
Abstract
Diabetes affects millions of people worldwide. This devastating disease dramatically increases the risk of developing cardiovascular disorders. A hallmark metabolic abnormality in diabetes is hyperglycemia, which contributes to the pathogenesis of cardiovascular complications. These cardiovascular complications are, at least in part, related to hyperglycemia-induced molecular and cellular changes in the cells making up blood vessels. Whereas the mechanisms mediating endothelial dysfunction during hyperglycemia have been extensively examined, much less is known about how hyperglycemia impacts vascular smooth muscle function. Vascular smooth muscle function is exquisitely regulated by many ion channels, including several members of the potassium (K+) channel superfamily and voltage-gated L-type Ca2+ channels. Modulation of vascular smooth muscle ion channels function by hyperglycemia is emerging as a key contributor to vascular dysfunction in diabetes. In this review, we summarize the current understanding of how diabetic hyperglycemia modulates the activity of these ion channels in vascular smooth muscle. We examine underlying mechanisms, general properties, and physiological relevance in the context of myogenic tone and vascular reactivity.
Similar content being viewed by others

Introduction
According to recent reports by the World Health Organization and the American Diabetes Association, diabetes is a prevalent health problem reaching epidemic proportions [1, 2]. Diabetes is a multifactorial metabolic disorder characterized by the inability of the body to produce, secrete, and/or use the hormone insulin, which regulates glucose homeostasis [3,4,5]. As a consequence of this insulin dysfunction, the concentration of extracellular glucose is significantly elevated, poorly controlled or both. Elevated extracellular glucose level or hyperglycemia, is a hallmark metabolic abnormality in diabetes and a major risk factor for cardiovascular diseases, including heart attacks, hypertension, and stroke [3, 5,6,7,8]. Hyperglycemia is also associated with decreased cognitive function, retinopathy and nephropathy [9]. These pathological conditions have in common a link between hyperglycemia and alterations in small-resistance artery function [10, 11].
Small-resistance arteries and arterioles (< 150 μm in diameter) are composed of connective tissue, endothelial cells lining the vessel lumen, and smooth muscle cells wrapping around the vessel. These blood vessels are critical for nutrient delivery as well as for control of blood flow, tissue perfusion, and total peripheral resistance. Small-resistance arteries respond to increases in intravascular pressure by constricting, a process known as the myogenic or vascular tone [12]. Myogenic tone is influenced by the sympathetic system and neurohumoral vasoactive signals that induce constriction or dilation to maintain the moment-to-moment control of blood flow and tissue perfusion, which is critical for organ function [13]. The myogenic tone is an intrinsic property of vascular smooth muscle cells [14,15,16]. These cells receive and integrate many inputs, including variations in intraluminal pressure, as well as vasoconstrictor and vasodilatory queues from endothelial cells and nerve terminals innervating the vessels, to regulate their function [14]. These inputs help fine-tune vascular smooth muscle excitability and contractile state, and thus have major influence over arterial diameter, myogenic tone and vascular reactivity.
Vascular smooth muscle excitability and contractile state is modulated by Rho-associated kinase (ROK) dependent mechanisms [17, 18], and the activity of several ion channels [14, 19]. Agonists- and pressure-mediated activation of ROK are involved in Ca2+ sensitization of vascular smooth muscle contraction via inhibition of myosin light chain phosphatase and induction of actin polymerization [17, 18, 20]. In addition, the integrated activity of various ion channels expressed in vascular smooth muscle facilitates the control of membrane potential (VM) and the magnitude of intracellular Ca2+ concentration ([Ca2+]i) [14, 19]. Voltage-gated (KV), Ca2+-activated (BKCa) and ATP-sensitive (KATP) K+ channels, ryanodine receptors (RyR), IP3 receptors (IP3R), and voltage-gated Ca2+ channels are critical for regulation of VM and [Ca2+]i [14]. Thus, changes in the functional expression of these channels may alter vascular smooth muscle function and significantly impact the regulation of myogenic tone and vascular reactivity during metabolic challenges and pathological states, such as hyperglycemia and diabetes.
Impaired endothelial function is a well-recognized early modification contributing to altered myogenic tone during diabetic hyperglycemia (see recent comprehensive reviews [3, 7, 21,22,23,24]), although the role of endothelial ion channels is less clear. Thus, we thought it important to provide an overview of our current understanding of how ion channels in endothelial cells are impacted by hyperglycemia. There is also increasing appreciation that alterations in vascular smooth muscle function may contribute to diabetic vasculopathies. Particularly, changes in the functional expression of ion channels in vascular smooth muscle during diabetic hyperglycemia, the mechanisms underlying those changes, and their functional implications have received much attention in recent years [3, 14, 25,26,27,28,29,30,31,32,33,34,35]. The main goal of this review is to examine preceding and emerging data on cellular and molecular mechanisms by which hyperglycemia alters the function of vascular smooth muscle ion channels. This is important because ion channels are (1) major regulators of vascular smooth muscle function, myogenic tone and vascular reactivity, (2) key targets for many drugs, and (3) the mechanisms by which they are modulated by hyperglycemia may reveal new opportunities for therapeutic intervention. We will focus our discussion on KV/BKCa/KATP K+ channels, RyR, IP3R and voltage-gated L-type CaV1.2 channels as the impact of hyperglycemia on their expression and function has been well characterized. We will describe general properties of these ion channels, underlying mechanisms by which hyperglycemia and diabetes alter their function and the patho-physiological relevance in the context of myogenic tone and vascular reactivity.
Myogenic tone and diabetic hyperglycemia
Accumulating evidence over the last 30 years has clearly established abnormal regulation of basal myogenic tone in both diabetic patients and different animal models of diabetes [3, 14, 18, 25,26,27,28,29, 32, 35,36,37,38]. Note that several animal models of diabetes have been developed to better understand diabetic complications, including vasculopathies [39, 40]. The advantages and drawbacks of the different models have been extensively described elsewhere [39, 40]. A comprehensive and integrative understanding of mechanisms underlying abnormal myogenic tone during diabetes has been challenging to reconcile. This is because studies have described either no change [35, 36, 41, 42], reduced [29, 43,44,45], or enhanced [31, 32, 37, 46,47,48,49,50] tone in both diabetic patients and animal models of diabetes (see Table 1 for details).
