in Drosophila, which generated many of the initial insights into in vivo mechanisms, studies of SYT7 have been primarily done in mice. As such, we discuss these data sets in more detail and compare them to recent studies on the Drosophila SYT7 homolog.
Drosophila SYT7 was not identified until the genome was sequenced in 2000 [99, 234, 235]. Syt7 resides on the small 4th chromosome in a genomic region that had been poorly characterized in the early 2000s and that lacked genetic toolkits to easily generate mutations. As such, the protein remained uncharacterized for many years. The first efforts to examine Drosophila SYT7 function used RNAi knockdown, RNA in situ hybridization and immunocytochemical studies with antibodies raised against the protein [100, 153]. These approaches demonstrated Syt7 mRNA was abundantly expressed in most neurons at levels similar to Syt1 and Syt4. Recent RNA profiling also indicates Syt7 mRNA is abundant in neurons (Fig. 3). Initial RNAi approaches that attempted to reduce the levels of SYT7 failed to show a synaptic phenotype [153]. SYT7 did not co-localize with SYT1 on SVs or enrich on synaptic plasma membranes with the t-SNARE Syntaxin on brain extracts fractionated from sucrose gradients (Fig. 5a) [100]. Similarly, CRISPR tagging of endogenous SYT7 with GFP or RFP showed the protein was present in presynaptic terminals but not localized to SVs [82]. Unlike an early study of mammalian SYT7 which identified the protein at the AZ plasma membrane [232], no enrichment of SYT7 at Drosophila AZs was observed. Subsequent work in mammals have reported SYT7 localization in multiple compartments, including the plasma membrane, DCVs, lysosomes, endosomes and other internal membrane compartments [100, 232, 236,237,238,239,240,241,242,243]. These diverse findings have made it unclear as to which membrane compartment contains SYT7, or if the protein is present in multiple locations that could participate in trafficking of distinct organelles.

Enhanced release and SV replenishment in Syt7 mutants. a Western analysis of Drosophila brain extracts separated on a 10–30% sucrose gradient. SYX1 labels the plasma membrane (left-most fraction) with SVs (n-SYB/SYT1) in intermediate fractions. SYT7 and SYT4 fractionate with other internal membrane compartments to distinct regions of the gradient. b Evoked release is increased in Syt7 null mutants (Syt7M1) and Syt7 heterozygotes. c Optical mapping of release probability at larval NMJs demonstrate Syt7 mutant AZs are shifted to higher Pr compared to controls. d Syt7 mutants undergo rapid depression during a stimulation train and recover their releasable SV pool more quickly than controls. Heterozygotes show an intermediate phenotype. Panel a modified from [100] and panels b–d modified from [82]
Further characterization of CRISPR-tagged SYT7 in Drosophila found the protein was expressed in a highly interconnected tubular membrane compartment in presynaptic boutons that made partial contact with numerous organelles, including endosomes, lysosomes and the plasma membrane [82]. Although SYT7 did not fully co-localize with any known compartmental marker, the protein was enriched within the peri-AZ (Fig. 3d, e), a synaptic domain surrounding AZs that contain proteins involved in endocytosis and protein sorting [82, 244,245,246,247,248,249,250]. Recent characterization of SYT7 localization using STORM in mouse hippocampal neurons also found the protein at the periphery of AZs [251], though it is unclear if this represents localization to the plasma membrane versus an internal SYT7 compartment. If SYT7 functions as the asynchronous Ca2+ sensor that mediates SV fusion with the plasma membrane, it should reside on SVs or at the AZ presynaptic membrane. SYT7 does not appear to be enriched on either of these compartments in Drosophila. Given the primary localization of SYT7 within the peri-AZ, functions beyond directly mediating fusion of SVs with the presynaptic plasma membrane must be considered.
Initial studies using Drosophila SYT7 RNAi knockdown and mouse Syt7 mutants did not suggest a role for SYT7 in asynchronous release [153, 252]. The first observation indicating SYT7 might function in this slower release pathway was based on morpholino knockdowns of zebrafish SYT7 that eliminated asynchronous release at NMJs [78]. This finding triggered a wave of new studies to re-evaluate SYT7’s role. Additional characterization of mammalian SYT7 using siRNA knockdown suggested the protein was indeed responsible for asynchronous release in excitatory and inhibitory neurons [84]. These phenotypes were found by knocking down SYT7 in SYT1 deficient neurons with enhanced asynchronous release, or in wildtype neurons during high frequency stimulation, making it easier to detect changes in the asynchronous pathway. Subsequent work in mouse Syt7 mutants supported a role for SYT7 as the asynchronous Ca2+ sensor [80, 83, 253,254,255,256]. Beyond asynchronous release, some studies found manipulations of SYT7 also disrupted facilitation, a form of short-term plasticity where SV release is enhanced during closely spaced stimuli [80, 81, 257]. These data supported a model that SYT7 acts as the Ca2+ sensor for both asynchronous release and facilitation in a subset of mammalian neurons.
Although much of the literature converged on the two Ca2+ sensor model for SV fusion, the role of SYT7 in asynchronous release remains controversial. Asynchronous release at several mammalian CNS synapses is anti-correlated with the levels of the synchronous Ca2+ sensors SYT1 and SYT2 and does not correlate with SYT7 levels [58]. In addition, another study found no defects in asynchronous release in mouse Syt7 mutants [76]. Mammalian SYT7 has also been suggested to play distinct roles in the SV cycle, including driving SV replenishment during high frequency stimulation [76], refilling of the readily releasable pool (RRP) [258], and targeting of SVs to specific endocytic pathways [77]. Such defects could potentially contribute to decreases in asynchronous fusion due to fewer SVs available for this phase of release. In addition to these roles, mammalian SYT7 regulates fusion of lysosomes with the plasma membrane in non-neuronal cells and DCV release in certain neuronal populations [83, 133, 134, 237, 240,241,242,243, 256, 259,260,261,262,263].
The discrepancies in mammalian studies make it difficult to determine if SYT7 has a conserved primary role at synapses, or if the protein has distinct functions in subpopulations of neurons. To examine conserved roles for SYT7 in Drosophila, CRISPR mutants in the locus were recently generated [82]. Consistent with mice lacking SYT7, Drosophila lacking the protein are viable and fertile, indicating SYT7 is not required for synaptic transmission or non-neuronal fusion events essential for survival. However, Syt7 mutants displayed numerous defects at NMJ synapses in 3rd instar larvae, including a large increase in evoked SV release, a larger RRP vesicle pool, and faster SV replenishment after strong stimulation (Fig. 5). Consistent with increased release, mutant terminals displayed depression during stimulation trains compared to controls that facilitated in the same extracellular [Ca2+]. Lowering extracellular Ca2+ to reduce release probability in Syt7 mutants restored facilitation, indicating the process itself is intact at synapses without SYT7. These release defects were dosage-sensitive, with Syt7/+ heterozygotes showing intermediate increases in SV fusion (Fig. 5b) and SYT7 overexpression suppressing evoked and spontaneous release. Similar dosage effects appear to be present at some mammalian synapses [264]. Optical imaging of evoked release in Drosophila Syt7 mutants demonstrated a higher release probability at single AZs compared to controls (Fig. 5c) [82]. The enhancement in evoked release resulted in less asynchronous fusion and facilitation compared to controls, as observed in mammals. However, increased release probability leads to faster SV depletion and reduces the number of SVs available for fusion via these pathways. These data suggest a model where the levels of SYT7 act as a dial to decrease or increase the amount of release to enable or mask facilitation mediated by an independent Ca2+ sensor, suggesting modulation of Syt7 transcription or SYT7 protein levels could play a central role in short-term plasticity.
To conclusively test if asynchronous release and facilitation requires SYT7, electrophysiological recordings were performed in Syt1, Syt7 double mutants [82]. In animals lacking both proteins, an enhancement of both evoked and spontaneous release compared to Syt1 null mutants alone was observed. In addition, double mutants still displayed synaptic facilitation. These results indicate SYT7 negatively regulates release probability for SVs destined for release through both the spontaneous and evoked pathways in the absence or presence of SYT1. Since evoked release remains in animals lacking both SYT isoforms, the popular two Ca2+ sensor model of SYT1 acting as the synchronous sensor and SYT7 as the sole asynchronous sensor is not applicable for Drosophila synapses. Similarly, the lack of SYT7 accumulation on SVs or at the AZ plasma membrane make it unlikely Ca2+ binding to SYT7 could directly trigger SV fusion since the protein is not physically present at release sites. Given similarity in release kinetics and Ca2+ sensitivity for asynchronous fusion between Drosophila and mammalian synapses [57, 75], it is perplexing that SYT7 could take over this role at mammalian synapses while a distinct Ca2+ sensor mediates asynchronous release in Drosophila. An alternative model is that SYT7 controls the number of SVs available for asynchronous release, but a distinct Ca2+-sensitive mechanism directly mediates fusion through this slower pathway.
Although more work is required to understand how SYT7 negatively regulates SV trafficking in Drosophila, some similarities and differences between invertebrate and mammalian systems can be noted. Lack of SYT7 results in defects in asynchronous release and facilitation, though the hypothesized mechanisms are very different (Ca2+ sensor for asynchronous fusion in mammals, negative regulator of release probability in Drosophila). To date, increases in SV release probability have not been reported in mammals. Both proteins also negatively regulate spontaneous release, as mini frequency is reduced when SYT7 is overexpressed in either mice or Drosophila [82, 84]. Similarly, spontaneous frequency is elevated in Syt1/Syt7 double mutants in both species [82, 84, 257]. This finding has been interpreted as SYT1 and SYT7 having redundant roles in clamping spontaneous fusion in mammals. In contrast, the Drosophila model argues more SVs are available for release that have to be clamped by SYT1 and/or CPX. SYT7 also regulates the size and recovery of SV pools after stimulation in Drosophila, as Syt7 mutants have a larger pool of releasable SVs and a faster refilling rate after stimulation (Fig. 5d). The enhanced replenishment of the RRP is still observed in the absence of endocytosis, suggesting it involves changes in movement between existing SV pools [82]. SYT7 overexpression causes the opposite phenotype, reducing the pool of fusogenic SVs and delaying replenishment. Several mammalian studies have identified a role for SYT7 in SV replenishment [76, 79, 191, 258], but the protein promotes refilling in contrast to Drosophila SYT7. One study identified a redundant role for mammalian SYT1 and SYT7 in maintaining the RRP [191], while SYT1 alone acts to positively regulate the RRP in Drosophila [57]. A few studies suggest STY7 controls SV endocytosis in mammals [77, 265], but steady-state endocytosis rates are not altered in Drosophila Syt7 mutants. Rather, SYT7 regulates how many and how rapidly SVs move between distinct pre-existing pools at Drosophila synapses. Although SYT7 is highly conserved at the sequence level across evolution, its function appears to have diverged more than SYT1. Given conflicting reports on mammalian SYT7 function, some of species differences may also reflect differential expression levels between SYT1 and SYT7 across diverse neuronal populations [58].
Although trafficking defects have now been characterized in Drosophila Syt7 mutants, how the protein negatively regulates SV release and replenishment from its peri-AZ location is unknown. EM analysis of Syt7 mutant NMJs did not reveal increased SV number or differences in SV distribution around AZs [82], suggesting the total SV pool is unaltered. These findings indicate loss of SYT7 alters the distribution and movement between existing SV pools that give rise to these phenotypes. Since SYT7 is not found on SVs or at AZs, it likely regulates SV release through an indirect mechanism. SYT7′s location within the peri-AZ positions it to interact with multiple endosomal and recycling compartments that could negatively impact SV trafficking. One model is that SYT7 hetero-oligomerizes with SYT1 to reduce fusogenicity of a SV pool or restrict SVs from entering the releasable pool. Although hetero-oligomerization between distinct SYT family members occurs in vitro [177, 225, 226, 266], the in vivo significance is unclear since most SYT proteins reside in distinct subcellular compartments. Given loss of SYT7 enhances release in the absence of SYT1, this mechanism seems unlikely to be the dominant pathway for SYT7 activity. Another model is that Ca2+ binding to SYT7 acts as a sponge to reduce fusion by limiting intracellular Ca2+ availability for SYT1 given the steep Ca2+ dependence of SV release. SYT7 might also regulate Ca2+ influx or extrusion pathways. The lack of SYT7 enrichment at fusion sites and the preservation of enhanced release in Syt1/Syt7 double mutants argues against these mechanisms. In addition, quantitative imaging of presynaptic Ca2+ levels with Fluo-4 AM at Syt7 mutant NMJs revealed a mild reduction in evoked Ca2+ influx rather than an elevation as predicted by this model [82]. Another potential mechanism would involve SVs being more efficiently recruited from the reserve pool (RP) in the absence of SYT7. The SV protein Synapsin helps maintain the RP by tethering SVs to actin filaments [267,268,269,270,271,272,273,274,275,276,277,278,279] and is known to interact with other peri-AZ proteins to regulate SV cycling [244, 280]. As such, SYT7 might modulate a Synapsin-dependent process that controls SV availability. Characterization of Synapsin/Syt7 double mutants could determine if SYT7 normally limits liberation of SVs from the RP.
SYT7 might also regulate SV distribution by directing sorting of newly generated SVs in a Ca2+-dependent manner. Studies of Cpx mutants, which have a dramatic increase in mini frequency, revealed segregation between SV recycling pathways following spontaneous versus evoked release [213]. Ca2+-independent recycling following spontaneous fusion resulted in a rapid re-accumulation of recycled SVs at AZs in the RRP. In contrast, SVs recycled following evoked release when cytosolic Ca2+ is elevated were slower to re-enter the AZ pool. SYT7 might act as the Ca2+-triggered recycling switch to redirect SVs to an internal pool and delay their return to the AZ. A final more speculative hypothesis for SYT7 function is that it acts similarly to more distantly related proteins like the ESYT family found at ER-plasma membrane contact sites that mediate lipid exchange between distinct membrane compartments [111, 281,282,283,284,285]. If so, SYT7 might function by bringing two presynaptic membrane compartments together in a Ca2+-dependent manner to regulate lipid exchange by phospholipid transporters. SYT7 binds to PIP2 with high affinity [231], and limiting plasma membrane PIP2 levels would be an attractive mechanism given this lipid enhances SV release through interactions with both UNC13 and SYT1 [197, 286,287,288,289,290,291].