In studies where myogenic tone was not different between non-diabetic and diabetic arteries, impaired arterial function in response to a given vasodilatory stimulus was still observed [35, 36, 41, 42]. In studies in which reduced basal myogenic tone during diabetes was observed, changes were correlated with decreased Ca2+ sensitization of the contractile machinery in smooth muscle [29, 43,44,45]. Accordingly, using cerebral vascular smooth muscle from Goto–Kakizaki (type-2 diabetes model) rats, reduced basal myogenic tone during diabetes was correlated with a decrease in focal adhesion kinase (FAK) autophosphorylation downstream of integrins [20, 45]. This breakdown in pressure-mediated FAK autophosphorylation appears to impair ROK signaling involved in myosin light chain phosphatase inhibition and actin polymerization [20, 45]. Intriguingly, a recent independent study using human internal mammary arteries from non-diabetic and diabetic patients found increased expression of contractile markers, as well as enhanced phosphorylation of proteins associated with ROK signaling activation and actin polymerization [51]. Although this study did not evaluate myogenic tone, one may speculate that human internal mammary arteries from diabetic patients may be either more constricted or hyperreactive to contractile agonists. Consistent with this possibility, a number of studies have shown ROK-mediated hyperreactivity of vascular smooth muscle cells in response to contractile stimuli in animal models of diabetes [48, 52, 53].
Conversely, studies showing increased myogenic tone in diabetes have described a number of mechanisms that include changes in functional expression of ion channels and signaling molecules controlling smooth muscle contractile state [31, 32, 37, 46,47,48,49,50]. The description of various mechanisms could result from the use of different species, animal models of diabetes, vascular beds and the severity of diabetes and hyperglycemic state. Intriguingly, a longitudinal study found distinct myogenic tone impairment with age during diabetes [54]. The study described no changes in myogenic tone in mesenteric arteries from dB/dB (type-2 diabetes model) mice compared to wild type control mice at 8 weeks of age [54]. However, myogenic tone was significantly elevated at 12 and 16 weeks of age [54]. These results were recapitulated in an independent study, which also demonstrated that the enhanced myogenic constriction in dB/dB mesenteric arteries was independent of endothelial influence [37], thus directly suggesting impairment of vascular smooth muscle function in diabetes. Moreover, although sex differences are well apparent in the context of diabetes [55, 56], how myogenic tone can be influenced by biological sex during this pathological condition is understudied. A recent study using STZ-treated Sprague Dawley rats found a significant change in the NO and endothelium-dependent hyperpolarizing factor (EDHF) signaling response in female mesenteric arteries compared to males [57], but no mechanisms or examination of effects on myogenic tone were provided. Together with the studies described above, these results suggest distinct myogenic/contractile responses during diabetes that may be influenced by age, biological sex, species, animal model of diabetes, vascular beds and degree of pathological state. These may be key confounding factors that should be taken into consideration when evaluating myogenic response during diabetes. Certainly, parallels and key information can be extracted from all these studies, which may be useful in identifying novel targets that could be exploited for improving the treatment of diabetic vasculopathy.
Similar to diabetes, the effects of acute and/or short-term hyperglycemia on myogenic tone are highly dependent on species, vascular bed, and the concentration of external glucose used (see Table 2 for details). Short-term glucose effects in vascular smooth muscle could also be influenced by sex, yet most studies have been performed using male tissue, thus highlighting the need to consider this biological variable in future experiments. Nonetheless, in posterior cerebral arteries from Wistar rats, equilibration of pressurized arteries in 44 mM d-glucose caused a reduction in myogenic tone over a range of pressures, compared to arteries equilibrated in 5 mM d-glucose [58]. The acute glucose effects on myogenic tone were prevented in endothelium-denuded arteries [58], thus indicating a role for an unknown endothelium-mediated vasodilatory signal. The mechanisms underlying the inhibition of myogenic tone during hyperglycemia in this model, however, are unknown. Conversely, in mesenteric arteries from Wistar rat, acute equilibration of pressurized arteries in 44 mM d-glucose did not change basal myogenic tone compared to control (5.5 mM d-glucose) [34]. However, myogenic tone in arteries exposed to 44 mM d-glucose was significantly elevated in response to activation of protein kinase C (PKC) or high K+ compared to controls [34]. These results suggest that elevated glucose potentiate responses that promote vascular smooth muscle contraction, such as PKC activation and membrane depolarization. In Sprague Dawley rat ophthalmic arteries, a glucose concentration-dependent effect was reported [43]. Accordingly, whereas 25 mM d-glucose significantly increased myogenic tone over a range of intraluminal pressures, 40 mM d-glucose had the opposite effect compared to control conditions (e.g. ~ 4 mM d-glucose) [43]. These differences were not observed in experiments using equimolar concentrations of mannitol and were abrogated in endothelial-denuded arteries [43]. These results suggest that endothelium-mediated signals drive glucose-induced differential changes in myogenic tone in rat ophthalmic arteries; the identity and mechanisms by which they act remain to be established. In C57BL6/J wild type mouse pressurized cerebral arteries, an acute elevation in external d-glucose from 10 to 20 mM caused robust constriction and increased tone [31, 32, 59]. Glucose-induced increased cerebral arterial tone in these studies was independent of endothelial function as a similar response was observed in endothelium-denuded arteries [31]. The mechanism involves activation of a novel signaling pathway resulting in regulation of ion channels and increased [Ca2+]i in vascular smooth muscle (see extended description in sections below) [31, 32, 59]. Similarly, acute elevations in glucose from 4 to 14 mM d-glucose cause a robust constriction of rat parenchymal arterioles from cortical brain slices [60]. This glucose-induced constriction was linked to inhibition of KV channels via a PKC-mediated mechanism [60]. These results suggest distinct regulation of myogenic tone in response to hyperglycemia in different vascular beds, animal models and hyperglycemic state. Given the importance of ion channels in both vascular endothelial and smooth muscle in controlling the myogenic response [14, 61], it is likely that their altered functional expression may contribute to changes in vascular response during diabetic hyperglycemia. In the next sections, we describe studies that have made direct links between changes in ion channels expression and function and changes in myogenic tone and vascular reactivity.
Endothelial ion channels and diabetic hyperglycemia
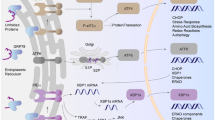
As cells lining the lumen of vessels, vascular endothelial cells are directly exposed to changes in blood glucose concentration. Glucose uptake in endothelial cells is mediated via the insulin-independent glucose transporter Glut1 [62, 63]. Therefore, high glucose levels in the external milieu may result in high glucose concentrations inside endothelial cells. This apparent lack of autoregulation could render endothelial cells more vulnerable to conditions, such as intermittent and chronic hyperglycemic states, which are known to alter endothelial function in different diabetic animal models and humans with diabetes [64,65,66]. In the context of myogenic tone regulation, endothelial dysfunction denotes the inability of the endothelium to promote vasodilation. Endothelial function is, at least in part, governed by a network of ion channels expressed in endothelial cells [61, 67]. These include (but not limited to) several types of Ca2+-activated K+ channels, a number of transient receptor potential (TRP) channels, store-operated channels and mitochondrial channels. These endothelial ion channels facilitate and/or promote the transmission of electrical signals and release of vasoactive signals to control vascular smooth muscle excitability and contractile state [61, 67]. Surprisingly, there is a limited understanding of how changes in endothelial ion channel functional expression contribute to endothelial dysfunction and impaired vasodilation during diabetic hyperglycemia. Only a handful of studies have addressed this issue, likely because of the difficulty in isolating native endothelial cells [68]. Figure 1 provides a summary of our current understanding of how diabetic hyperglycemia impacts endothelial ion channels.

Schematic of mechanisms by which diabetic hyperglycemia (HG) alters endothelial ion channels and vascular smooth muscle excitability. Diabetic hyperglycemia distinctively modulates the activity of various ion channels in endothelial cells to alter ROS production, intracellular Ca2+ concentration and other signaling pathways that impair endothelium-dependent regulation of vascular smooth muscle cells. ?? denotes unknown mechanisms by which HG alters a particular target. Red dotted lines denotes reduced effects. MEP myoendothelial projections. Arrows = activation, lines with bars = inhibition
Endothelial TRP channels and diabetic hyperglycemia
Endothelial cells express several members of the TRP superfamily of cationic channels, including surface membrane TRPC (canonical), TRPV (vanilloid), and TRPM (melastatin) channels [69, 70]. Among them, TRPV4 channels have emerged as key regulators of endothelial cell function and modulators of myogenic tone and vascular reactivity due to their prominent Ca2+ permeability [61, 70, 71]. These channels localize to the myoendothelial projections (sections of close interaction between endothelial and smooth muscle cells) [72] where they can physically and functionally interact with small (KCa2.X) and intermediate (KCa3.X) conductance, Ca2+-activated K+ channels (SK and IK, respectively) in vessels from many species, including humans [71, 73, 74]. Activation of SK/IK channels by Ca2+ influx via TRPV4 channels is thought to induce endothelial cell membrane hyperpolarization and vasodilation [71]. It is suspected that this could be a generalized mechanism controlling vascular tone and vasodilation in many blood vessels [71, 74,75,76,77].
A recent study found that exposing primary culture bovine retinal endothelial cells to 25 mM external d-glucose for 72 h causes a significant reduction in the expression and function of TRPV4 channels compared to control conditions (e.g. 5 mM external d-glucose in the same cells) [78]. This change in glucose-induced downregulation of TRPV4 functional expression was not observed when cells were incubated with equimolar concentrations of mannitol, ruling out osmotic effects [78]. A reduction in TRPV4 expression was also described in diabetic rat retinal arterioles from a type-1 diabetic model (streptozotocin—STZ) [78], and rat primary cultured mesenteric endothelial cells [74]. A similar decrease in SK channel expression was also observed in mesenteric endothelial cells from STZ-treated rats [74]. This reduction in TRPV4 and SK channel expression may contribute to impair endothelium membrane hyperpolarization that is typically transmitted to the adjacent smooth muscle cells via gap junctions at the myoendothelial projections or alter the release of vasodilatory factors. Consistent with this, downregulation of both TRPV4 and SK channel was correlated with impaired acetylcholine-induced relaxation in mesenteric arteries from STZ mice [74]. Intriguingly, a recent study found reduced interaction (rather than decreased expression) between TRPV4 and SK channels in endothelial cells from hypertensive patients and animal models of hypertension that correlated with impaired vasodilation [79]. The impaired vasodilation was corrected in animals treated with JNc-440, a small molecule that enhanced the interaction between TRPV4 and SK channels in mesenteric endothelial cells [79]. Whether this structural change in TRPV4 and SK channel interaction also occurs in diabetic endothelial cells and if it could be rectified with the use of the JNc-440 to improve endothelial function during diabetes are exciting possibilities. Moreover, it will be important to define the mechanisms by which hyperglycemia alters TRPV4 and SK channel expression, distribution and interaction, and whether TRPV4 and SK channel function are actually altered in native endothelial cells leading to endothelial dysfunction.
TRPM7, TRPC1 and TRPV1 are also expressed in endothelial cells [69, 70], and their role in these cells has been studied in the context of diabetic hyperglycemia [80,81,82]. An upregulation in the expression of TRPM7 in response to high glucose (30 mM d-glucose) incubation for 72 h was described in human umbilical vein endothelial cells (HUVEC) [80]. Increased TRPM7 expression was correlated with enhanced reactive oxygen species (ROS) signaling and cytotoxicity [80]. However, the mechanisms underlying glucose-induced TRPM7 upregulation, their link to endothelial cell injury and how these changes may influence vascular tone are unknown. In bovine aortic endothelial cells, TRPC1, but not TRPC3, TRPC4 and TRPC6, expression was significantly upregulated when cells were cultured in 25 mM d-glucose for 72 h [81]. These changes were associated with sustained Ca2+ entry in endothelial cell. Yet, the physiological implications of this change and the mechanisms mediating glucose-induced TRPC1 upregulation have not been established. Conversely, culture of porcine ileal endothelial cells in 30 mM external d-glucose for 12 h resulted in a significant reduction in the expression of TRPV1 channels that was correlated with increased ROS production, reduced NO synthesis, and impaired acetylcholine-induced vasodilation [82]. Similar results were observed in dB/dB mice [82]. Intriguingly, all of these glucose-induced changes were prevented/ameliorated by capsaicin, a well-known TRPV1 agonist [82]. The capsaicin effects were mediated by increased functional expression of the uncoupling protein 2 (UPC2) [82], an antioxidant protein that regulates ROS production [83]. These results raise the possibility that the use of dietary capsaicin, via its action on endothelial TRPV1 channels, could be considered to improve endothelial function, including vasodilation, during diabetic hyperglycemia. In summary, diabetic hyperglycemia may have distinct effects on endothelial TRP channels that contribute to altering endothelial function.
Endothelial MCU and diabetic hyperglycemia
The mitochondrial Ca2+ uniporter (MCU) located in the inner mitochondrial membrane is a key ion channel mediating Ca2+ uptake in this organelle [84]. MCU function can modulate [Ca2+]i and redox signaling [84], which can have profound implications for myogenic tone and vascular reactivity regulation. A recent study described a significant augmentation in MCU expression in HUVEC cells upon increasing concentrations of external d-glucose from 5 to 30 mM for 72 h [85]. Glucose-induced MCU upregulation was correlated with increased cytosolic and mitochondrial Ca2+ as well as enhanced ROS and mitochondrial superoxide production [85]. These changes were prevented in the presence of an MCU inhibitor [85]. Thus, data suggest that changes in MCU function could contribute to alter endothelial Ca2+ and ROS signaling during diabetic hyperglycemia. Yet, further studies are necessary to convincingly establish functional implications of these hyperglycemia-induced changes in endothelial MCU function in the context of myogenic tone and vascular reactivity regulation.