Unlike structure–function analysis of Drosophila SYT1, little is known about SYT7’s required molecular interactions. However, it is unlikely the C2 domains of SYT1 and SYT7 regulate SV trafficking through similar mechanisms. In contrast to SYT1, Ca2+ binding to the SYT7 C2A domain appears to play the major role in regulating SV release in mammals [81, 84, 231]. The C2 domains of SYT1 and SYT7 are not interchangeable in vivo as SYT1/SYT7 chimeras cannot rescue Syt1 mutant phenotypes [207]. One difference between isoforms is the HB helix in the C2B domain that negatively regulates SV fusion in SYT1 is not present in Drosophila or mammalian SYT7 proteins [82, 207]. Another difference maps to the primary SNARE-binding surface on the SYT1 C2B domain. Four of the five essential residues required for this interaction in SYT1 are not conserved in SYT7 [62, 82]. Indeed, SYT7 has amino acid substitutions at two of these residues that precisely match previously isolated Syt1 mutants that block SNARE binding and abolish SYT1 function in SV release [62]. These observations indicate SYT7 does not interact with the SNARE complex or does so through a distinct mechanism from SYT1. Beyond these changes, numerous other nonsynonymous amino acids within the C2 domains are conserved only in SYT1 or SYT7 family members, suggesting extensive divergence during evolution from the last common SYT ancestor. Together, these data indicate SYT1 and SYT7 regulate SV trafficking through distinct mechanisms and from separate membrane compartments. More studies are required to determine mechanistically how SYT7 contributes to SV cycling in both Drosophila and mammals, as well as to determine why asynchronous release defects are found in Syt7 mutants.
Synaptotagmin 4 regulates retrograde signaling and is transferred between synaptic compartments by exosomes
The mammalian SYT4 protein was identified in searches for novel SYT genes prior to the advent of genome sequencing [292, 293]. Early studies found SYT4 expressed throughout the mouse brain, but revealed the protein lacked Ca2+-dependent lipid binding in vitro. Another family member, SYT11, was also identified that failed to bind Ca2+ [294]. The failure to bind Ca2+ mapped to an aspartate to serine substitution in a key Ca2+ binding aspartate residue (D3) in the C2A domain of SYT4 and SYT11. The mammalian Syt4 gene was independently identified as an activity-regulated intermediate early gene induced by seizure activity [295]. These two initial observations led to a model that activity-dependent upregulation of a Ca2+-insensitive SYT isoform might have a neuroprotective role [296]. By oligomerizing with SYT1 and reducing the ability of the heterooligomer to bind Ca2+, SYT4 was hypothesized to reduce SV release following excessive neuronal hyperactivity. This model assumed SYT4 was a resident SV protein like SYT1.
Drosophila encodes a single SYT4 family member and initial studies suggested SYT4 overexpression could reduce SV release [177], consistent with mammalian models for its function. Another early study hypothesized a different role where SYT4 could substitute for SYT1 function in SV release in Drosophila [297]. Both of these hypotheses turned out to be wrong when subsequent work found SYT4 was not a SV protein [100]. Antibodies specific to SYT4 revealed a prominent localization of the protein in puncta within the postsynaptic compartment of muscles at the Drosophila NMJ (Fig. 3c) [97, 100, 104]. This localization suggested SYT4 might instead regulate membrane trafficking within postsynaptic compartments. Drosophila SYT4 was also found to bind Ca2+ unlike its mammalian orthologs [97, 104, 298]. Ca2+-regulated membrane trafficking is known to occur within postsynaptic compartments and can use similar vesicular trafficking components to those found in presynaptic terminals [100, 144, 299,300,301,302]. In Drosophila, retrograde signaling mediated through the release of multiple secreted factors is essential for development and plasticity of the NMJ where it links neuronal activity and synaptic growth regulation [14, 22, 97, 105, 303,304,305,306,307,308]. To determine if SYT4 regulates postsynaptic vesicular trafficking similar to the role of SYT1 in presynaptic terminals, Syt4 null mutants were generated in Drosophila [104]. Animals lacking SYT4 were viable and fertile, indicating the protein was not essential for viability. However, electrophysiological and imaging studies supported a role for SYT4 as a postsynaptic Ca2+ sensor for retrograde signaling at NMJs [95, 97, 104, 105, 305].
Several synaptic phenotypes were identified in Syt4 mutants. Drosophila NMJs display a unique form of presynaptic plasticity following high frequency stimulation. Within a minute after strong stimulation of the motor nerve, a robust increase in the frequency of spontaneous release is observed (Fig. 6a) [95, 97, 104, 106, 309]. This enhanced mini rate lasts for several minutes and requires postsynaptic Ca2+ influx to trigger release of a retrograde signal from the muscle that increases cAMP levels in presynaptic boutons [104]. The elevation in spontaneous release requires presynaptic PKA phosphorylation of CPX to reduce its clamping function [95]. This activity-dependent increase in spontaneous release is eliminated in Syt4 mutants (Fig. 6b) [104], suggesting SYT4 is either required to directly mediate release of an unknown retrograde signal or to enable the process through other mechanisms. In addition to the acute effects on presynaptic release, postsynaptic Ca2+ influx also drives SYT4-dependent retrograde signaling that promotes synaptic differentiation and synaptic growth [97, 104, 105]. Embryos lacking SYT4 have delayed synapse formation and mutant 3rd instar larvae have fewer synaptic boutons [97, 104]. Syt4 mutants also disrupt activity-dependent formation of “ghost” boutons that represent nascent presynaptic outgrowths that lack postsynaptic specializations that rapidly form in response to exceptionally strong presynaptic stimulation [106]. SYT4 overexpression in postsynaptic muscles could also induce overgrowth of presynaptic terminals [97]. Together, these data suggested Ca2+-dependent retrograde vesicular trafficking supported by SYT4 induced acute changes in synaptic output and triggered synapse-specific growth, providing a link between short-term synaptic plasticity and activity-dependent synaptic rewiring.

SYT4 controls retrograde signaling at Drosophila NMJs. a Drosophila NMJs display a robust form of activity-dependent presynaptic plasticity mediated by increased spontaneous release after high frequency stimulation (HFS). Optical imaging of spontaneous SV releases rates are shown before and after stimulation. Note the different y-axis scale on the post-stimulation map. b The enhanced spontaneous release observed at control NMJs after stimulation is abolished in Syt4 mutants. Arrows denote the timing of HFS to the motor axon. c Screen for muscle RNAi knockdown of loci that disrupt postsynaptic membrane expression of a SYT4-pHlourin (SYT4-pH) construct. Control SYT4-pH localization to the postsynaptic membrane is shown on the left. Syx4‐RNAi reduces membrane SYT4-pH and causes a redistribution of the protein to cytoplasmic puncta within the muscle (right). The motoneuron is stained with anti-HRP (red). a Modified from [95], b modified from [104], and c modified from [105]
The initial studies of SYT4 function assumed Syt4 mRNA was transcribed and the protein produced in postsynaptic muscles. A major surprise in the field was the discovery that Syt4 is only transcribed in presynaptic motoneurons and that the SYT4 protein is transmitted to muscles through the release of presynaptic exosomes [106]. Indeed, endogenously CRISPR-tagged SYT4-GFP expression is eliminated from the postsynaptic compartment following RNAi knockdown of the Syt4 gene presynaptically but not postsynaptically. SYT4 is one of the several proteins that undergo trans-synaptic delivery in extracellular vesicles through a pathway regulated by presynaptic Rab11 [107, 310, 311]. This finding parallels trans-synaptic delivery of Drosophila and mammalian ARC proteins that regulate synaptic plasticity [312,313,314,315]. Such a mechanism could allow presynaptic activity to regulate the levels of local retrograde signaling by controlling delivery of essential postsynaptic components required for the process. However, the observation generated a host of new questions on how SYT4 is loaded into exosomes and delivered. In addition, how the protein is unpackaged in the postsynaptic cell and what membrane compartment it ultimately occupies are unknown. Although the original model envisioned SYT4-dependent fusion of a vesicular compartment with the postsynaptic membrane that released retrograde signals, SYT4’s presence in exosomes provided other candidate mechanisms for how it might contribute to retrograde signaling.
To identify components of the SYT4 retrograde signaling pathway that function in the postsynaptic compartment, rather than in presynaptic exosome delivery, a UAS-SYT4-pHlourin (SYT4-pH) construct was generated and expressed directly in postsynaptic cells with a muscle GAL4 driver to visualize its trafficking to the plasma membrane [105]. A targeted screen for muscle genes that disrupted postsynaptic surface expression of SYT4-pH using RNAi knockdown was then performed. This approach identified multiple candidates that altered SYT4 membrane delivery, including the postsynaptic t-SNARE SYX4 (Fig. 6c) [105]. Generation of Syx4 mutants in Drosophila showed the protein was required for delivery of both SYT4 and the well-known postsynaptic transmembrane protein Neuroligin1 (NLG1) to the postsynaptic membrane. Syx4 mutants displayed reduced synaptic growth and a loss of activity-induced enhancement of spontaneous release [105], similar to Syt4 mutants [95, 97, 104, 305]. These findings suggested NLG1 might function within the SYT4 signaling pathway, with alterations in postsynaptic membrane content rather than release of diffusible retrograde signals mediating trans-synaptic communication. Beyond SYX4, several interactors that disrupted postsynaptic surface expression of SYT4-pH were known regulators of endocytosis. Given this data, one hypothesis is SYT4′s role in retrograde signaling is not to trigger release of a diffusible retrograde signal, but rather to endocytose or deliver a postsynaptic membrane protein in a Ca2+-regulated manner that initiates a presynaptic response. Similar to SYT1, SYT4 interacts with known regulators of endocytosis like AP-2 [98, 177, 292]. Although these studies found SYX4 and SYT4 act in a common pathway, SYX4 also supported other forms of retrograde signaling that negatively regulated neurotransmitter release by decreasing Ca2+ channel abundance and Ca2+ release cooperativity at presynaptic AZs [316]. These phenotypes were not observed in Syt4 mutants, indicating the t-SNARE SYX4 supports additional postsynaptic trafficking steps independent of SYT4 in Drosophila.
SYX4 is the sole Drosophila homolog of the mammalian SYX3 and SYX4 postsynaptic t-SNARE subfamily [105, 235, 316]. Postsynaptic vesicle trafficking is also regulated by Ca2+ in mammals, with postsynaptic SNAREs and SYTs controlling long-term potentiation (LTP) and activity-dependent AMPA receptor trafficking [97, 104, 105, 143,144,145, 148, 171, 299,300,301,302, 305, 317,318,319,320,321,322,323,324,325,326]. Mammalian homologs of SYT4 and SYX3/SYX4 have been implicated in multiple postsynaptic membrane-trafficking steps in dendrites, including regulating exocytosis of the neurotrophin BDNF [143,144,145, 148, 171, 319, 321,322,323, 325, 327, 328]. These data suggest SYT4, along with SYX3 and SYX4, may represent a conserved pathway that contributes to membrane trafficking and retrograde signaling within postsynaptic compartments. Beyond retrograde trafficking, mammalian SYT4 and SYT11 have been implicated in other trafficking steps that have not been characterized in Drosophila, including DCV maturation and release, DCV fusion pore dynamics, endosomal trafficking and endocytosis [134, 138, 139, 143, 171, 239, 323, 326, 329].
Going forward, many open questions remain for how SYT4 regulates trafficking of membrane organelles at synapses. One major question surrounds the role of Ca2+ binding. Unlike the lack of Ca2+ binding by mammalian SYT4 and SYT11, Drosophila SYT4 binds Ca2+ and its C2 domain Ca2+ binding properties are essential for retrograde signaling [97, 104, 298]. These differences are quite perplexing, as Drosophila SYT4 contains the conserved aspartate to serine substitution in the key D3 residue that defines this family, yet is still able to bind Ca2+. Mutation of this residue back to an aspartate in the mammalian homologs restore Ca2+ binding, indicating the on/off nature of this interaction is tied specifically to this residue [294]. In addition, all the remaining Ca2+ binding aspartate residues in C2A and C2B are conserved across evolution in the SYT4 family, unlike the higher divergence seen in other SYT family members that act through Ca2+ independent mechanisms. Finally, membrane organelles containing mammalian SYT4 or SYT11 can undergo Ca2+-regulated fusion [171, 323]. These findings suggest a potential in vivo ability of mammalian SYT4 and SYT11 to bind Ca2+ that is not captured in in vitro studies. One possibility is that the serine residue in the Ca2+ binding pocket mediates lower affinity Ca2+ interactions that are difficult to detect in vitro. Another option is the serine residue is a site of phosphorylation that would reintroduce stronger negative charge in the Ca2+ binding pocket, allowing SYT4 to function in a phosphorylation-controlled manner in vivo. Mutagenesis of this serine in Drosophila has demonstrated the residue is critical for SYT4 function in vivo [97]. The specific conservation of serine at this site in the SYT4 family across ~ 800 million years of evolution suggests it has functional significance, but more work is required to define its role in supporting Ca2+ binding in vivo.
Additional questions on SYT4 function revolve around the discovery that the protein is found on exosomes in Drosophila. There are no current studies indicating the mammalian homologs are present on exosomes. SYT4 positive compartments co-localize with DCV markers and BDNF in mammals [143, 171, 330], while BDNF has been found presynaptically and in exosomes from brain extracts [331,332,333]. It is currently unclear if SYT4 will emerge as a common component of exosomes similar to ARC, or if this delivery mechanism is unique to Drosophila. There is also much to be done on how SYT4 is loaded and unloaded from exosomes. Given the requirement for SYT4 C2 domain interactions, it seems likely these domains would orient towards the cytoplasm within the postsynaptic compartment to bind Ca2+ and regulate trafficking. How SYT4-containing exosomes transit the synaptic cleft and are taken up by the postsynaptic cell is unknown. Once inside the muscle, how do SYT4 proteins transit to a specific postsynaptic compartment to regulate synaptic plasticity? What potential retrograde signaling molecules are present in these compartments? Alternatively, does SYT4 act primarily at the postsynaptic membrane to control endocytosis of trans-synaptic signaling components? Future work on this family should provide important insights into similarities between Ca2+-regulated trafficking steps in pre- versus post-synaptic compartments.
Role of the remaining SYT isoforms
Beyond the abundant SYT family members (SYT1, SYT4, SYT7), four additional Syt genes are expressed at lower levels or in specific neuronal subpopulations in Drosophila [99, 100]. Syt-α and Syt-β contain highly conserved C2 domain Ca2+ binding residues that indicate a role as Ca2+ sensors. Antisera raised against the two proteins demonstrate they are expressed in multiple neuropeptide releasing neurons in both the CNS and PNS [100]. Syt-α and Syt-β expression are also controlled by a master regulator of neuroendocrine neurons, the Dimmed transcription factor [108,109,110, 334,335,336]. These data are consistent with Syt-α and Syt-β residing on DCVs to mediate Ca2+-dependent release of neuropeptides, though genetic analysis will be required to define their function. It is unclear if the more abundant SYT isoforms (SYT1, SYT4, SYT7) also serve as Ca2+-sensors for DCV fusion in Drosophila, given their established role for this step in many mammalian neurons. With the limited expression of Syt-α and Syt-β in specific neuronal subpopulations, and the presence of DCVs in most Drosophila neurons, it is likely the more abundant SYT isoforms also participate in DCV trafficking and fusion.
The remaining two SYT isoforms, SYT12 and SYT14 lack conserved Ca2+ binding residues and are unlikely to function as Ca2+ sensors. RNA profiling and in situ analysis indicate these genes are expressed at very low levels (Fig. 3b). Syt12 mRNA was not detected using in situ to Drosophila embryos, while RNA profiling showed low expression in the larval and adult brain. One study of mammalian SYT12 found it resides on SVs and acts as a PKA substrate to control spontaneous SV release by multimerizing with SYT1 and negatively regulating its function [337]. Knockout Syt12 mice are viable and show no changes in baseline synaptic transmission [338]. A disruption of presynaptic cAMP-dependent mossy-fiber LTP in the hippocampal CA3 region was the only defect found in Syt12 mutants, suggesting a link to PKA regulation of release in this brain area. It is unlikely Drosophila SYT12 is a ubiquitous component of SVs given the low abundance, but it may regulate membrane trafficking in a small population of CNS neurons. The Drosophila Syt14 gene is present at low levels throughout the embryonic CNS based on in situ analysis [100]. RNA profiling found low to moderate expression of Syt14 in the CNS of larvae and adults as well, with weaker expression in salivary glands and testis. The only link to SYT14 function comes from a single report suggesting Syt14 mutations may cause autosomal recessive spinocerebellar ataxia in humans [339]. Further work will be required to define the expression patterns and subcellular locations for these remaining SYT proteins and determine if and how they participate in membrane trafficking in Drosophila neurons.
Relevance of Synaptotagmin dysfunction to human disease
Specific mutations in several SYT family members have been identified in human neurological disorders that affect both the CNS and PNS. In addition, several SYTs are linked more loosely to pathogenesis in neurological disorders arising from other primary causes. The first mutations identified in human SYTs were found in the SYT2 C2B domain in patients presenting with non-progressive peripheral motor neuropathies with unusual similarities to Lambert–Eaton myasthenic syndrome (LEMS) [189, 190, 340, 341]. LEMS is an autoimmune disorder commonly linked to auto-antibodies against the presynaptic Ca2+ channel that disrupt NMJ function [342]. SYT2 mutations disrupt motor function and cause peripheral muscle weakness due to defective acetylcholine release at NMJs, but have no obvious effect on cognition. The phenotypes are consistent with data from mice indicating SYT2 is abundantly expressed in the PNS, with SYT1 mediating SV fusion for the majority of CNS neurons [101]. This group of human SYT2 mutations are dominantly inherited and run through family lineages in patient populations, with an affected individual harboring one normal copy and one mutated version of SYT2. All mutations identified to date cluster in specific residues lining the C2B Ca2+ binding pocket, suggesting they are likely to dominantly disrupt C2B domain interactions with membrane lipids. Loss of a single copy of the orthologous SYT1 protein does not dramatically alter neurotransmitter release in Drosophila or mouse models, suggesting the phenotype is not due to haplo-insufficiency but rather acts by poisoning the fusion machinery [49, 62, 75, 156, 189, 340]. Indeed, homozygous mutations in human SYT2 resulting in complete biallelic loss of the protein have been identified in five families that result in a more severe congenital myasthenic syndrome than in autosomal dominant patients [343]. In this study, heterozygous carries of SYT2 null alleles had no obvious phenotypes, confirming dominant mutations in SYT2 patients are not caused by a simple loss of function haplo-insufficiency model. The less severe phenotypes in patients harboring SYT2 dominant mutations indicate SYT2 function is not abolished in these patients. In addition, SYT1 may play a partially redundant role in acetylcholine release at NMJs, consistent with phenotypes in Syt2 knockout mice [101].
Recently, similar heterozygote dominant mutations in the C2B Ca2+ binding site of human SYT1 have been identified, resulting in a severe neurodevelopmental disorder with infant hypotonia, hyperkinetic movement disorder, profound developmental delay, cognitive dysfunction, and failure to develop language [344, 345]. These mutations are not heritable due to their phenotypic severity and instead arise as spontaneous mutations in the germline. Similar to SYT2 autosomal dominant mutations, these patients maintain residual synaptic transmission, as complete loss of SYT1 function would be lethal. From a clinical perspective, it is essential to define how these dominant-negative C2B mutations disrupt the fusion machinery and to identify potential mechanisms to increase SV release in these backgrounds to bypass the disruption. Future work will be required to determine if the C2B Ca2+-binding pocket, the location of all mutations identified to date, is a privileged site for alleles that can dominantly disrupt SV release.
Drosophila contains a single ortholog of the human SYT1 and SYT2 proteins, and mutations corresponding to several of the dominant-negative disease alleles have been modeled [62, 189, 340, 346]. Given SYT1 mediates all synchronous SV release in Drosophila, the phenotypes of these alleles are expected to be more severe than individual human SYT1 or SYT2 mutants. Indeed, overexpression of several of these alleles, including an aspartate to alanine substitution in the C2B domain D2 Ca2+ binding residue, cause lethality when overexpressed in Drosophila [189]. This mutation did not rescue any Syt1 null phenotypes, indicating the amino acid change completely abolished the ability of the protein to function. How do these mutant SYT1 and SYT2 proteins dominantly interfere with neurotransmitter release? The studies in Drosophila suggest mutant SYT alleles are likely to multimerize with endogenous SYT1 or SYT2 and disrupt the normal function of a SYT oligomer in a dominant fashion. Another model based on studies in mice suggests loss of Ca2+-dependent lipid binding is the primary mechanism [347]. How this property alone would trigger a dominant phenotype, given loss of function of a single copy of SYT1 or SYT2 does not disrupt release, is less clear unless the mutant was also able to disrupt the wildtype SYT1 proteins encoded from the non-affected chromosome. As described above, a genetic screen to find second-site mutations that reduce the dominant disruption of SV fusion caused by C2B Ca2+ binding mutants was performed in Drosophila that identified interactions required for the pathogenic mechanism [62]. This study found these C2B mutants require oligomerization and SNARE binding, as well as Ca2+-independent lipid interactions through the C2B polybasic region, to dominantly poison the endogenous SV fusion machinery. These interactions likely position mutant SYT proteins near release sites where they dominantly disrupt fusion even in the presence of a normal copy of the protein. Two candidate mechanisms for the dominant effect seem most likely. In one model, a defect in the ability of the C2B domain loops to penetrate lipid bilayers upon Ca2+ influx would poison the fusion machinery by oligomerizing with the wildtype copy of SYT1. Another possibility is that the disease alleles result in neutralization of negative charge or increased hydrophobicity of the C2B loops, resulting in spurious activation of Ca2+-independent lipid interactions by the mutant protein. This could bring a SYT oligomer to membranes at inappropriate times, or act to clog release sites as a monomer only that blocks the activity of the normal allele. With genome sequencing becoming more common, it is likely more cases of SYT-mediated neurological disorders will be found given a disruptive amino acid change in the C2B Ca2+-binding pocket in a single copy can cause disease.
Beyond neurological disorders caused by SYT1 and SYT2 mutations, the remaining links to disease are still somewhat speculative and relatively understudied. Disruptions of SYT7 have been suggested as a risk factor for bipolar disorder (BD) in humans, with mutant Syt7 mice displaying manic and depression like behaviors [251, 348]. A study of BD patients showed reduced plasma levels of Syt7 mRNA, and several SNPs in the Syt7 gene have been identified in BD individuals [251, 348]. SYT7 proteins levels are also decreased in some variants of Alzheimer’s disease [349] and a role in lysosomal trafficking and membrane repair is implicated in muscular dystrophy [261, 262]. A few studies have associated SYT11 with schizophrenia and Parkinson’s disease (PD), and SYT11 can accumulate in dopaminergic neurons lacking Parkin [350,351,352,353]. SYT14 mutations have been reported to cause autosomal recessive spinocerebellar ataxia with psychomotor retardation in humans [339, 354], but little else is known about the protein. Dysregulation of expression of several SYTs have also been reported in models of neurodegenerative disorders, including Parkinson’s, Alzheimer’s, and Amyotrophic Lateral Sclerosis (ALS) [355,356,357,358,359,360,361,362,363,364,365,366,367]. Overall, the mechanisms by which other SYT isoforms participate in disease pathogenesis is largely unknown. However, given the broad role of SYT proteins in regulating intracellular membrane trafficking and fusion within neurons, it is likely more links to brain diseases will be identified going forward.
References
Katz BS (1969) The Release of Neural Transmitter Substances (Sherrington Lecture): Bernard S. Katz: 9780853230601: Amazon.com: Books. Liverpool University Press, Liverpool
Sudhof TC (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27:509–547. https://doi.org/10.1146/annurev.neuro.26.041002.131412
Ghelani T, Sigrist SJ (2018) Coupling the structural and functional assembly of synaptic release sites. Front Neuroanat 12:81. https://doi.org/10.3389/fnana.2018.00081
Südhof TC (2012) The presynaptic active zone. Neuron 75:11–25. https://doi.org/10.1016/j.neuron.2012.06.012
Zhai RG, Bellen HJ (2004) The architecture of the active zone in the presynaptic nerve terminal. Physiology 19:262–270. https://doi.org/10.1152/physiol.00014.2004
Emperador-Melero J, Kaeser PS (2020) Assembly of the presynaptic active zone. Curr Opin Neurobiol 63:95–103. https://doi.org/10.1016/j.conb.2020.03.008
Maritzen T, Haucke V (2018) Coupling of exocytosis and endocytosis at the presynaptic active zone. Neurosci Res 127:45–52. https://doi.org/10.1016/j.neures.2017.09.013
Ackermann F, Waites CL, Garner CC (2015) Presynaptic active zones in invertebrates and vertebrates. EMBO Rep 16:923–938. https://doi.org/10.15252/embr.201540434
Biederer T, Kaeser PS, Blanpied TA (2017) Transcellular nanoalignment of synaptic function. Neuron 96:680–696. https://doi.org/10.1016/j.neuron.2017.10.006
Gramlich MW, Klyachko VA (2019) Nanoscale organization of vesicle release at central synapses. Trends Neurosci 42:425–437. https://doi.org/10.1016/j.tins.2019.03.001
Kittel RJ, Hallermann S, Thomsen S, Wichmann C, Sigrist SJ, Heckmann M (2006) Active zone assembly and synaptic release. Biochem Soc Trans 34:939–941. https://doi.org/10.1042/BST0340939
Ehmann N, Owald D, Kittel RJ (2018) Drosophila active zones: from molecules to behaviour. Neurosci Res 127:14–24. https://doi.org/10.1016/j.neures.2017.11.015
Van Vactor D, Sigrist SJ (2017) Presynaptic morphogenesis, active zone organization and structural plasticity in Drosophila. Curr Opin Neurobiol 43:119–129. https://doi.org/10.1016/j.conb.2017.03.003
Harris KP, Littleton JT (2015) Transmission, development, and plasticity of synapses. Genetics 201:345–375. https://doi.org/10.1534/genetics.115.176529
Catterall WA, Few AP (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59:882–901. https://doi.org/10.1016/j.neuron.2008.09.005
Guerrier C, Holcman D (2018) The first 100 nm inside the pre-synaptic terminal where calcium diffusion triggers vesicular release. Front Synaptic Neurosci 10:23. https://doi.org/10.3389/fnsyn.2018.00023
Katz B, Miledi R (1967) The timing of calcium action during neuromuscular transmission. J Physiol 189:535–544. https://doi.org/10.1113/jphysiol.1967.sp008183
Augustine GJ, Charlton MP, Smith SJ (1985) Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J Physiol 367:163–181. https://doi.org/10.1113/jphysiol.1985.sp015819
Sabatini BL, Regehr WG (1996) Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384:170–172. https://doi.org/10.1038/384170a0
Meinrenken CJ, Gerard J, Borst G, Sakmann B (2003) Local routes revisited: the space and time dependence of the Ca2+ signal for phasic transmitter release at the rat calyx of Held. J Physiol 547:665–689. https://doi.org/10.1113/jphysiol.2002.032714
Körber C, Kuner T (2016) Molecular machines regulating the release probability of synaptic vesicles at the active zone. Front Synaptic Neurosci 8:5. https://doi.org/10.3389/fnsyn.2016.00005
Akbergenova Y, Cunningham KL, Zhang YV, Weiss S, Littleton JT (2018) Characterization of developmental and molecular factors underlying release heterogeneity at Drosophila synapses. eLife. https://doi.org/10.7554/eLife.38268
Melom JE, Akbergenova Y, Gavornik JP, Littleton JT (2013) Spontaneous and evoked release are independently regulated at individual active zones. J Neurosci 33:17253–17263. https://doi.org/10.1523/JNEUROSCI.3334-13.2013
Kaeser PS, Regehr WG (2014) Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76:333–363. https://doi.org/10.1146/annurev-physiol-021113-170338
Goda Y, Stevens CF (1994) Two components of transmitter release at a central synapse. Proc Natl Acad Sci USA 91:12942–12946. https://doi.org/10.1073/pnas.91.26.12942