Endothelial store-operated channels and diabetic hyperglycemia
Store-operated Ca2+ entry (SOCE) is a mechanism leading to sustained Ca2+ entry due to activation of surface membrane receptors and subsequent reduction of Ca2+ concentration in the sarco/endoplasmic reticulum (S/ER) [86]. The underlying proteins for SOCE in many cells, including endothelial cells, have been identified as the S/ER Ca2+ concentration ([Ca2+]S/ER) sensor STIM and the surface membrane Ca2+ channel Orai [87, 88]. It is now recognized that a reduction in [Ca2+]S/ER promotes the dynamic clustering of STIM to stimulate the activity of Orai channels leading to SOCE [87, 88]. Endothelial cells express both STIM and Orai channels [89]. Although the role of SOCE mediated by STIM and Orai in endothelial cells is not well established, both proteins have been associated with proliferation, migration and angiogenesis [89]. Additionally, a role for STIM and Orai in mechanisms underlying endothelial dysfunction and vascular complications is emerging [90]. In the context of hyperglycemia, a significant increase in SOCE has been reported in HUVEC cultured for 96 h in 30 mM d-glucose compared to control (e.g. 5 mM d-glucose) [91]. Similar results were reported in independent experiments using endothelial cells from different vascular beds and species exposed to 25 mM d-glucose for 72 h, as well as samples from type-2 diabetic patients and animal models of diabetes, specifically the Akita mouse [92]. The increase in SOCE by elevated glucose was shown to be mediated by ROS signaling [91], and/or by a distinct upregulation in the expression (not increase store-operated channel activation) of the different STIM and Orai isoforms expressed in endothelial cells [92]. However, the impact of STIM and Orai over-expression and increased SOCE on endothelial-dependent vasodilation is unclear. One possibility is that increased SOCE during diabetic hyperglycemia stimulates the activity of the Ca2+-dependent phosphatase calcineurin to regulate gene expression via activation of the transcription factor nuclear factor of activated T cells (NFAT) [92]. Consistent with this possibility, a recent study found elevated NFAT signaling in the Akita diabetic mice, and NFAT inhibition improved endothelial function in this animal model [93]. Thus, NFAT-dependent changes in gene expression could contribute to the alteration of endothelial function, including endothelium-dependent vasodilation. Future studies should further expand on these and other intriguing possibilities.
Further considerations
Endothelial cells express many other ion channels that are critical for their functional regulation. Examples include IP3R that are involved in myoendothelial projection Ca2+ signals triggering vasodilation [94], inward rectifying K+ (Kir) channels that boost vasodilation upon activation [95, 96], and TRPA1 channels that contribute to Ca2+ signaling in the myoendothelial projections to promote vasodilation in response to ROS signaling [97]. How these channels are impacted by hyperglycemia is unclear. The accumulation of ROS producing advanced glycation end products (AGEs) is an additional element contributing to endothelial dysfunction during prolonged diabetic hyperglycemia [98, 99]. AGE is a biochemical modification triggered in response to elevated glucose levels that attaches to different structures and proteins to modify their function [100]. AGEs bind to different cell surface receptors, including the receptor for advanced glycation end products (RAGE) [100]. The AGE/RAGE complex can trigger activation of several pathways that modulate myriad cellular events including endocytosis, cell signaling and gene expression [100]. Intriguingly, recent findings suggest that acute AGEs exposure impairs Ca2+ signaling in endothelial cells [101]. Ca2+ signaling in endothelial cells are important for their function as well as myogenic tone and vascular reactivity regulation [61]. Thus, AGE-mediated alterations in endothelial Ca2+ signaling may play a fundamental role in the development of endothelial dysfunction during diabetic hyperglycemia. Yet, the underlying mechanisms by which AGEs impair Ca2+ signaling in endothelial cells are not well understood. Thus, these knowledge gaps could be further explored in future research. In summary, the functional expression of several endothelial ion channels is altered by diabetic hyperglycemia via multiple mechanisms, and this may contribute to endothelial dysfunction.
Vascular smooth muscle ion channels and diabetic hyperglycemia
Vascular smooth muscle excitability and contractile state is governed by a sophisticated interplay between different ionic conductances [14, 19, 102]. Among them, KV, BKCa and KATP channels, RyR, IP3R and voltage-gated Ca2+ channels are critical for regulation of VM and [Ca2+]i [14]. Interest in the mechanisms by which glucose mediates changes in vascular smooth muscle function have heightened in recent years. This is because initial studies examining the effects of hyperglycemia described a significant increase in [Ca2+]i in several vascular beds, including rat tail arteries [103], and mouse cerebral arteries [31, 32, 59]. In addition, accumulating epidemiological and functional studies also suggest vascular smooth muscle dysfunction during diabetic hyperglycemia [3, 14, 25,26,27,28,29,30,31,32,33,34]. Changes in ion channels functional expression are likely to contribute to alterations in vascular smooth muscle function during diabetic hyperglycemia. In the next sections, we will discuss how ion channels are modulated by diabetic hyperglycemia and the implications for vascular smooth muscle function.
Effects of hyperglycemia on L-type Ca2+ channels
L-type Ca2+ channels are comprised of a pore-forming CaV1.2 α1C subunit and auxiliary β3 (predominant isoform in vascular smooth muscle) [104], α2δ and γ subunits that modulate channel function [105]. As stated above, Ca2+ influx via these channels plays a prominent role in vascular smooth muscle contraction and the level of the myogenic tone in resistance arteries [14, 19]. Therefore, changes in their expression and/or function could significantly influence vascular smooth muscle excitability.
In the context of diabetic hyperglycemia, although there are reports indicating diminished or unaltered L-type Ca2+ channel activity [106,107,108,109,110], the majority of the studies report a significant increase in channel activity in different experimental diabetic models and stages of hyperglycemia [31, 32, 48, 59, 111,112,113,114,115]. These differences are likely a reflection of the use of different vascular beds, cultured versus freshly isolated cells, and presumably the magnitude and duration of the hyperglycemic state. Moreover, the discrepancies in L-type Ca2+ channel activity could be associated to the specific animal model and concomitant metabolic abnormalities, such as hypercholesterolemia [116, 117] and hyperinsulinemia, [118] which could affect channel function. Table 3 summarizes the effects of diabetic hyperglycemia on vascular smooth muscle L-type Ca2+ channels.
A recent series of studies systematically and comprehensively explored the immediate effects of acute elevations in extracellular glucose on vascular L-type Ca2+ channel activity [31, 32, 59, 112, 115]. These studies have consistently shown that application of elevated glucose (20 mM d-glucose) caused a robust increase in the whole-cell L-type Ca2+ current density and in the frequency of localized persistent Ca2+ influx events via L-type Ca2+ channels (persistent CaV1.2 sparklets [119]) in freshly dissociated human adipose, rat and mouse cerebral, and mouse mesenteric and femoral vascular smooth muscle [31, 32, 59, 112, 115]. The increase in the frequency of persistent CaV1.2 sparklets is important because this gating modality can significantly amplify Ca2+ influx and therefore promote vascular smooth muscle contraction [120, 121].