Katz B, Miledi R (1969) Spontaneous and evoked activity of motor nerve endings in calcium Ringer. J Physiol 203:689–706
Kavalali ET (2015) The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16:5–16. https://doi.org/10.1038/nrn3875
Ramirez DMO, Kavalali ET (2011) Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol 21:275–282. https://doi.org/10.1016/j.conb.2011.01.007
Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324. https://doi.org/10.1038/362318a0
Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395:347–353. https://doi.org/10.1038/26412
Acuna C, Guo Q, Burré J, Sharma M, Sun J, Südhof TC (2014) Microsecond dissection of neurotransmitter release: SNARE-complex assembly dictates speed and Ca2+ sensitivity. Neuron 82:1088–1100. https://doi.org/10.1016/j.neuron.2014.04.020
Rizo J, Rosenmund C (2008) Synaptic vesicle fusion. Nat Struct Mol Biol 15:665–674. https://doi.org/10.1038/nsmb.1450
Wickner W, Rizo J (2017) A cascade of multiple proteins and lipids catalyzes membrane fusion. Mol Biol Cell 28:707–711. https://doi.org/10.1091/mbc.E16-07-0517
Südhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477. https://doi.org/10.1126/science.1161748
Ma C, Lijing Su, Seven AB, Yibin Xu, Rizo J (2013) Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339:421–425. https://doi.org/10.1126/science.1230473
Bykhovskaia M, Jagota A, Gonzalez A, Vasin A, Littleton JT (2013) Interaction of the complexin accessory helix with the C-terminus of the SNARE complex: molecular-dynamics model of the fusion clamp. Biophys J 105:679–690. https://doi.org/10.1016/j.bpj.2013.06.018
Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT (2012) Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J Neurosci 32:18234–18245. https://doi.org/10.1523/JNEUROSCI.3212-12.2012
Zhou Q, Zhou P, Wang AL, Dick Wu, Zhao M, Südhof TC, Brunger AT (2017) The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548:420–425. https://doi.org/10.1038/nature23484
Trimbuch T, Rosenmund C (2016) Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci 17:118–125. https://doi.org/10.1038/nrn.2015.16
Brunger AT, Choi UB, Lai Y, Leitz J, White KI, Zhou Q (2019) The pre-synaptic fusion machinery. Curr Opin Struct Biol 54:179–188. https://doi.org/10.1016/j.sbi.2019.03.007
Huntwork S, Littleton JT (2007) A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci 10:1235–1237. https://doi.org/10.1038/nn1980
Malsam J, Bärfuss S, Trimbuch T, Zarebidaki F, Sonnen A-P, Wild K, Scheutzow A et al (2020) Complexin suppresses spontaneous exocytosis by capturing the membrane-proximal regions of VAMP2 and SNAP25. Cell Rep 32:107926. https://doi.org/10.1016/j.celrep.2020.107926
Tang J, Maximov A, Shin O-H, Dai H, Rizo J, Südhof TC (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126:1175–1187. https://doi.org/10.1016/j.cell.2006.08.030
Chapman ER (2008) How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem 77:615–641. https://doi.org/10.1146/annurev.biochem.77.062005.101135
Brunger AT, Leitz J, Zhou Q, Choi UB, Lai Y (2018) Ca2+-triggered synaptic vesicle fusion initiated by release of inhibition. Trends Cell Biol 28:631–645. https://doi.org/10.1016/j.tcb.2018.03.004
Yoshihara M, Adolfsen B, Littleton JT (2003) Is synaptotagmin the calcium sensor? Curr Opin Neurobiol 13:315–323
Schupp M, Malsam J, Ruiter M, Scheutzow A, Wierda KDB, Söllner TH, Sørensen JB (2016) Interactions between SNAP-25 and synaptotagmin-1 are involved in vesicle priming, clamping spontaneous and stimulating evoked neurotransmission. J Neurosci 36:11865–11880. https://doi.org/10.1523/JNEUROSCI.1011-16.2016
Bai H, Xue R, Bao H, Zhang L, Yethiraj A, Cui Q, Chapman ER (2016) Different states of synaptotagmin regulate evoked versus spontaneous release. Nat Commun 7:10971. https://doi.org/10.1038/ncomms10971
Lee J, Guan Z, Akbergenova Y, Littleton JT (2013) Genetic analysis of synaptotagmin C2 domain specificity in regulating spontaneous and evoked neurotransmitter release. J Neurosci 33:187–200. https://doi.org/10.1523/JNEUROSCI.3214-12.2013
Ramakrishnan S, Bera M, Coleman J, Krishnakumar SS, Pincet F, Rothman JE (2019) Synaptotagmin oligomers are necessary and can be sufficient to form a Ca2+-sensitive fusion clamp. FEBS Lett 593:154–162. https://doi.org/10.1002/1873-3468.13317
Littleton JT, Stern M, Perin M, Bellen HJ (1994) Calcium dependence of neurotransmitter release and rate of spontaneous vesicle fusions are altered in Drosophila synaptotagmin mutants. Proc Natl Acad Sci USA 91:10888–10892
Kochubey O, Schneggenburger R (2011) Synaptotagmin increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron 69:736–748. https://doi.org/10.1016/j.neuron.2011.01.013
Vevea JD, Chapman ER (2020) Acute disruption of the synaptic vesicle membrane protein synaptotagmin 1 using knockoff in mouse hippocampal neurons. eLife. https://doi.org/10.7554/eLife.56469
Courtney NA, Bao H, Briguglio JS, Chapman ER (2019) Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin. Nat Commun 10:4076. https://doi.org/10.1038/s41467-019-12015-w
Pang ZP, Sun J, Rizo J, Maximov A, Südhof TC (2006) Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J 25:2039–2050. https://doi.org/10.1038/sj.emboj.7601103
Chang S, Trimbuch T, Rosenmund C (2018) Synaptotagmin-1 drives synchronous Ca2+-triggered fusion by C2B-domain-mediated synaptic-vesicle-membrane attachment. Nat Neurosci 21:33–40. https://doi.org/10.1038/s41593-017-0037-5
Yoshihara M, Littleton JT (2002) Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron 36:897–908
Turecek J, Regehr WG (2019) Neuronal regulation of fast synaptotagmin isoforms controls the relative contributions of synchronous and asynchronous release. Neuron 101:938-949.e4. https://doi.org/10.1016/j.neuron.2019.01.013
Nishiki T-I, Augustine GJ (2004) Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci 24:8542–8550. https://doi.org/10.1523/JNEUROSCI.2545-04.2004
Xu J, Mashimo T, Südhof TC (2007) Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54:567–581. https://doi.org/10.1016/j.neuron.2007.05.004
Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79:717–727. https://doi.org/10.1016/0092-8674(94)90556-8
Guan Z, Bykhovskaia M, Jorquera RA, Sutton RB, Akbergenova Y, Littleton JT (2017) A synaptotagmin suppressor screen indicates SNARE binding controls the timing and Ca2+ cooperativity of vesicle fusion. eLife. https://doi.org/10.7554/eLife.28409
Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ (1993) Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell 74:1125–1134. https://doi.org/10.1016/0092-8674(93)90733-7
Liu H, Dean C, Arthur CP, Dong M, Chapman ER (2009) Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci 29:7395–7403. https://doi.org/10.1523/JNEUROSCI.1341-09.2009
Rozov A, Bolshakov AP, Valiullina-Rakhmatullina F (2019) The ever-growing puzzle of asynchronous release. Front Cell Neurosci 13:28. https://doi.org/10.3389/fncel.2019.00028
Chanaday NL, Kavalali ET (2018) Presynaptic origins of distinct modes of neurotransmitter release. Curr Opin Neurobiol 51:119–126. https://doi.org/10.1016/j.conb.2018.03.005
Pang ZP, Südhof TC (2010) Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol 22:496–505. https://doi.org/10.1016/j.ceb.2010.05.001
Fioravante D, Regehr WG (2011) Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21:269–274. https://doi.org/10.1016/j.conb.2011.02.003
Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. https://doi.org/10.1146/annurev.physiol.64.092501.114547
Volynski KE, Krishnakumar SS (2018) Synergistic control of neurotransmitter release by different members of the synaptotagmin family. Curr Opin Neurobiol 51:154–162. https://doi.org/10.1016/j.conb.2018.05.006
Kochubey O, Lou X, Schneggenburger R (2011) Regulation of transmitter release by Ca(2+) and synaptotagmin: insights from a large CNS synapse. Trends Neurosci 34:237–246. https://doi.org/10.1016/j.tins.2011.02.006
Chung ChiHye, Raingo J (2013) Vesicle dynamics: how synaptic proteins regulate different modes of neurotransmission. J Neurochem 126:146–154. https://doi.org/10.1111/jnc.12245
Fesce R (1999) The kinetics of nerve-evoked quantal secretion . Philos Trans R Soc Lond Ser B Biol Sci 354:319–329. https://doi.org/10.1098/rstb.1999.0383
Publicover SJ (1985) Calmodulin, synchronous and asynchronous release of neurotransmitter. Comp Biochem Physiol A Comp Physiol 82:7–11. https://doi.org/10.1016/0300-9629(85)90696-6
Yoshihara M, Guan Z, Littleton JT (2010) Differential regulation of synchronous versus asynchronous neurotransmitter release by the C2 domains of synaptotagmin 1. Proc Natl Acad Sci USA 107:14869–14874. https://doi.org/10.1073/pnas.1000606107
Liu H, Bai H, Hui E, Yang L, Evans CS, Wang Z, Kwon SE, Chapman ER (2014) Synaptotagmin 7 functions as a Ca2+-sensor for synaptic vesicle replenishment. eLife 3:e01524. https://doi.org/10.7554/eLife.01524
Li YC, Chanaday NL, Wei Xu, Kavalali ET (2017) Synaptotagmin-1- and synaptotagmin-7-dependent fusion mechanisms target synaptic vesicles to kinetically distinct endocytic pathways. Neuron 93:616-631.e3. https://doi.org/10.1016/j.neuron.2016.12.010
Wen H, Linhoff MW, McGinley MJ, Li G-L, Corson GM, Mandel G, Brehm P (2010) Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc Natl Acad Sci USA 107:13906–13911. https://doi.org/10.1073/pnas.1008598107
Chen C, Satterfield R, Young SM, Jonas P (2017) Triple function of synaptotagmin 7 ensures efficiency of high-frequency transmission at central GABAergic synapses. Cell reports 21:2082–2089. https://doi.org/10.1016/j.celrep.2017.10.122
Turecek J, Regehr WG (2018) Synaptotagmin 7 mediates both facilitation and asynchronous release at granule cell synapses. J Neurosci 38:3240–3251. https://doi.org/10.1523/JNEUROSCI.3207-17.2018
Jackman SL, Turecek J, Belinsky JE, Regehr WG (2016) The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529:88–91. https://doi.org/10.1038/nature16507
Guan Z, Quiñones-Frías MC, Akbergenova Y, Littleton JT (2020) Drosophila Synaptotagmin 7 negatively regulates synaptic vesicle release and replenishment in a dosage-dependent manner. eLife. https://doi.org/10.7554/eLife.55443
Huson V, Regehr WG (2020) Diverse roles of Synaptotagmin-7 in regulating vesicle fusion. Curr Opin Neurobiol 63:42–52. https://doi.org/10.1016/j.conb.2020.02.006
Bacaj T, Dick Wu, Yang X, Morishita W, Zhou P, Wei Xu, Malenka RC, Südhof TC (2013) Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80:947–959. https://doi.org/10.1016/j.neuron.2013.10.026
Littleton JT, Barnard RJ, Titus SA, Slind J, Chapman ER, Ganetzky B (2001) SNARE-complex disassembly by NSF follows synaptic-vesicle fusion. Proc Natl Acad Sci USA 98:12233–12238. https://doi.org/10.1073/pnas.221450198
Rizo J (2018) Mechanism of neurotransmitter release coming into focus. Protein Sci 27:1364–1391. https://doi.org/10.1002/pro.3445
Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B (1998) Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron 21:401–413
Zhao M, Shenping Wu, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, Brunger AT (2015) Mechanistic insights into the recycling machine of the SNARE complex. Nature 518:61–67. https://doi.org/10.1038/nature14148
White KI, Zhao M, Choi UB, Pfuetzner RA, Brunger AT (2018) Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife. https://doi.org/10.7554/eLife.38888
Burgalossi A, Jung S, Meyer G, Jockusch WJ, Jahn O, Taschenberger H, O’Connor VM et al (2010) SNARE protein recycling by αSNAP and βSNAP supports synaptic vesicle priming. Neuron 68:473–487
Kawasaki F, Mattiuz AM, Ordway RW (1998) Synaptic physiology and ultrastructure in comatose mutants define an in vivo role for NSF in neurotransmitter release. J Neurosci 18:10241–10249
Peled ES, Newman ZL, Isacoff EhudY (2014) Evoked and spontaneous transmission favored by distinct sets of synapses. Curr Biol 24:484–493. https://doi.org/10.1016/j.cub.2014.01.022
Peled ES, Isacoff EhudY (2011) Optical quantal analysis of synaptic transmission in wild-type and rab3-mutant Drosophila motor axons. Nat Neurosci 14:519–526. https://doi.org/10.1038/nn.2767
Newman ZL, Hoagland A, Aghi K, Worden K, Levy SL, Son JH, Lee LP, Isacoff EY (2017) Input-Specific plasticity and homeostasis at the Drosophila larval neuromuscular junction. Neuron 93:1388-1404.e10. https://doi.org/10.1016/j.neuron.2017.02.028
Cho RW, Buhl LK, Volfson D, Tran A, Li F, Akbergenova Y, Littleton JT (2015) Phosphorylation of complexin by PKA regulates activity-dependent spontaneous neurotransmitter release and structural synaptic plasticity. Neuron 88:749–761. https://doi.org/10.1016/j.neuron.2015.10.011
Craxton M (2010) A manual collection of Syt, Esyt, Rph3a, Rph3al, Doc2, and Dblc2 genes from 46 metazoan genomes–an open access resource for neuroscience and evolutionary biology. BMC Genom 11:37. https://doi.org/10.1186/1471-2164-11-37
Barber CF, Jorquera RA, Melom JE, Littleton JT (2009) Postsynaptic regulation of synaptic plasticity by synaptotagmin 4 requires both C2 domains. J Cell Biol 187:295–310. https://doi.org/10.1083/jcb.200903098
Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Südhof TC (1995) Ca(2+)-dependent and -independent activities of neural and non-neural synaptotagmins. Nature 375:594–599. https://doi.org/10.1038/375594a0
Adolfsen B, Littleton JT (2001) Genetic and molecular analysis of the synaptotagmin family. Cell Mol Life Sci 58:393–402. https://doi.org/10.1007/PL00000865
Adolfsen B, Saraswati S, Yoshihara M, Littleton JT (2004) Synaptotagmins are trafficked to distinct subcellular domains including the postsynaptic compartment. J Cell Biol 166:249–260. https://doi.org/10.1083/jcb.200312054
Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Südhof TC (2006) Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci 26:13493–13504. https://doi.org/10.1523/JNEUROSCI.3519-06.2006
DiAntonio A, Parfitt KD, Schwarz TL (1993) Synaptic transmission persists in synaptotagmin mutants of Drosophila. Cell 73:1281–1290. https://doi.org/10.1016/0092-8674(93)90356-u