In one study, glucose-induced potentiation of L-type Ca2+ channel activity was prevented in WKY rat cerebral smooth muscle incubated in the presence of Salidroside, an active ingredient in Rhodiola rosea [115]. This compound has broad bioactive effects that can regulate inflammation, apoptosis, and the production of ROS signaling [122]. Considering that ROS is increased by elevated glucose [123], and ROS signaling can modulate L-type Ca2+ channel activity [124,125,126], it is tempting to speculate that glucose-induced ROS contributes to potentiation of L-type Ca2+ channels and that salidroside prevents this change in vascular smooth muscle by inhibiting ROS production. In additional independent studies, elevated glucose effects on the potentiation of L-type Ca2+ channel activity were found to require glucose transport into the cell and metabolization [31, 59, 112]. Indeed, application of the nonpermeable mannitol or non-metabolizable L-glucose had no effect on channel activity (Fig. 2). Moreover, vascular smooth muscle cells pre-incubated with indinavir, a broad glucose transporter inhibitor, ablated the elevated glucose-induced potentiation of L-type Ca2+ channel activity [31], which is consistent with the idea that facilitative glucose transport may contribute to modulating vascular reactivity [127]. Using cerebral arteries as a model system, glucose-induced potentiation of L-type Ca2+ channels was correlated with enhanced myogenic tone through an endothelium-independent mechanism [31, 32, 59]. Similar acute glucose-induced changes in myogenic tone have been observed in mesenteric arteries [128]. These results highlight the physiological relevance of the glucose effects on vascular smooth muscle L-type Ca2+ channels. Moreover, results raise a need to understand the mechanisms by which glucose regulate L-type Ca2+ channel activity in vascular smooth muscle and perhaps other excitable and non-excitable cells.

Schematic of a model for regulation of L-type Ca2+ channels and K+ channels during hyperglycemia and diabetes. In this model, the extent of Ca2+ influx via L-type Ca2+ channels play a key role in regulating excitation–contraction and excitation-transcription coupling. Under normoglycemic conditions, K+ channels, including KV1.X, KV2.X and BKCa channels, provide negative feedback control of pressure-induced membrane depolarization to constrain the activity of L-type Ca2+ channels, reduce global [Ca2+]i and limit vascular smooth muscle contraction (see Ref. [19] for original work describing this concept). L-type Ca2+ channels are closely associated with signaling proteins, such as PKA, PP2B and AC5, through their scaffold by AKAP5. During hyperglycemic conditions, glucose is transported into the cells via a glucose transporter (GLUT). Inside the cell, metabolized glucose results in the production of nucleotides (NUC), such as ATP and UTP. These NUC are released to the extracellular space where they activate purinergic receptors coupled to Gs proteins (i.e. P2Y11). Activation of P2Y11 promotes AC5 activity and localized cAMP production. This cAMP microdomain can enable a pool of PKA that is intimately associated with L-type Ca2+ channels to increase CaV1.2 phosphorylation at S1928, which will potentiate channel activity. Hyperactive L-type Ca2+ channels result in increased global [Ca2+]i and contraction of vascular smooth muscle. The global increase in [Ca2+]i also activates AKAP5-anchored PP2B resulting in dephosphorylation of the transcription factor NFATc3, which allows its nuclear translocation. Once in the nucleus, NFATc3 can regulate the expression of many genes, including K+ channels. Data have shown that glucose-induced NFATc3 activation leads to the selective suppression KV2.1 and BKCa β1 subunits. The reduction in the expression of these subunits decreases KV currents and BKCa channel function, thus impairing the negative feedback membrane potential hyperpolarization. Ultimately these changes cause membrane potential depolarization, further Ca2+ influx via L-type Ca2+ channels and vascular smooth muscle contraction, thus creating a feedforward mechanism that could perpetuate the system under chronic hyperglycemic conditions. Green lines represent activation pathways and red lines represent inhibition pathways. Δψ = change in membrane potential. Model based on work from Refs. [31, 32, 59, 112, 165, 200, 210].
How does glucose stimulate vascular L-type Ca2+ channel activity?
Increased PKC activity in vascular smooth muscle has been extensively reported during diabetic hyperglycemia [129,130,131,132]. PKC activity is also known to be required for persistent vascular L-type Ca2+ channel events as well as for increased channel activity during other pathological conditions, such as hypertension [119, 133,134,135,136,137]. Thus, it was reasonable to hypothesize that enhanced PKC activity during an elevated glucose challenge would underlie enhanced L-type Ca2+ channel activity. Yet, when using vascular smooth muscle cells from PKC knockout mice, it was found that glucose was still able to stimulate L-type Ca2+ channel activity [112]. Rather, glucose-induced potentiation of L-type Ca2+ channels was prevented in wild type and PKC knockout cells that were pre-treated with a protein kinase A (PKA) inhibitor [112]. Moreover, glucose-induced constriction was also prevented in pressurized arteries exposed to a PKA inhibitor [31]. These results suggest that glucose-induced vascular L-type Ca2+ channel potentiation leading to vasoconstriction is mediated by PKA activity (Fig. 2) [31, 112]. These observations were unexpected as a role for PKA in modulating vascular L-type Ca2+ channels has not been previously established [138]. Moreover, PKA activity has been usually linked to relaxation of vascular smooth muscle, and therefore vasodilation [139,140,141,142,143]. The observation that glucose mediates vascular L-type Ca2+ channel potentiation, vascular smooth muscle contraction and vasoconstriction via a PKA-dependent pathway could be reconciled if elevated glucose is predominantly stimulating a subpopulation of PKA molecules near L-type Ca2+ channels while avoiding the activation of other PKA pools associated with signaling pathways underlying vascular smooth muscle relaxation. In other words, hyperglycemia would mainly activate PKA molecules that are compartmentalized with L-type Ca2+ channels, thus facilitating their potentiation.
Compartmentalization of proteins like PKA can be facilitated by scaffold proteins, such as A-kinase anchoring proteins (AKAPs). In doing so, AKAPs fine-tune signal transduction in multiple cells by positioning signaling generators, regulatory enzymes, and effector proteins in close proximity to a specific substrate [144,145,146]. The AKAP5 isoform (murine AKAP150 and human AKAP79) interacts with adenylyl cyclase (AC), PKA, PKC and CaV1.2 [147,148,149,150,151,152]. Therefore, this scaffold could mediate compartmentalization of PKA and the glucose-induced, PKA-mediated stimulation of vascular L-type Ca2+ channels. Consistent with this possibility, super-resolution localization maps generated with ground state depletion microscopy, and complementary proximity ligation assay confirmed close spatial association between PKA and CaV1.2 in native human adipose and mouse cerebral vascular smooth muscle cells that were dependent on expression of AKAP5 [31]. Indeed, it was found that genetic depletion of AKAP5 increased the distance between pools of PKA and CaV1.2 clusters and prevented the stimulation of L-type Ca2+ channels by glucose in mouse cerebral smooth muscle [31]. Moreover, the use of genetically modified mice expressing an AKAP5 that could not bind PKA or PKC confirmed an essential role for compartmentalized PKA, but not compartmentalized PKC, in mediating the glucose effects on L-type Ca2+ channel activity and cerebral artery reactivity [31]. Together with prior studies on the role of PKA in vascular smooth muscle relaxation [139,140,141,142,143], findings suggest unappreciated mechanisms by which PKA may differentially regulate the contractile state of these cells (Fig. 2).