Loewen CA, Mackler JM, Reist NE (2001) Drosophila synaptotagmin I null mutants survive to early adulthood. Genesis 31:30–36. https://doi.org/10.1002/gene.10002
Yoshihara M, Adolfsen B, Galle KT, Littleton JT (2005) Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science 310:858–863. https://doi.org/10.1126/science.1117541
Harris KP, Zhang YV, Piccioli ZD, Perrimon N, Littleton JT (2016) The postsynaptic t-SNARE Syntaxin 4 controls traffic of Neuroligin 1 and Synaptotagmin 4 to regulate retrograde signaling. eLife. https://doi.org/10.7554/eLife.13881
Korkut C, Li Y, Koles K, Brewer C, Ashley J, Yoshihara M, Budnik V (2013) Regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron 77:1039–1046. https://doi.org/10.1016/j.neuron.2013.01.013
Walsh RB, Becalska AN, Zunitch MJ, Wang S, Isaac B, Yeh A, Koles K, Rodal AA (2019) Opposing functions for retromer and Rab11 in extracellular vesicle cargo traffic at synapses. BioRxiv. https://doi.org/10.1101/645713
Park D, Li P, Dani A, Taghert PH (2014) Peptidergic cell-specific synaptotagmins in Drosophila: localization to dense-core granules and regulation by the bHLH protein DIMMED. J Neurosci 34:13195–13207. https://doi.org/10.1523/JNEUROSCI.2075-14.2014
Park D, Hadžić T, Yin P, Rusch J, Abruzzi K, Rosbash M, Skeath JB, Panda S, Sweedler JV, Taghert PH (2011) Molecular organization of Drosophila neuroendocrine cells by Dimmed. Curr Biol 21:1515–1524. https://doi.org/10.1016/j.cub.2011.08.015
Hamanaka Y, Park D, Yin P, Annangudi SP, Edwards TN, Sweedler J, Meinertzhagen IA, Taghert PH (2010) Transcriptional orchestration of the regulated secretory pathway in neurons by the bHLH protein DIMM. Curr Biol 20:9–18. https://doi.org/10.1016/j.cub.2009.11.065
Min S-W, Chang W-P, Südhof TC (2007) E-Syts, a family of membranous Ca2+-sensor proteins with multiple C2 domains. Proc Natl Acad Sci USA 104:3823–3828. https://doi.org/10.1073/pnas.0611725104
Kikuma K, Li X, Kim D, Sutter D, Dickman DK (2017) Extended synaptotagmin localizes to presynaptic ER and promotes neurotransmission and synaptic growth in drosophila. Genetics 207:993–1006. https://doi.org/10.1534/genetics.117.300261
Shin O-H, Han W, Wang Y, Südhof TC (2005) Evolutionarily conserved multiple C2 domain proteins with two transmembrane regions (MCTPs) and unusual Ca2+ binding properties. J Biol Chem 280:1641–1651. https://doi.org/10.1074/jbc.M407305200
Genç Ö, Dickman DK, Ma W, Tong A, Fetter RD, Davis GW (2017) MCTP is an ER-resident calcium sensor that stabilizes synaptic transmission and homeostatic plasticity. eLife. https://doi.org/10.7554/eLife.22904
Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K (1999) Drosophila UNC-13 is essential for synaptic transmission. Nat Neurosci 2:965–971. https://doi.org/10.1038/14764
Liu KSY, Siebert M, Mertel S, Knoche E, Wegener S, Wichmann C, Matkovic T et al (2011) RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 334:1565–1569. https://doi.org/10.1126/science.1212991
Smith MK, Wakimoto BT (2007) Complex regulation and multiple developmental functions of misfire, the Drosophila melanogaster ferlin gene. BMC Dev Biol 7:21. https://doi.org/10.1186/1471-213X-7-21
Serano J, Rubin GM (2003) The Drosophila synaptotagmin-like protein bitesize is required for growth and has mRNA localization sequences within its open reading frame. Proc Natl Acad Sci USA 100:13368–13373. https://doi.org/10.1073/pnas.1835727100
Pang ZP, Bacaj T, Yang X, Zhou P, Wei Xu, Südhof TC (2011) Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron 70:244–251. https://doi.org/10.1016/j.neuron.2011.03.011
Yao J, Gaffaney JD, Kwon SE, Chapman ER (2011) Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell 147:666–677. https://doi.org/10.1016/j.cell.2011.09.046
Groffen AJ, Martens S, Arazola RD, Niels Cornelisse L, Lozovaya N, de Jong APH, Goriounova NA et al (2010) Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science (New York, NY) 327:1614–1618
Courtney NA, Briguglio JS, Bradberry MM, Greer C, Chapman ER (2018) Excitatory and inhibitory neurons utilize different ca2+ sensors and sources to regulate spontaneous release. Neuron 98:977-991.e5. https://doi.org/10.1016/j.neuron.2018.04.022
Südhof TC, Baumert M, Perin MS, Jahn R (1989) A synaptic vesicle membrane protein is conserved from mammals to Drosophila. Neuron 2:1475–1481
Ullrich B, Südhof TC (1995) Differential distributions of novel synaptotagmins: comparison to synapsins. Neuropharmacology 34:1371–1377. https://doi.org/10.1016/0028-3908(95)00132-P
Elíes J, Yáñez M, Pereira TMC, Gil-Longo J, MacDougall DA, Campos-Toimil M (2020) An update to calcium binding proteins. Adv Exp Med Biol 1131:183–213. https://doi.org/10.1007/978-3-030-12457-1_8
McCue HV, Haynes LP, Burgoyne RD (2010) The diversity of calcium sensor proteins in the regulation of neuronal function. Cold Spring Harbor Perspect Biol 2:a004085. https://doi.org/10.1101/cshperspect.a004085
Burgoyne RD, Helassa N, McCue HV, Haynes LP (2019) Calcium sensors in neuronal function and dysfunction. Cold Spring Harbor Perspect Biol. https://doi.org/10.1101/cshperspect.a035154
de Jong APH, Fioravante D (2014) Translating neuronal activity at the synapse: presynaptic calcium sensors in short-term plasticity. Front Cell Neurosci 8:356. https://doi.org/10.3389/fncel.2014.00356
Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB et al (2015) Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525:62–67. https://doi.org/10.1038/nature14975
Kuo W, Herrick DZ, Ellena JF, Cafiso DS (2009) The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. J Mol Biol 387:284–294. https://doi.org/10.1016/j.jmb.2009.01.064
Loewen CA, Lee S-M, Shin Y-K, Reist NE (2006) C2B polylysine motif of synaptotagmin facilitates a Ca2+-independent stage of synaptic vesicle priming in vivo. Mol Biol Cell 17:5211–5226. https://doi.org/10.1091/mbc.E06-07-0622
Wang S, Li Y, Ma C (2016) Synaptotagmin-1 C2B domain interacts simultaneously with SNAREs and membranes to promote membrane fusion. eLife. https://doi.org/10.7554/eLife.14211
Fukuda M, Kanno E, Satoh M, Saegusa C, Yamamoto A (2004) Synaptotagmin VII is targeted to dense-core vesicles and regulates their Ca2+-dependent exocytosis in PC12 cells. J Biol Chem 279:52677–52684. https://doi.org/10.1074/jbc.M409241200
Zhang Z, Zhang Z, Jackson MB (2010) Synaptotagmin IV modulation of vesicle size and fusion pores in PC12 cells. Biophys J 98:968–978. https://doi.org/10.1016/j.bpj.2009.11.024
Moghadam PK, Jackson MB (2013) The functional significance of synaptotagmin diversity in neuroendocrine secretion. Front Endocrinol 4:124. https://doi.org/10.3389/fendo.2013.00124
Cao P, Yang X, Südhof TC (2013) Complexin activates exocytosis of distinct secretory vesicles controlled by different synaptotagmins. J Neurosci 33:1714–1727. https://doi.org/10.1523/JNEUROSCI.4087-12.2013
Papke JB, Moore-Dotson JM, Watson DJ, Wedell CD, French LR, Rendell SR, Harkins AB (2012) Titration of synaptotagmin I expression differentially regulates release of norepinephrine and neuropeptide Y. Neuroscience 218:78–88. https://doi.org/10.1016/j.neuroscience.2012.05.020
Zhang Z, Yao Wu, Zhao Wang F, Dunning M, Rehfuss J, Ramanan D, Chapman ER, Jackson MB (2011) Release mode of large and small dense-core vesicles specified by different synaptotagmin isoforms in PC12 cells. Mol Biol Cell 22:2324–2336. https://doi.org/10.1091/mbc.E11-02-0159
Wang CT, Grishanin R, Earles CA, Chang PY, Martin TF, Chapman ER, Jackson MB (2001) Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science 294:1111–1115. https://doi.org/10.1126/science.1064002
Martin TF (1994) The molecular machinery for fast and slow neurosecretion. Curr Opin Neurobiol 4:626–632. https://doi.org/10.1016/0959-4388(94)90002-7
Sørensen JB, Fernández-Chacón R, Südhof TC, Neher E (2003) Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+. J General Physiol 122:265–276. https://doi.org/10.1085/jgp.200308855
Walch-Solimena C, Takei K, Marek KL, Midyett K, Südhof TC, De Camilli P, Jahn R (1993) Synaptotagmin: a membrane constituent of neuropeptide-containing large dense-core vesicles. J Neurosci 13:3895–3903
Dean C, Huisheng Liu F, Dunning M, Chang PY, Jackson MB, Chapman ER (2009) Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci 12:767–776. https://doi.org/10.1038/nn.2315
Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Wei Xu, Chen Lu, Malenka RC, Südhof TC (2017) Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 544:316–321. https://doi.org/10.1038/nature21720
Dean C, Liu H, Staudt T, Stahlberg MA, Vingill S, Bückers J, Kamin D et al (2012) Distinct subsets of Syt-IV/BDNF vesicles are sorted to axons versus dendrites and recruited to synapses by activity. J Neurosci 32:5398–5413. https://doi.org/10.1523/JNEUROSCI.4515-11.2012
Sumi T, Harada K (2020) Mechanism underlying hippocampal long-term potentiation and depression based on competition between endocytosis and exocytosis of AMPA receptors. Sci Rep 10:14711. https://doi.org/10.1038/s41598-020-71528-3
Wu X, Shaoqin Hu, Kang X, Wang C (2020) Synaptotagmins: beyond presynaptic neurotransmitter release. Neuroscientist 26:9–15. https://doi.org/10.1177/1073858419844497
Awasthi A, Ramachandran B, Ahmed S, Benito E, Shinoda Yo, Nitzan N, Heukamp A et al (2019) Synaptotagmin-3 drives AMPA receptor endocytosis, depression of synapse strength, and forgetting. Science. https://doi.org/10.1126/science.aav1483
Littleton JT, Bellen HJ, Perin MS (1993) Expression of synaptotagmin in Drosophila reveals transport and localization of synaptic vesicles to the synapse. Development 118:1077–1088
Littleton JT, Bai J, Vyas B, Desai R, Baltus AE, Garment MB, Carlson SD, Ganetzky B, Chapman ER (2001) synaptotagmin mutants reveal essential functions for the C2B domain in Ca2+-triggered fusion and recycling of synaptic vesicles in vivo. J Neurosci 21:1421–1433
Broadie K, Bellen HJ, DiAntonio A, Littleton JT, Schwarz TL (1994) Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proc Natl Acad Sci USA 91:10727–10731
Lee J, Littleton JT (2015) Transmembrane tethering of synaptotagmin to synaptic vesicles controls multiple modes of neurotransmitter release. Proc Natl Acad Sci USA 112:3793–3798. https://doi.org/10.1073/pnas.1420312112
Saraswati S, Adolfsen B, Littleton JT (2007) Characterization of the role of the Synaptotagmin family as calcium sensors in facilitation and asynchronous neurotransmitter release. Proc Natl Acad Sci USA 104:14122–14127. https://doi.org/10.1073/pnas.0706711104
DiAntonio A, Schwarz TL (1994) The effect on synaptic physiology of synaptotagmin mutations in Drosophila. Neuron 12:909–920. https://doi.org/10.1016/0896-6273(94)90342-5
Reist NE, Buchanan J, Li J, DiAntonio A, Buxton EM, Schwarz TL (1998) Morphologically docked synaptic vesicles are reduced in synaptotagmin mutants of Drosophila. J Neurosci 18:7662–7673
Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE (2002) The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418:340–344. https://doi.org/10.1038/nature00846
Poskanzer KE, Marek KW, Sweeney ST, Davis GW (2003) Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature 426:559–563. https://doi.org/10.1038/nature02184
Wolfes AC, Dean C (2020) The diversity of synaptotagmin isoforms. Curr Opin Neurobiol 63:198–209. https://doi.org/10.1016/j.conb.2020.04.006
Gundersen CB (2020) Fast, synchronous neurotransmitter release: past, present and future. Neuroscience 439:22–27. https://doi.org/10.1016/j.neuroscience.2019.04.030
Bornschein G, Schmidt H (2018) Synaptotagmin ca2+ sensors and their spatial coupling to presynaptic cav channels in central cortical synapses. Front Mol Neurosci 11:494. https://doi.org/10.3389/fnmol.2018.00494
Park Y, Ryu J-K (2018) Models of synaptotagmin-1 to trigger Ca2+ -dependent vesicle fusion. FEBS Lett 592:3480–3492. https://doi.org/10.1002/1873-3468.13193
Südhof TC (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80:675–690. https://doi.org/10.1016/j.neuron.2013.10.022
Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490:201–207. https://doi.org/10.1038/nature11320
Mackler JM, Reist NE (2001) Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. J Comp Neurol 436:4–16
Paddock BE, Wang Z, Biela LM, Chen K, Getzy MD, Striegel A, Richmond JE, Chapman ER, Featherstone DE, Reist NE (2011) Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo. J Neurosci 31:2248–2257. https://doi.org/10.1523/JNEUROSCI.3153-09.2011
Paddock BE, Striegel AR, Hui E, Chapman ER, Reist NE (2008) Ca2+-dependent, phospholipid-binding residues of synaptotagmin are critical for excitation-secretion coupling in vivo. J Neurosci 28:7458–7466. https://doi.org/10.1523/JNEUROSCI.0197-08.2008
Striegel AR, Biela LM, Evans CS, Wang Z, Delehoy JB, Bryan Sutton R, Chapman ER, Reist NE (2012) Calcium binding by synaptotagmin’s C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J Neurosci 32:1253–1260. https://doi.org/10.1523/JNEUROSCI.4652-11.2012
Mace KE, Biela LM, Sares AG, Reist NE (2009) Synaptotagmin I stabilizes synaptic vesicles via its C2A polylysine motif. Genesis 47:337–345
Marek KW, Davis GW (2002) Transgenically encoded protein photoinactivation (FlAsH-FALI): acute inactivation of synaptotagmin I. Neuron 36:805–813
Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC (2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410:41–49. https://doi.org/10.1038/35065004
Dean C, Mark Dunning F, Liu H, Bomba-Warczak E, Martens H, Bharat V, Ahmed S, Chapman ER (2012) Axonal and dendritic synaptotagmin isoforms revealed by a pHluorin-syt functional screen. Mol Biol Cell 23:1715–1727. https://doi.org/10.1091/mbc.E11-08-0707