AKAP5-anchored PKA regulation of L-type Ca2+ channel activity and vascular reactivity upon increased glucose was found to be mediated by direct phosphorylation of the pore-forming CaV1.2 subunit at serine 1928 (S1928) (Fig. 2) [31]. This amino acid residue is a highly conserved PKA phosphorylation site [153], which has been linked with PKA-dependent regulation of L-type Ca2+ channels in neurons and cardiac cells [151, 154,155,156,157]. In cardiac myocytes, however, the functional relevance of S1928 phosphorylation is unclear [158,159,160], as PKA regulation of cardiac L-type Ca2+ channels was normal in cells from knockin mice with disrupted phosphorylation of S1928 (e.g. S1928A mice) [158] or 21 other putative PKA sites [159]. In stark contrast, S1928 plays a key role on AKAP5-anchored PKA-dependent regulation of vascular CaV1.2 [31]. Accordingly, ablation of AKAP5 or disruption of the interaction between AKAP5 and PKA prevented glucose-induced increase in S1928 phosphorylation [31]. Elevated glucose failed to potentiate L-type Ca2+ channel activity and increase global [Ca2+]i in vascular smooth muscle from S1928A mice [31]. In addition, no glucose-induced constriction was observed in S1928A cerebral arteries [31]. The complete loss of glucose effects on L-type Ca2+ channel activity and vascular reactivity just by the modification of a single CaV1.2 amino acid was surprising since elevated glucose levels may also suppress the function of K+ channels (see below) [60, 128, 161, 162], which will depolarize vascular smooth muscle membrane potential leading to an increase in the open probability of L-type Ca2+ channels and an increase in global [Ca2+]i [14, 163]. Using mathematical modeling that facilitates quantification of the relative role of many elements that interact nonlinearly to regulate arterial myocyte excitability [164], it was predicted that preventing the potentiation of L-type Ca2+ channels in a similar manner as that observed in S1928A experiments would prevent/ameliorate changes in global [Ca2+]i despite concomitant modifications in K+ channel function and corresponding membrane depolarization [165]. Indeed, these predictions were confirmed experimentally [31, 165]. These results confirmed a predominant role for CaV1.2 S1928 phosphorylation as a primary mechanism underlying [Ca2+]i and vasoconstriction in response to elevated glucose. Altogether, these findings uncovered an important molecular link by which increased CaV1.2 S1928 phosphorylation triggers the potentiation of vascular L-type Ca2+ channels leading to vasoconstriction upon glucose-induced AKAP5-anchored PKA activation. Whether CaV1.2 S1928 phosphorylation may be relevant for vascular L-type Ca2+ channel regulation by other stimuli remains to be examined. Findings also highlight an unappreciated dichotomy in S1928 regulation between cardiac and vascular CaV1.2 that suggest unique tissue-specific regulation. We speculate that this differential S1928 regulation of cardiac and vascular CaV1.2 may be associated with the specific expression of different splice variants, distinct phosphorylation profiles, post-translational modifications of the CaV1.2 subunit and interacting partners or unique interactome profile. Indeed, a recent report discovered that PKA-dependent CaV1.2 regulation in the heart was mediated by Rad, a small Ras-like G protein, through an indirect interaction via the accessory CaV β subunit [166]. Whether Rad regulates CaV1.2 in vascular smooth muscle and during diabetic hyperglycemia remains to be established.
How does glucose activate PKA to stimulate vascular L-type Ca2+ channel activity?
The observation that elevated glucose stimulates vascular smooth muscle L-type Ca2+ channel activity via a PKA dependent pathway raised an important question: How does glucose activate PKA? The classic molecular machinery stimulating activity of an effector protein, such as PKA implicates upstream production of second messengers [e.g. cyclic adenosine monophosphate (cAMP)], as well as activation of regulatory enzymes (e.g. AC) and signaling generators [e.g. G protein-coupled receptors (GPCRs)] [144, 145]. Uncovering the identities of proteins that are activated in response to elevated glucose is important as it may identify novel signaling pathways and additional mechanisms for signaling compartmentalization that could help explain the intriguing effects of glucose-induced PKA activity on vascular smooth muscle contractility.
Glucose has been shown to stimulate cAMP synthesis in yeast and murine pancreatic β cells [167, 168], thus raising the possibility that a similar glucose-induced cAMP production could be observed in vascular smooth muscle. cAMP signaling has been extensively examined in vascular smooth muscle using conventional biochemical approaches [169]. However, these approaches do not provide spatiotemporal information about cAMP signaling that is critical for cellular regulation [170], including those in vascular smooth muscle [169]. To overcome this issue, investigators are beginning to express different FRET biosensors capable of measuring cAMP nanodomains in cultured vascular smooth muscle [32, 59, 171, 172]. One such FRET biosensors is the Epac1-camps-based FRET sensor typically known as ICUE3 [173]. The ICUE3 sensor can be targeted to different organelles and subcellular compartments [170], thus facilitating examination of cAMP signaling spatiotemporal dynamics in response to physiological and pathological stimuli, such as elevated glucose. Accordingly, using a membrane-targeted ICUE3 (but not nucleus-targeted ICUE3) biosensor expressed in unpassaged mouse aortic vascular smooth muscle cells, it was found that elevated glucose was able to induce a subtle, yet significant increase in cAMP synthesis that was further amplified by the broad AC activator forskolin [32, 59]. These results suggested localized cAMP synthesis upon elevated glucose, which may be essential for activation of a PKA pool that could trigger contraction rather than relaxation (Fig. 2). Consistent with this view, glucose caused contraction of vascular smooth muscle and vasoconstriction, while forskolin, which will engage all PKA pools, led to relaxation of vascular smooth muscle and vasodilation [31, 32].