Kaempf N, Kochlamazashvili G, Puchkov D, Maritzen T, Bajjalieh SM, Kononenko NL, Haucke V (2015) Overlapping functions of stonin 2 and SV2 in sorting of the calcium sensor synaptotagmin 1 to synaptic vesicles. Proc Natl Acad Sci USA 112:7297–7302. https://doi.org/10.1073/pnas.1501627112
Soekmadji C, Angkawidjaja C, Kelly LE (2012) Ca2+ regulates the Drosophila Stoned-A and Stoned-B proteins interaction with the C2B domain of Synaptotagmin-1. PLoS ONE 7:e38822. https://doi.org/10.1371/journal.pone.0038822
Jarousse N, Wilson JD, Arac D, Rizo J, Kelly RB (2003) Endocytosis of synaptotagmin 1 is mediated by a novel, tryptophan-containing motif. Traffic 4:468–478. https://doi.org/10.1034/j.1600-0854.2003.00101.x
Jarousse N, Kelly RB (2001) The AP2 binding site of synaptotagmin 1 is not an internalization signal but a regulator of endocytosis. J Cell Biol 154:857–866. https://doi.org/10.1083/jcb.200103040
Poskanzer KE, Fetter RD, Davis GW (2006) Discrete residues in the c(2)b domain of synaptotagmin I independently specify endocytic rate and synaptic vesicle size. Neuron 50:49–62. https://doi.org/10.1016/j.neuron.2006.02.021
Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER (1999) Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature 400:757–760. https://doi.org/10.1038/23462
Kelly LE, Marie A, Phillips. (2005) Molecular and genetic characterization of the interactions between the Drosophila stoned-B protein and DAP-160 (intersectin). Biochem J 388:195–204. https://doi.org/10.1042/BJ20041797
Fergestad T, Broadie K (2001) Interaction of stoned and synaptotagmin in synaptic vesicle endocytosis. J Neurosci 21:1218–1227
Das D, Bao H, Courtney KC, Lanxi Wu, Chapman ER (2020) Resolving kinetic intermediates during the regulated assembly and disassembly of fusion pores. Nat Commun 11:231. https://doi.org/10.1038/s41467-019-14072-7
Lynch KL, Gerona RRL, Kielar DM, Martens S, McMahon HT, Martin TFJ (2008) Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell 19:5093–5103. https://doi.org/10.1091/mbc.e08-03-0235
Bendahmane M, Bohannon KP, Bradberry MM, Rao TC, Schmidtke MW, Abbineni PS, Chon NL et al (2018) The synaptotagmin C2B domain calcium-binding loops modulate the rate of fusion pore expansion. Mol Biol Cell 29:834–845. https://doi.org/10.1091/mbc.E17-11-0623
Rao TC, Passmore DR, Peleman AR, Das M, Chapman ER, Anantharam A (2014) Distinct fusion properties of synaptotagmin-1 and synaptotagmin-7 bearing dense core granules. Mol Biol Cell 25:2416–2427. https://doi.org/10.1091/mbc.E14-02-0702
Lai Y, Diao J, Liu Y, Ishitsuka Y, Zengliu Su, Schulten K, Ha T, Shin Y-K (2013) Fusion pore formation and expansion induced by Ca2+ and synaptotagmin 1. Proc Natl Acad Sci USA 110:1333–1338. https://doi.org/10.1073/pnas.1218818110
Zhang Z, Hui E, Chapman ER, Jackson MB (2009) Phosphatidylserine regulation of Ca2+-triggered exocytosis and fusion pores in PC12 cells. Mol Biol Cell 20:5086–5095. https://doi.org/10.1091/mbc.E09-08-0691
Zhu D, Zhou W, Liang T, Yang F, Zhang R-Y, Zheng-Xing Wu, Tao Xu (2007) Synaptotagmin I and IX function redundantly in controlling fusion pore of large dense core vesicles. Biochem Biophys Res Commun 361:922–927. https://doi.org/10.1016/j.bbrc.2007.07.083
Wang C-T, Bai J, Chang PY, Chapman ER, Jackson MB (2006) Synaptotagmin-Ca2+ triggers two sequential steps in regulated exocytosis in rat PC12 cells: fusion pore opening and fusion pore dilation. J Physiol 570:295–307. https://doi.org/10.1113/jphysiol.2005.097378
Bai J, Wang C-T, Richards DA, Jackson MB, Chapman ER (2004) Fusion pore dynamics are regulated by synaptotagmin*t-SNARE interactions. Neuron 41:929–942. https://doi.org/10.1016/s0896-6273(04)00117-5
Herrmann DN, Horvath R, Sowden JE, Gonzalez M, Sanchez-Mejias A, Guan Z, Whittaker RG et al (2014) Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet 95:332–339. https://doi.org/10.1016/j.ajhg.2014.08.007
Whittaker RG, Herrmann DN, Bansagi B, Hasan BAS, Lofra RM, Logigian EL, Sowden JE et al (2015) Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology 85:1964–1971. https://doi.org/10.1212/WNL.0000000000002185
Bacaj T, Dick Wu, Burré J, Malenka RC, Liu X, Südhof TC (2015) Synaptotagmin-1 and -7 are redundantly essential for maintaining the capacity of the readily-releasable pool of synaptic vesicles. PLoS Biol 13:e1002267. https://doi.org/10.1371/journal.pbio.1002267
Striegel AR, Biela LM, Evans CS, Wang Z, Delehoy JB, Sutton RB, Chapman ER, Reist NE (2012) Calcium binding by synaptotagmin’s C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J Neurosci 32:1253–1260
Shields MC, Bowers MR, Kramer HL, Fulcer MM, Perinet LC, Metz MJ, Reist NE (2020) The role of the C2A domain of synaptotagmin 1 in asynchronous neurotransmitter release. PLoS ONE 15:e0232991. https://doi.org/10.1371/journal.pone.0232991
Bowers MR, Reist NE (2020) The C2A domain of synaptotagmin is an essential component of the calcium sensor for synaptic transmission. PLoS ONE 15:e0228348. https://doi.org/10.1371/journal.pone.0228348
Bowers MR, Reist NE (2020) Synaptotagmin: mechanisms of an electrostatic switch. Neurosci Lett 722:134834. https://doi.org/10.1016/j.neulet.2020.134834
Vrljic M, Strop P, Ernst JA, Bryan Sutton R, Chu S, Brunger AT (2010) Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat Struct Mol Biol 17:325–331. https://doi.org/10.1038/nsmb.1764
Bai J, Tucker WC, Chapman ER (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol 11:36–44. https://doi.org/10.1038/nsmb709
Hui E, Gaffaney JD, Wang Z, Johnson CP, Evans CS, Chapman ER (2011) Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol 18:813–821. https://doi.org/10.1038/nsmb.2075
Chapman ER, Jahn R (1994) Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes. Autonomous function of a single C2-homologous domain. J Biol Chem 269:5735–5741
Davletov BA, Südhof TC (1994) Ca(2+)-dependent conformational change in synaptotagmin I. J Biol Chem 269:28547–28550
Davletov BA, Südhof TC (1993) A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem 268:26386–26390
Ubach J, Lao Y, Fernandez I, Arac D, Südhof TC, Rizo J (2001) The C2B domain of synaptotagmin I is a Ca2+-binding module. Biochemistry 40:5854–5860. https://doi.org/10.1021/bi010340c
Tucker WC, Weber T, Chapman ER (2004) Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304:435–438. https://doi.org/10.1126/science.1097196
Fernández-Chacón R, Shin O-H, Königstorfer A, Matos MF, Meyer AC, Garcia J, Gerber SH, Rizo J, Südhof TC, Rosenmund C (2002) Structure/function analysis of Ca2+ binding to the C2A domain of synaptotagmin 1. J Neurosci 22:8438–8446
Araç D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Südhof TC, Rizo J (2006) Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol 13:209–217. https://doi.org/10.1038/nsmb1056
Shin O-H, Jun Xu, Rizo J, Südhof TC (2009) Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc Natl Acad Sci USA 106:16469–16474. https://doi.org/10.1073/pnas.0908798106
Xue M, Craig TK, Shin O-H, Li L, Brautigam CA, Tomchick DR, Südhof TC, Rosenmund C, Rizo J (2010) Structural and mutational analysis of functional differentiation between synaptotagmins-1 and -7. PLoS ONE. https://doi.org/10.1371/journal.pone.0012544
Buhl LK, Jorquera RA, Akbergenova Y, Huntwork-Rodriguez S, Volfson D, Littleton JT (2013) Differential regulation of evoked and spontaneous neurotransmitter release by C-terminal modifications of complexin. Mol Cell Neurosci 52:161–172. https://doi.org/10.1016/j.mcn.2012.11.009
Cho RW, Song Y, Littleton JT (2010) Comparative analysis of Drosophila and mammalian complexins as fusion clamps and facilitators of neurotransmitter release. Mol Cell Neurosci 45:389–397. https://doi.org/10.1016/j.mcn.2010.07.012
Cho RW, Kümmel D, Li F, Baguley SW, Coleman J, Rothman JE, Littleton JT (2014) Genetic analysis of the Complexin trans-clamping model for cross-linking SNARE complexes in vivo. Proc Natl Acad Sci USA 111:10317–10322. https://doi.org/10.1073/pnas.1409311111
Iyer J, Wahlmark CJ, Kuser-Ahnert GA, Kawasaki F (2013) Molecular mechanisms of COMPLEXIN fusion clamp function in synaptic exocytosis revealed in a new Drosophila mutant. Mol Cell Neurosci 56:244–254. https://doi.org/10.1016/j.mcn.2013.06.002
Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C (2009) Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron 64:367–380. https://doi.org/10.1016/j.neuron.2009.09.043
Sabeva N, Cho RW, Vasin A, Agustin Gonzalez J, Littleton JT, Bykhovskaia M (2017) Complexin mutants reveal partial segregation between recycling pathways that drive evoked and spontaneous neurotransmission. J Neurosci 37:383–396. https://doi.org/10.1523/JNEUROSCI.1854-16.2016
Hua Y, Scheller RH (2001) Three SNARE complexes cooperate to mediate membrane fusion. Proc Natl Acad Sci USA 98:8065–8070. https://doi.org/10.1073/pnas.131214798
van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R (2010) One SNARE complex is sufficient for membrane fusion. Nat Struct Mol Biol 17:358–364. https://doi.org/10.1038/nsmb.1748
Mohrmann R, de Wit H, Verhage M, Neher E, Sørensen JB (2010) Fast vesicle fusion in living cells requires at least three SNARE complexes. Science (New York, NY) 330:502–505
Shi L, Shen Q-T, Kiel A, Wang J, Wang H-W, Melia TJ, Rothman JE, Pincet F (2012) SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science 335:1355–1359. https://doi.org/10.1126/science.1214984
Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H et al (2006) Molecular anatomy of a trafficking organelle. Cell 127:831–846. https://doi.org/10.1016/j.cell.2006.10.030
Wu Yi, He Y, Bai J, Ji S-R, Tucker WC, Chapman ER, Sui S-F (2003) Visualization of synaptotagmin I oligomers assembled onto lipid monolayers. Proc Natl Acad Sci USA 100:2082–2087. https://doi.org/10.1073/pnas.0435872100
Desai RC, Vyas B, Earles CA, Littleton JT, Kowalchyck JA, Martin TF, Chapman ER (2000) The C2B domain of synaptotagmin is a Ca(2+)-sensing module essential for exocytosis. J Cell Biol 150:1125–1136. https://doi.org/10.1083/jcb.150.5.1125
Chapman ER, Desai RC, Davis AF, Tornehl CK (1998) Delineation of the oligomerization, AP-2 binding, and synprint binding region of the C2B domain of synaptotagmin. J Biol Chem 273:32966–32972
Fukuda M, Mikoshiba K (2000) Distinct self-oligomerization activities of synaptotagmin family. Unique calcium-dependent oligomerization properties of synaptotagmin VII. J Biol Chem 275:28180–28185. https://doi.org/10.1074/jbc.M001376200
Zanetti MN, Bello OD, Wang J, Coleman J, Cai Y, Sindelar CV, Rothman JE, Krishnakumar SS (2016) Ring-like oligomers of Synaptotagmins and related C2 domain proteins. eLife. https://doi.org/10.7554/eLife.17262
Wang J, Li F, Bello OD, Sindelar CV, Pincet F, Krishnakumar SS, Rothman JE (2017) Circular oligomerization is an intrinsic property of synaptotagmin. eLife. https://doi.org/10.7554/eLife.27441
Wang DG, Takeuchi H, Gao J, Zhang Z, Hirata M (2015) Hetero-oligomerization of C2 domains of phospholipase C-related but catalytically inactive protein and synaptotagmin-1. Adv Biol Regul 57:120–129. https://doi.org/10.1016/j.jbior.2014.09.001
Fukuda M, Mikoshiba K (2000) Calcium-dependent and -independent hetero-oligomerization in the synaptotagmin family. J Biochem 128:637–645. https://doi.org/10.1093/oxfordjournals.jbchem.a022796
Bello OD, Jouannot O, Chaudhuri A, Stroeva E, Coleman J, Volynski KE, Rothman JE, Krishnakumar SS (2018) Synaptotagmin oligomerization is essential for calcium control of regulated exocytosis. Proc Natl Acad Sci USA 115:E7624–E7631. https://doi.org/10.1073/pnas.1808792115
Wang P, Chicka MC, Bhalla A, Richards DA, Chapman ER (2005) Synaptotagmin VII is targeted to secretory organelles in PC12 cells, where it functions as a high-affinity calcium sensor. Mol Cell Biol 25:8693–8702. https://doi.org/10.1128/MCB.25.19.8693-8702.2005
Bhalla A, Tucker WC, Chapman ER (2005) Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell 16:4755–4764. https://doi.org/10.1091/mbc.E05-04-0277
Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER (2005) Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci USA 102:5210–5214. https://doi.org/10.1073/pnas.0500941102
Voleti R, Tomchick DR, Südhof TC, Rizo J (2017) Exceptionally tight membrane-binding may explain the key role of the synaptotagmin-7 C2A domain in asynchronous neurotransmitter release. Proc Natl Acad Sci USA 114:E8518–E8527. https://doi.org/10.1073/pnas.1710708114
Sugita S, Han W, Butz S, Liu X, Fernández-Chacón R, Lao Y, Südhof TC (2001) Synaptotagmin VII as a plasma membrane Ca(2+) sensor in exocytosis. Neuron 30:459–473. https://doi.org/10.1016/s0896-6273(01)00290-2
Sugita S, Shin O-H, Han W, Lao Ye, Südhof TC (2002) Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J 21:270–280. https://doi.org/10.1093/emboj/21.3.270
Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME et al (2000) Comparative genomics of the eukaryotes. Science 287:2204–2215. https://doi.org/10.1126/science.287.5461.2204
Littleton JT (2000) A genomic analysis of membrane trafficking and neurotransmitter release in Drosophila. J Cell Biol 150:F77-82
Monterrat C, Grise F, Benassy MN, Hémar A, Lang J (2007) The calcium-sensing protein synaptotagmin 7 is expressed on different endosomal compartments in endocrine, neuroendocrine cells or neurons but not on large dense core vesicles. Histochem Cell Biol 127:625–632. https://doi.org/10.1007/s00418-007-0271-0
Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW (2000) Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol 148:1141–1149. https://doi.org/10.1083/jcb.148.6.1141
Schonn J-S, Maximov A, Lao Ye, Südhof TC, Sørensen JB (2008) Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci USA 105:3998–4003. https://doi.org/10.1073/pnas.0712373105
Mendez JA, Bourque M-J, Fasano C, Kortleven C, Trudeau L-E (2011) Somatodendritic dopamine release requires synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J Biol Chem 286:23928–23937. https://doi.org/10.1074/jbc.M111.218032