The production of cAMP is mediated by AC isoforms. Nine membrane-bound AC isoforms have been identified [174]. In vascular smooth muscle, AC3, AC5 and AC6 are abundantly expressed [172, 175, 176]. AC6 and, to an extent, AC3 have been linked with β adrenergic regulation of the activity of various K+ channels that contributes to relaxation of vascular smooth muscle [172, 175]. Unsurprisingly, alterations in AC signaling has been reported in vascular smooth muscle upon elevated glucose and diabetes, which may contribute to cell proliferation, increased oxidative stress, impaired relaxation, and enhanced expression of contractile genes [51, 177,178,179]. Accordingly, basal AC activity was reduced in cultured A10 cell lines and rat aorta exposed to 26 mM d-glucose for 3–4 days, compared to control (5.5 d-glucose) [177]. The glucose-induced reduction in AC activity was subsequently associated with ROS production, particularly superoxide anion (O2−), as treatment with antioxidants prevented the changes in AC function [178]. In additional independent experiments, a reduction in AC6 expression was observed in rat mesenteric arteries [179]. The functional implications for this glucose-induced reduction in AC activity were mostly linked to changes in vascular smooth muscle proliferation and migration [129], although there is recent evidence of altered agonist-induced relaxation in isolated mesenteric artery rings [179]. Important for mechanisms underlying glucose activation of PKA, it was found that AC5 activity, but not AC6, was necessary for glucose-induced cAMP synthesis, L-type Ca2+ channel stimulation and vasoconstriction in mouse cerebral vascular smooth muscle/arteries (Fig. 2) [32]. Mathematical modeling and subsequent experimental validation confirmed that genetic ablation of AC5 normalized global [Ca2+]i and vascular reactivity in response to elevated glucose [32]. These result suggest a detrimental role for AC5 in vascular smooth muscle upon elevated glucose. Intriguingly, results are similar to those observed in S1928A vascular smooth muscle cells [31, 165]. We thus speculate that in the absence of AC5 activity, cAMP/PKA-mediated phosphorylation of CaV1.2 S1928 upon elevated glucose is unlikely to occur. It can be also speculated that AC5 and AC6/AC3 may have distinctive, yet critical roles in vascular smooth muscle function. Moreover, the specific activation of AC5 leading to localized AC5-dependent cAMP synthesis can provide two extra layers of compartmentalization that could facilitate PKA-dependent regulation of vascular smooth muscle excitability. Thus, results in this section highlight how changes in AC activity may modulate activation of different molecular pathways to alter vascular smooth muscle function in response to diabetic hyperglycemia.
AC signaling generation is triggered by the activation of Gs protein-coupled receptors (GsPCR) [145]. Within the vasculature, elevated glucose can stimulate autocrine release of nucleotides (e.g. ATP, UTP) into the extracellular space [59, 180, 181]. This can then activate P2Y receptors in vascular smooth muscle leading to increased [Ca2+]i and changes in vascular smooth muscle contractile state [181,182,183,184]. While purinergic regulation of tone has been mostly associated with activation of Gq/i-linked P2Y2/4/6 receptors [185], the P2Y11 is the only purinergic receptor coupled to Gs, which can stimulate AC/PKA activity [186, 187]. P2Y11 receptors, however, have also been shown to hetero-oligomerize with P2Y1 and P2Y6 receptors and this structural arrangement can modulate P2Y11 activity and the underlying cellular response [188,189,190,191,192]. Thus, a role for P2Y11 in glucose-induced cAMP synthesis leading to PKA activation could be masked by the influence of other P2Y receptors.
In comprehensive experiments that address the aforementioned possibility, the expression for this purinergic receptor and its close proximity to PKA were confirmed in freshly dissociated human adipose vascular smooth muscle cells [59]. In these cells, the ICUE3 sensor revealed that elevated glucose and the selective P2Y11 agonist NF546 stimulated cAMP production to about the same magnitude, even when both stimuli were applied simultaneously [59]. The glucose/NF546-induced production of cAMP was blocked by the selective P2Y11 antagonist NF340 but not by selective inhibitors of P2Y1 and P2Y6 [59]. Moreover, results correlated with PKA-dependent enhancement of S1928 phosphorylation and L-type Ca2+ current potentiation by elevated glucose and NF546 [59]. These effects were blocked by the P2Y11, but not P2Y1 and P2Y6, antagonist [59]. These results are relevant because they suggest a key role for P2Y11 receptor function in mediating the elevated glucose effects in vascular smooth muscle that is independent of their potential hetero-oligomerization with P2Y1 or P2Y6 receptors. Comparable findings were observed using a robust approach in mouse cerebral artery vascular smooth muscle. Even though the P2Y11 gene has not been found at the expected location in the mouse genome, there has been recent rodent annotations for the P2Y11 gene (e.g. XM_008766009.2 and XM_0130655917.2) and a growing number of studies using pharmacological approaches that hint at the presence of at least a P2Y11-like receptor in mice [193]. Therefore, a P2Y11-like receptor may underlie the glucose response in murine vascular smooth muscle. Altogether, these findings indicate a role for P2Y11/P2Y11-like receptors as the signaling generators triggering the localized cAMP synthesis upon elevated glucose (Fig. 2). This glucose-induced local cAMP production can then activate a specific pool of PKA near CaV1.2 to regulate L-type Ca2+ channel activity and vascular reactivity. Moreover, although a partnership between P2Y11 and AC5 remains to be established, we propose the assembly of a previously unrecognized macromolecular signaling complex formed by P2Y11, AC5, AKAP5-anchored PKA and CaV1.2 (Fig. 2) [31, 32, 59, 112]. The formation and activation of this macromolecular complex could mediate glucose signaling in vascular smooth muscle and perhaps other excitable and non-excitable cells, which may have broad clinical and therapeutic implications.
Effects of hyperglycemia on K+ channels
K+ channels regulate vascular smooth muscle membrane potential and therefore has a major impact on the control of [Ca2+]i, myogenic tone and vascular reactivity [14]. Activation of K+ channels relaxes vascular smooth muscle, whereas their inhibition leads to contraction [194]. Vascular smooth muscle cells express a number of isoforms from various types of K+ channels, including KV, BKCa and KATP. In this section, we discuss the effects of hyperglycemia on the function of KV, BKCa and KATP channels and the physiological implications (Fig. 3). Table 4 provides a summary of different studies showing alterations in K+ channels activity and potential underlying mechanisms in response to diabetic hyperglycemia.