Shin O-H, Rizo J, Südhof TC (2002) Synaptotagmin function in dense core vesicle exocytosis studied in cracked PC12 cells. Nat Neurosci 5:649–656. https://doi.org/10.1038/nn869
Zhao H, Ito Y, Chappel J, Andrews NW, Teitelbaum SL, Ross FP (2008) Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Dev Cell 14:914–925. https://doi.org/10.1016/j.devcel.2008.03.022
Czibener C, Sherer NM, Becker SM, Pypaert M, Hui E, Chapman ER, Mothes W, Andrews NW (2006) Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J Cell Biol 174:997–1007. https://doi.org/10.1083/jcb.200605004
Flannery AR, Czibener C, Andrews NW (2010) Palmitoylation-dependent association with CD63 targets the Ca2+ sensor synaptotagmin VII to lysosomes. J Cell Biol 191:599–613. https://doi.org/10.1083/jcb.201003021
Winther ÅME, Vorontsova O, Rees KA, Näreoja T, Sopova E, Jiao W, Shupliakov O (2015) An endocytic scaffolding protein together with synapsin regulates synaptic vesicle clustering in the Drosophila neuromuscular junction. J Neurosci 35:14756–14770. https://doi.org/10.1523/JNEUROSCI.1675-15.2015
Dornan S, Jackson AP, Gay NJ (1997) Alpha-adaptin, a marker for endocytosis, is expressed in complex patterns during Drosophila development. Mol Biol Cell 8:1391–1403. https://doi.org/10.1091/mbc.8.8.1391
Rodal AA, Motola-Barnes RN, Littleton JT (2008) Nervous wreck and Cdc42 cooperate to regulate endocytic actin assembly during synaptic growth. J Neurosci 28:8316–8325. https://doi.org/10.1523/JNEUROSCI.2304-08.2008
Koh T-W, Korolchuk VI, Wairkar YP, Jiao W, Evergren E, Pan H, Zhou Yi et al (2007) Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J Cell Biol 178:309–322. https://doi.org/10.1083/jcb.200701030
Gerth F, Jäpel M, Pechstein A, Kochlamazashvili G, Lehmann M, Puchkov D, Onofri F et al (2017) Intersectin associates with synapsin and regulates its nanoscale localization and function. Proc Natl Acad Sci USA 114:12057–12062. https://doi.org/10.1073/pnas.1715341114
Rodal AA, Blunk AD, Akbergenova Y, Jorquera RA, Buhl LK, Littleton JT (2011) A presynaptic endosomal trafficking pathway controls synaptic growth signaling. J Cell Biol 193:201–217. https://doi.org/10.1083/jcb.201009052
O’Connor-Giles KM, Ho LL, Ganetzky B (2008) Nervous wreck interacts with thick veins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron 58:507–518. https://doi.org/10.1016/j.neuron.2008.03.007
Wang Q-W, Si-Yao Lu, Liu Y-N, Chen Y, Wei H, Shen W, Chen Y-F et al (2020) Synaptotagmin-7 deficiency induces mania-like behavioral abnormalities through attenuating GluN2B activity. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.2016416117
Maximov A, Südhof TC (2005) Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48:547–554. https://doi.org/10.1016/j.neuron.2005.09.006
Luo F, Bacaj T, Südhof TC (2015) Synaptotagmin-7 Is essential for Ca2+-triggered delayed asynchronous release but not for Ca2+-dependent vesicle priming in retinal ribbon synapses. J Neurosci 35:11024–11033. https://doi.org/10.1523/JNEUROSCI.0759-15.2015
Weber JP, Toft-Bertelsen TL, Mohrmann R, Delgado-Martinez I, Sørensen JB (2014) Synaptotagmin-7 is an asynchronous calcium sensor for synaptic transmission in neurons expressing SNAP-23. PLoS ONE 9:e114033. https://doi.org/10.1371/journal.pone.0114033
Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC (2007) A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450:676–682. https://doi.org/10.1038/nature06308
MacDougall DD, Lin Z, Chon NL, Jackman SL, Lin H, Knight JD, Anantharam A (2018) The high-affinity calcium sensor synaptotagmin-7 serves multiple roles in regulated exocytosis. J General Physiol 150:783–807. https://doi.org/10.1085/jgp.201711944
Luo F, Südhof TC (2017) Synaptotagmin-7-mediated asynchronous release boosts high-fidelity synchronous transmission at a central synapse. Neuron 94:826-839.e3. https://doi.org/10.1016/j.neuron.2017.04.020
Durán E, Montes MÁ, Jemal I, Satterfield R, Young S, de Toledo GÁ (2018) Synaptotagmin-7 controls the size of the reserve and resting pools of synaptic vesicles in hippocampal neurons. Cell Calcium 74:53–60. https://doi.org/10.1016/j.ceca.2018.06.004
Jaiswal JK, Chakrabarti S, Andrews NW, Simon SM (2004) Synaptotagmin VII restricts fusion pore expansion during lysosomal exocytosis. PLoS Biol 2:E233. https://doi.org/10.1371/journal.pbio.0020233
Reddy A, Caler EV, Andrews NW (2001) Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106:157–169. https://doi.org/10.1016/s0092-8674(01)00421-4
Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW (2003) Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol 162:543–549. https://doi.org/10.1083/jcb.200305131
Barzilai-Tutsch H, Dewulf M, Lamaze C, Browne GB, Pines M, Halevy O (2018) A promotive effect for halofuginone on membrane repair and synaptotagmin-7 levels in muscle cells of dysferlin-null mice. Hum Mol Genet 27:2817–2829. https://doi.org/10.1093/hmg/ddy185
Tsuboi T, Fukuda M (2007) Synaptotagmin VII modulates the kinetics of dense-core vesicle exocytosis in PC12 cells. Genes Cells 12:511–519. https://doi.org/10.1111/j.1365-2443.2007.01070.x
Beed P, Ray S, Velasquez LM, Stumpf A, Parthier D, Swaminathan A, Nitzan N et al (2020) Species-specific differences in synaptic transmission and plasticity. Sci Rep 10:16557. https://doi.org/10.1038/s41598-020-73547-6
Virmani T, Han W, Liu X, Südhof TC, Kavalali ET (2003) Synaptotagmin 7 splice variants differentially regulate synaptic vesicle recycling. EMBO J 22:5347–5357. https://doi.org/10.1093/emboj/cdg514
Osborne SL, Herreros J, Bastiaens PI, Schiavo G (1999) Calcium-dependent oligomerization of synaptotagmins I and II. Synaptotagmins I and II are localized on the same synaptic vesicle and heterodimerize in the presence of calcium. J Biol Chem 274:59–66. https://doi.org/10.1074/jbc.274.1.59
Akbergenova Y, Bykhovskaia M (2010) Synapsin regulates vesicle organization and activity-dependent recycling at Drosophila motor boutons. Neuroscience 170:441–452. https://doi.org/10.1016/j.neuroscience.2010.07.021
Sun J, Bronk P, Liu X, Han W, Südhof TC (2006) Synapsins regulate use-dependent synaptic plasticity in the calyx of Held by a Ca2+/calmodulin-dependent pathway. Proc Natl Acad Sci USA 103:2880–2885. https://doi.org/10.1073/pnas.0511300103
Shupliakov O, Haucke V, Pechstein A (2011) How synapsin I may cluster synaptic vesicles. Semin Cell Dev Biol 22:393–399. https://doi.org/10.1016/j.semcdb.2011.07.006
Akbergenova Y, Bykhovskaia M (2007) Synapsin maintains the reserve vesicle pool and spatial segregation of the recycling pool in Drosophila presynaptic boutons. Brain Res 1178:52–64. https://doi.org/10.1016/j.brainres.2007.08.042
Fornasiero EF, Raimondi A, Guarnieri FC, Orlando M, Fesce R, Benfenati F, Valtorta F (2012) Synapsins contribute to the dynamic spatial organization of synaptic vesicles in an activity-dependent manner. J Neurosci 32:12214–12227. https://doi.org/10.1523/JNEUROSCI.1554-12.2012
Bykhovskaia M (2011) Synapsin regulation of vesicle organization and functional pools. Semin Cell Dev Biol 22:387–392. https://doi.org/10.1016/j.semcdb.2011.07.003
Gitler D, Cheng Q, Greengard P, Augustine GJ (2008) Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci 28:10835–10843. https://doi.org/10.1523/JNEUROSCI.0924-08.2008
Baldelli P, Fassio A, Valtorta F, Benfenati F (2007) Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci 27:13520–13531. https://doi.org/10.1523/JNEUROSCI.3151-07.2007
Humeau Y, Doussau F, Vitiello F, Greengard P, Benfenati F, Poulain B (2001) Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J Neurosci 21:4195–4206
Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ (1998) Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci 1:29–35. https://doi.org/10.1038/229
Ryan TA, Li L, Chin LS, Greengard P, Smith SJ (1996) Synaptic vesicle recycling in synapsin I knock-out mice. J Cell Biol 134:1219–1227. https://doi.org/10.1083/jcb.134.5.1219
Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC (1995) Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375:488–493. https://doi.org/10.1038/375488a0
Milovanovic D, Yumei Wu, Bian X, De Camilli P (2018) A liquid phase of synapsin and lipid vesicles. Science 361:604–607. https://doi.org/10.1126/science.aat5671
Hur JH, Lee S-H, Kim A-Y, Koh YH (2018) Regulation of synaptic architecture and synaptic vesicle pools by Nervous wreck at Drosophila Type 1b glutamatergic synapses. Exp Mol Med 50:e462. https://doi.org/10.1038/emm.2017.303
Bian X, Saheki Y, De Camilli P (2018) Ca2+ releases E-Syt1 autoinhibition to couple ER-plasma membrane tethering with lipid transport. EMBO J 37:219–234. https://doi.org/10.15252/embj.201797359
Idevall-Hagren O, Lü A, Xie B, De Camilli P (2015) Triggered Ca2+ influx is required for extended synaptotagmin 1-induced ER-plasma membrane tethering. EMBO J 34:2291–2305. https://doi.org/10.15252/embj.201591565
Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P (2013) PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 153:1494–1509. https://doi.org/10.1016/j.cell.2013.05.026
Saheki Y (2017) Endoplasmic reticulum—plasma membrane crosstalk mediated by the extended synaptotagmins. Adv Exp Med Biol 997:83–93. https://doi.org/10.1007/978-981-10-4567-7_6
Saheki Y, Bian X, Schauder CM, Sawaki Y, Surma MA, Klose C, Pincet F, Reinisch KM, De Camilli P (2016) Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat Cell Biol 18:504–515. https://doi.org/10.1038/ncb3339
Walter AM, Müller R, Tawfik B, Wierda KD, Pinheiro PS, Nadler A, McCarthy AW et al (2017) Phosphatidylinositol 4,5-bisphosphate optical uncaging potentiates exocytosis. eLife. https://doi.org/10.7554/eLife.30203
van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R (2012) Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem 287:16447–16453. https://doi.org/10.1074/jbc.M112.343418
Radhakrishnan A, Stein A, Jahn R, Fasshauer D (2009) The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J Biol Chem 284:25749–25760. https://doi.org/10.1074/jbc.M109.042499
Martin TFJ (2015) PI(4,5)P2-binding effector proteins for vesicle exocytosis. Biochem Biophys Acta 1851:785–793. https://doi.org/10.1016/j.bbalip.2014.09.017
Schiavo G, Gu QM, Prestwich GD, Söllner TH, Rothman JE (1996) Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc Natl Acad Sci USA 93:13327–13332. https://doi.org/10.1073/pnas.93.23.13327
Shin O-H, Jun Lu, Rhee J-S, Tomchick DR, Pang ZP, Wojcik SM, Camacho-Perez M et al (2010) Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol 17:280–288. https://doi.org/10.1038/nsmb.1758
Ullrich B, Li C, Zhang JZ, McMahon H, Anderson RG, Geppert M, Südhof TC (1994) Functional properties of multiple synaptotagmins in brain. Neuron 13:1281–1291. https://doi.org/10.1016/0896-6273(94)90415-4
Mizuta M, Inagaki N, Nemoto Y, Matsukura S, Takahashi M, Seino S (1994) Synaptotagmin III is a novel isoform of rat synaptotagmin expressed in endocrine and neuronal cells. J Biol Chem 269:11675–11678
von Poser C, Ichtchenko K, Shao X, Rizo J, Südhof TC (1997) The evolutionary pressure to inactivate. A subclass of synaptotagmins with an amino acid substitution that abolishes Ca2+ binding. J Biol Chem 272:14314–14319
Vician L, Lim IK, Ferguson G, Tocco G, Baudry M, Herschman HR (1995) Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. Proc Natl Acad Sci USA 92:2164–2168. https://doi.org/10.1073/pnas.92.6.2164
Ferguson GD, Vician L, Herschman HR (2001) Synaptotagmin IV: biochemistry, genetics, behavior, and possible links to human psychiatric disease. Mol Neurobiol 23:173–185. https://doi.org/10.1385/MN:23:2-3:173
Robinson IM, Ranjan R, Schwarz TL (2002) Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature 418:336–340. https://doi.org/10.1038/nature00915
Wang Z, Chapman ER (2010) Rat and Drosophila synaptotagmin 4 have opposite effects during SNARE-catalyzed membrane fusion. J Biol Chem 285:30759–30766. https://doi.org/10.1074/jbc.M110.137745
Kennedy MJ, Ehlers MD (2011) Mechanisms and function of dendritic exocytosis. Neuron 69:856–875. https://doi.org/10.1016/j.neuron.2011.02.032
Ehlers MD (2013) Dendritic trafficking for neuronal growth and plasticity. Biochem Soc Trans 41:1365–1382. https://doi.org/10.1042/BST20130081
Lledo PM, Zhang X, Südhof TC, Malenka RC, Nicoll RA (1998) Postsynaptic membrane fusion and long-term potentiation. Science 279:399–403. https://doi.org/10.1126/science.279.5349.399
Jurado S, Goswami D, Zhang Y, Miñano Molina AJ, Südhof TC, Malenka RC (2013) LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 77:542–558. https://doi.org/10.1016/j.neuron.2012.11.029
Ball RW, Warren-Paquin M, Tsurudome K, Liao EH, Elazzouzi F, Cavanagh C, An B-S, Wang T-T, White JH, Haghighi AP (2010) Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron 66:536–549. https://doi.org/10.1016/j.neuron.2010.04.011
Berke B, Wittnam J, McNeill E, Van Vactor DL, Keshishian H (2013) Retrograde BMP signaling at the synapse: a permissive signal for synapse maturation and activity-dependent plasticity. J Neurosci 33:17937–17950. https://doi.org/10.1523/JNEUROSCI.6075-11.2013
Piccioli ZD, Littleton JT (2014) Retrograde BMP signaling modulates rapid activity-dependent synaptic growth via presynaptic LIM kinase regulation of cofilin. J Neurosci 34:4371–4381. https://doi.org/10.1523/JNEUROSCI.4943-13.2014
McCabe BD, Guillermo Marqués A, Haghighi P, Fetter RD, Lisa Crotty M, Haerry TE, Goodman CS, O’Connor MB (2003) The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39:241–254. https://doi.org/10.1016/s0896-6273(03)00426-4
Budnik V, Zhong Y, Wu CF (1990) Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci 10:3754–3768
Guan Z, Saraswati S, Adolfsen B, Littleton JT (2005) Genome-wide transcriptional changes associated with enhanced activity in the Drosophila nervous system. Neuron 48:91–107. https://doi.org/10.1016/j.neuron.2005.08.036
Yoshihara M, Ueda A, Zhang D, Deitcher DL, Schwarz TL, Kidokoro Y (1999) Selective effects of neuronal-synaptobrevin mutations on transmitter release evoked by sustained versus transient Ca2+ increases and by cAMP. J Neurosci 19:2432–2441
Blanchette CR, Rodal AA (2020) Mechanisms for biogenesis and release of neuronal extracellular vesicles. Curr Opin Neurobiol 63:104–110. https://doi.org/10.1016/j.conb.2020.03.013
Budnik V, Ruiz-Cañada C, Wendler F (2016) Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17:160–172. https://doi.org/10.1038/nrn.2015.29
Mattaliano MD, Montana ES, Parisky KM, Troy Littleton J, Griffith LC (2007) The Drosophila ARC homolog regulates behavioral responses to starvation. Mol Cell Neurosci 36:211–221. https://doi.org/10.1016/j.mcn.2007.06.008
Ashley J, Cordy B, Lucia D, Fradkin LG, Budnik V, Thomson T (2018) Retrovirus-like Gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell 172:262-274.e11. https://doi.org/10.1016/j.cell.2017.12.022
Pastuzyn ED, Day CE, Kearns RB, Kyrke-Smith M, Taibi AV, McCormick J, Yoder N et al (2018) The neuronal gene arc encodes a repurposed retrotransposon Gag protein that mediates intercellular RNA transfer. Cell 172:275-288.e18. https://doi.org/10.1016/j.cell.2017.12.024
Shepherd JD (2018) Arc—an endogenous neuronal retrovirus? Semin Cell Dev Biol 77:73–78. https://doi.org/10.1016/j.semcdb.2017.09.029
Harris KP, Troy Littleton J, Stewart BA (2018) Postsynaptic syntaxin 4 negatively regulates the efficiency of neurotransmitter release. J Neurogenet 32:221–229. https://doi.org/10.1080/01677063.2018.1501372
Leschik J, Eckenstaler R, Endres T, Munsch T, Edelmann E, Richter K, Kobler O et al (2019) Prominent postsynaptic and dendritic exocytosis of endogenous BDNF vesicles in BDNF-GFP knock-in mice. Mol Neurobiol 56:6833–6855. https://doi.org/10.1007/s12035-019-1551-0
Madrigal MP, Portalés A, SanJuan MP, Jurado S (2018) Postsynaptic SNARE proteins: role in synaptic transmission and plasticity. Neuroscience. https://doi.org/10.1016/j.neuroscience.2018.11.012
Kennedy MJ, Davison IG, Robinson CG, Ehlers MD (2010) Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell 141:524–535. https://doi.org/10.1016/j.cell.2010.02.042
Tao HW, Poo M (2001) Retrograde signaling at central synapses. Proc Natl Acad Sci USA 98:11009–11015. https://doi.org/10.1073/pnas.191351698
Arendt KL, Zhang Y, Jurado S, Malenka RC, Südhof TC, Chen Lu (2015) Retinoic acid and LTP recruit postsynaptic AMPA receptors using distinct SNARE-dependent mechanisms. Neuron 86:442–456. https://doi.org/10.1016/j.neuron.2015.03.009
Jurado S (2014) The dendritic SNARE fusion machinery involved in AMPARs insertion during long-term potentiation. Front Cell Neurosci 8:407. https://doi.org/10.3389/fncel.2014.00407
Shimojo M, Madara J, Pankow S, Liu X, Yates J, Südhof TC, Maximov A (2019) Synaptotagmin-11 mediates a vesicle trafficking pathway that is essential for development and synaptic plasticity. Genes Dev 33:365–376. https://doi.org/10.1101/gad.320077.118
Bin N-R, Ma Ke, Harada H, Tien C-W, Bergin F, Sugita K, Luyben TT et al (2018) Crucial role of postsynaptic syntaxin 4 in mediating basal neurotransmission and synaptic plasticity in hippocampal CA1 neurons. Cell Rep 23:2955–2966. https://doi.org/10.1016/j.celrep.2018.05.026
Gu Yi, Huganir RL (2016) Identification of the SNARE complex mediating the exocytosis of NMDA receptors. Proc Natl Acad Sci USA 113:12280–12285. https://doi.org/10.1073/pnas.1614042113
Zhang G, Bai H, Zhang H, Dean C, Qiang Wu, Li J, Guariglia S, Meng Q, Cai D (2011) Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron 69:523–535. https://doi.org/10.1016/j.neuron.2010.12.036
Mohanasundaram P, Shanmugam MM (2010) Role of syntaxin 4 in activity-dependent exocytosis and synaptic plasticity in hippocampal neurons. Sci Signal 3:7. https://doi.org/10.1126/scisignal.3144jc7
López-Benito S, Sánchez-Sánchez J, Brito V, Calvo L, Lisa S, Torres-Valle M, Palko ME et al (2018) Regulation of BDNF Release by ARMS/Kidins220 through Modulation of Synaptotagmin-IV Levels. J Neurosci 38:5415–5428. https://doi.org/10.1523/JNEUROSCI.1653-17.2018
Arthur CP, Dean C, Pagratis M, Chapman ER, Stowell MHB (2010) Loss of synaptotagmin IV results in a reduction in synaptic vesicles and a distortion of the Golgi structure in cultured hippocampal neurons. Neuroscience 167:135–142. https://doi.org/10.1016/j.neuroscience.2010.01.056
Bharat V, Siebrecht M, Burk K, Ahmed S, Reissner C, Kohansal-Nodehi M, Steubler V, Zweckstetter M, Ting JT, Dean C (2017) Capture of dense core vesicles at synapses by JNK-dependent phosphorylation of synaptotagmin-4. Cell Rep 21:2118–2133. https://doi.org/10.1016/j.celrep.2017.10.084
Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED et al (2012) BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol 196:775–788. https://doi.org/10.1083/jcb.201201038
Marell PS, Blohowiak SE, Evans MD, Georgieff MK, Kling PJ, Tran PV (2019) Cord blood-derived exosomal CNTN2 and BDNF: potential molecular markers for brain health of neonates at risk for iron deficiency. Nutrients. https://doi.org/10.3390/nu11102478
Mustapic M, Eitan E, Werner JK, Berkowitz ST, Lazaropoulos MP, Tran J, Goetzl EJ, Kapogiannis D (2017) Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci 11:278. https://doi.org/10.3389/fnins.2017.00278
Diesner M, Predel R, Neupert S (2018) Neuropeptide mapping of dimmed cells of adult drosophila brain. J Am Soc Mass Spectrom 29:890–902. https://doi.org/10.1007/s13361-017-1870-1
Luo J, Liu Y, Nässel DR (2017) Transcriptional reorganization of Drosophila motor neurons and their muscular junctions toward a neuroendocrine phenotype by the bHLH protein dimmed. Front Mol Neurosci 10:260. https://doi.org/10.3389/fnmol.2017.00260
Hadžić T, Park D, Abruzzi KC, Yang L, Trigg JS, Rohs R, Rosbash M, Taghert PH (2015) Genome-wide features of neuroendocrine regulation in Drosophila by the basic helix-loop-helix transcription factor DIMMED. Nucleic Acids Res 43:2199–2215. https://doi.org/10.1093/nar/gku1377
Maximov A, Shin O-H, Liu X, Südhof TC (2007) Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol 176:113–124. https://doi.org/10.1083/jcb.200607021
Kaeser-Woo YJ, Younts TJ, Yang X, Zhou P, Dick Wu, Castillo PE, Südhof TC (2013) Synaptotagmin-12 phosphorylation by cAMP-dependent protein kinase is essential for hippocampal mossy fiber LTP. J Neurosci 33:9769–9780. https://doi.org/10.1523/JNEUROSCI.5814-12.2013
Doi H, Yoshida K, Yasuda T, Fukuda M, Fukuda Y, Morita H, Ikeda S-I et al (2011) Exome sequencing reveals a homozygous SYT14 mutation in adult-onset, autosomal-recessive spinocerebellar ataxia with psychomotor retardation. Am J Hum Genet 89:320–327. https://doi.org/10.1016/j.ajhg.2011.07.012
Montes-Chinea NI, Guan Z, Coutts M, Vidal C, Courel S, Rebelo AP, Abreu L, Stephan Zuchner J, Littleton T, Saporta MA (2018) Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype. Neurol Genet 4:e282. https://doi.org/10.1212/NXG.0000000000000282
Mironovich O, Dadali E, Malmberg S, Markova T, Ryzhkova O, Poliakov A (2020) Identification of a novel de novo variant in the SYT2 gene causing a rare type of distal hereditary motor neuropathy. Genes. https://doi.org/10.3390/genes11111238
Lorenzoni PJ, Scola RH, Kay CSK, Werneck LC, Horvath R, Lochmüller H (2018) How to Spot congenital myasthenic syndromes resembling the lambert-eaton myasthenic syndrome? A brief review of clinical, electrophysiological, and genetics features. NeuroMol Med 20:205–214. https://doi.org/10.1007/s12017-018-8490-1
Donkervoort S, Mohassel P, Laugwitz L, Zaki MS, Kamsteeg E-J, Maroofian R, Chao KR et al (2020) Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome. Am J Med Genet Part A 182:2272–2283. https://doi.org/10.1002/ajmg.a.61765
Baker K, Gordon SL, Melland H, Bumbak F, Scott DJ, Jiang TJ, Owen D et al (2018) SYT1-associated neurodevelopmental disorder: a case series. Brain J Neurol 141:2576–2591. https://doi.org/10.1093/brain/awy209
Baker K, Gordon SL, Grozeva D, van Kogelenberg M, Roberts NY, Pike M, Blair E et al (2015) Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J Clin Investig 125:1670–1678. https://doi.org/10.1172/JCI79765
Shields MC, Bowers MR, Fulcer MM, Bollig MK, Rock PJ, Sutton BR, Vrailas-Mortimer AD et al (2017) Drosophila studies support a role for a presynaptic synaptotagmin mutation in a human congenital myasthenic syndrome. PLoS ONE 12:e0184817. https://doi.org/10.1371/journal.pone.0184817
Bradberry MM, Courtney NA, Dominguez MJ, Lofquist SM, Knox AT, Bryan Sutton R, Chapman ER (2020) Molecular basis for synaptotagmin-1-associated neurodevelopmental disorder. Neuron 107:52-64.e7. https://doi.org/10.1016/j.neuron.2020.04.003
Shen W, Wang Q-W, Liu Y-N, Marchetto MC, Linker S, Si-Yao Lu, Chen Y et al (2020) Synaptotagmin-7 is a key factor for bipolar-like behavioral abnormalities in mice. Proc Natl Acad Sci USA 117:4392–4399. https://doi.org/10.1073/pnas.1918165117
Barthet G, Jordà-Siquier T, Rumi-Masante J, Bernadou F, Müller U, Mulle C (2018) Presenilin-mediated cleavage of APP regulates synaptotagmin-7 and presynaptic plasticity. Nat Commun 9:4780. https://doi.org/10.1038/s41467-018-06813-x
Wang C, Kang X, Zhou Li, Chai Z, Qihui Wu, Huang R, Huadong Xu et al (2018) Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson’s disease-like pathology. Nat Commun 9:81. https://doi.org/10.1038/s41467-017-02593-y
Huynh DP, Scoles DR, Nguyen D, Pulst SM (2003) The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum Mol Genet 12:2587–2597. https://doi.org/10.1093/hmg/ddg269
Inoue S, Imamura A, Okazaki Y, Yokota H, Arai M, Hayashi N, Furukawa A, Itokawa M, Oishi M (2007) Synaptotagmin XI as a candidate gene for susceptibility to schizophrenia. Am J Med Genet Part B Neuropsychiatr Genet 144B:332–340. https://doi.org/10.1002/ajmg.b.30465
Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC (2016) The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun 7:11803. https://doi.org/10.1038/ncomms11803
Quintero-Rivera F, Chan A, Donovan DJ, Gusella JF, Ligon AH (2007) Disruption of a synaptotagmin (SYT14) associated with neurodevelopmental abnormalities. Am J Med Genet Part A 143A:558–563. https://doi.org/10.1002/ajmg.a.31618
Shariful IM, Nolte H, Jacob W, Ziegler AB, Pütz S, Grosjean Y, Szczepanowska K et al (2016) Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum Mol Genet 25:5365–5382. https://doi.org/10.1093/hmg/ddw352
Rodin DI, Schwarzman AL, Sarantseva SV (2015) Expression of human amyloid precursor protein in Drosophila melanogaster nerve cells causes a decrease in presynaptic gene mRNA levels. Genet Mol Res 14:9225–9232. https://doi.org/10.4238/2015.August.10.2
Machamer JB, Woolums BM, Fuller GG, Lloyd TE (2018) FUS causes synaptic hyperexcitability in Drosophila dendritic arborization neurons. Brain Res 1693:55–66. https://doi.org/10.1016/j.brainres.2018.03.037
Bonifacino T, Musazzi L, Milanese M, Seguini M, Marte A, Gallia E, Cattaneo L, Onofri F, Popoli M, Bonanno G (2016) Altered mechanisms underlying the abnormal glutamate release in amyotrophic lateral sclerosis at a pre-symptomatic stage of the disease. Neurobiol Dis 95:122–133. https://doi.org/10.1016/j.nbd.2016.07.011
Nizzardo M, Taiana M, Rizzo F, Aguila Benitez J, Nijssen J, Allodi I, Melzi V et al (2020) Synaptotagmin 13 is neuroprotective across motor neuron diseases. Acta Neuropathol 139:837–853. https://doi.org/10.1007/s00401-020-02133-x
Shi Z, Zhang K, Zhou H, Jiang L, Xie B, Wang R, Xia W et al (2020) Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer’s Disease. Aging Cell 19:e13125. https://doi.org/10.1111/acel.13125
Bereczki E, Branca RM, Francis PT, Pereira JB, Baek J-H, Hortobágyi T, Winblad B, Ballard C, Lehtiö J, Aarsland D (2018) Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain J Neurol 141:582–595. https://doi.org/10.1093/brain/awx352
Öhrfelt A, Brinkmalm A, Dumurgier J, Brinkmalm G, Hansson O, Zetterberg H, Bouaziz-Amar E, Hugon J, Paquet C, Blennow K (2016) The pre-synaptic vesicle protein synaptotagmin is a novel biomarker for Alzheimer’s disease. Alzheimer’s Res Therapy 8:41. https://doi.org/10.1186/s13195-016-0208-8
Laßek M, Weingarten J, Acker-Palmer A, Bajjalieh SM, Muller U, Volknandt W (2014) Amyloid precursor protein knockout diminishes synaptic vesicle proteins at the presynaptic active zone in mouse brain. Curr Alzheimer Res 11:971–980. https://doi.org/10.2174/1567205011666141107152458
Tratnjek L, Zivin M, Glavan G (2013) Up-regulation of Synaptotagmin IV within amyloid plaque-associated dystrophic neurons in Tg2576 mouse model of Alzheimer’s disease. Croatian Med J 54:419–428. https://doi.org/10.3325/cmj.2013.54.419
Glavan G, Schliebs R, Zivin M (2009) Synaptotagmins in neurodegeneration. Anat Rec 292:1849–1862. https://doi.org/10.1002/ar.21026
Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Kaye J, Manczak M (2005) Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimer’s Dis 7:103–117 (discussion 173)
Kabayama H, Tokushige N, Takeuchi M, Kabayama M, Fukuda M, Mikoshiba K (2017) Parkin promotes proteasomal degradation of synaptotagmin IV by accelerating polyubiquitination. Mol Cell Neurosci 80:89–99. https://doi.org/10.1016/j.mcn.2017.02.006
Leader DP, Krause SA, Pandit A, Davies SA, Dow JAT (2018) FlyAtlas 2: a new version of the Drosophila melanogaster expression atlas with RNA-Seq, miRNA-Seq and sex-specific data. Nucleic Acids Res 46:D809–D815. https://doi.org/10.1093/nar/gkx976
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MQF and JTL wrote and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Quiñones-Frías, M.C., Littleton, J.T. Function of Drosophila Synaptotagmins in membrane trafficking at synapses. Cell. Mol. Life Sci. 78, 4335–4364 (2021). https://doi.org/10.1007/s00018-021-03788-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03788-9