Schematic of mechanisms by which diabetic hyperglycemia (HG) alters vascular smooth muscle K+ channels. Multiple pathways have been described by which diabetic hyperglycemia can modulate the activity of K+ channels. Impaired K+ channels function has a significant impact on intracellular Ca2+ levels and contractile state of vascular smooth muscle, which will modulate arterial diameter and neurovascular coupling. Arrows = activation, lines with bars = inhibition, double question marks denotes unknown mechanisms by which HG alters a particular target, and Δψ = change in membrane potential
K V channels
Vascular smooth muscle cells express a wide variety of KV subunits, including KV1 (KV1.1, KV1.2, KV1.3, KV1.5, KV1.6), KV2 (KV2.1), KV7 (KV 7.1–5) and member of the silent KV subunits (KV9.3) [14, 195,196,197,198]. It has long been recognized that the function of several of these KV subunits can be distinctively modified by acute and short-term chronic hyperglycemia [14, 60, 128, 161, 162, 199,200,201,202]. Acute (5–10 min) elevations in glucose from 4–5 mM d-glucose to 15–20 mM d-glucose was found to inhibit KV channel activity in vascular smooth muscle from rat mesenteric arteries and cerebral arterioles [60, 128, 162]. Acute glucose-mediated inhibition of KV channels appears to involve PKC signaling [60, 162], specifically those associated with PKCα and PKCβ isoforms (Fig. 3) [128]. Glucose-induced KV channel inhibition was subsequently correlated with vascular smooth muscle membrane depolarization and increased vasoconstriction (or myogenic tone) in rat mesenteric arteries (ex vivo) and cerebral arteriole (in situ) [60, 128, 162]. KCl− and agonist-induced vasoconstriction were also elevated in rat mesenteric arteries [128]. Moreover, acute changes in extracellular glucose concentration were related to immediate in vivo alterations in neurovascular coupling [60], which is the modification of local perfusion due to changes in neuronal activity [203]. Glucose could modulate neurovascular coupling either by direct effects in the vasculature via mechanisms described above and/or by altering astrocytic function. However, how glucose stimulates PKC signaling, how PKC ultimately inhibits KV channels and the mechanisms by which elevated glucose could impair neurovascular coupling are either unclear or unknown, thus opening new lines of research.
Chronic hyperglycemia (≥ 24 h incubation) has also been shown to cause a reduction in KV channel activity in vascular smooth muscle [161, 199,200,201,202, 204]. Studies have found a decrease in KV current density in rat coronary vascular smooth muscle incubated for 24 h in 23 mM d-glucose [161]. Glucose-induced reduction in KV channel function was shown to be mediated by superoxide and peroxynitrite production with no apparent change in the expression of KV1.2 and KV1.5 subunits (other KV subunits were not examined) [201, 202]. In independent experiments where rat coronary intact arteries rather than isolated vascular smooth muscle were incubated for 48 h in 23 mM d-glucose, a reduction in KV function was also observed [199]. This change in KV function was correlated with downregulation in the expression of KV1.2 and KV1.5 mRNA and protein levels [199]. The decreased functional expression of KV1.2/1.5 in response to elevated glucose was found to be mediated by the production of AGE and specifically the activation of RAGE, as inhibiting RAGE with an anti-RAGE antibody prevented both the reduction in KV1.2/1.5 protein levels and KV current component (Fig. 3) [199]. How glucose-induced AGE/RAGE signaling leads to decreased functional expression of KV1.2/1.5-mediated KV currents is currently unclear. Interestingly, AGE activity has been correlated with the stimulation of PKC in different diabetic settings [51, 205, 206]. Thus, it could be speculated that glucose-induced AGE signaling may trigger activation of PKC to regulate functional expression of KV subunits during diabetic hyperglycemia. In additional experiments using vascular smooth muscle from rat coronary arteries incubated for 20 h in 30 mM d-glucose, the reduction in KV channel function was also associated, at least in part, with a reduction in KV7 subunit functional expression [204]. The mechanisms for impaired KV7 function were correlated with downregulation of KCNQ1 and KCNQ2 expression [204]. Finally, in vascular smooth muscle from cerebral arteries incubated for 48 h in 20 mM d-glucose, a reduction in KV current density was associated with selective downregulation of the KV2 subunit mRNA and protein levels via a mechanism that requires anchoring of the phosphatase calcineurin by AKAP5 (see further discussion below) (Fig. 2) [200]. These glucose-mediated changes in KV function did not seem to alter resting vessel diameter, but rather impair vasoactive responses that could proceed via engagement of KV channels in some studies [201, 202]. In others, the reduction in KV function was correlated with increased vascular smooth muscle hypercontractility and enhanced myogenic tone [200, 204]. Altogether, results suggest that alterations in KV1.X, KV2.X and KV7.X subunit functional expression via various mechanisms during hyperglycemia may contribute to impair vascular smooth muscle function, myogenic tone and vascular reactivity. How acute and chronic hyperglycemia may affect other KV subunits is unclear, thus opening opportunities for additional research.
BK Ca channels
BKCa channels are abundantly expressed in vascular smooth muscle cells where they provide negative tonic feedback regulation of myogenic tone [14]. They are composed of a pore-forming α subunit and accessory β1 and γ subunits, which regulate Ca2+ sensitivity of the channel [14]. BKCa channels can be activated by membrane depolarization and localized increases in intracellular Ca2+ signals known as Ca2+ sparks [14, 105, 207]. This Ca2+ signals are produced by the opening of ryanodine receptors (RyR) located in the membrane of the sarcoplasmic reticulum [14, 105, 208]. Intriguingly, acute elevations in extracellular glucose do not seem to affect BKCa channel function in HEK cells expressing the pore-forming α subunit [209]. However, similar experiments examining the acute effects of elevated glucose in native vascular smooth muscle cells appear to be missing.
On the other hand, incubation of vascular smooth muscle in elevated glucose for as short as 48 h seems to induce a significant reduction in the activity of BKCa channels (see Table 4) [209, 210]. Accordingly, chronic incubation (72 h) of rat coronary vascular smooth muscle in 22 mM d-glucose resulted in a reduction in BKCa channel activity [209]. This glucose-induced inhibition of BKCa channel activity was reported to be mediated by increased oxidation of the BKCa α subunit at cysteine 911 by hydrogen peroxide (Fig. 3 and Table 4) [209]. In mouse cerebral vascular smooth muscle, a reduction in BKCa channel activity during chronic glucose incubation (20 mM d-glucose for 48 h) stemmed from decreased Ca2+ sensitivity due to downregulation of the BKCa β1 subunit function (Figs. 2, 3) [210]. BKCa β1 downregulation during chronic hyperglycemia could be the result of increased protein degradation and/or impaired transcriptional expression of the subunit [210, 211]. Regardless of the mechanism, downregulation of BKCa β1 expression leads to a reduction in BKCa channel activity, which is likely to alter vascular smooth muscle function during chronic diabetic hyperglycemia. Intriguingly, no change in BKCa β1 expression was observed in cultured mouse aortic smooth muscle cells incubated for one week in 25 mM d-glucose compared to control conditions (5.5 mM d-glucose) [51]. The differences are likely due to the use of different vascular beds and culturing conditions. Together, results suggest that short-term chronic exposure of vascular smooth muscle to elevated glucose may alter the functional expression of BKCa channel subunits. Yet, the physiological implications of these changes remain to be established.
K ATP channels
KATP channels are octameric protein complexes containing 4 pore-forming Kir6.X subunits and 4 accessory sulfonylurea receptor (SUR), likely SUR2B, subunits [14, 212]. These channels are expressed in vascular smooth muscle where they can be regulated by many signaling pathways and molecules, including ATP and ADP [14, 212, 213]. Indeed, whereas elevations in ADP concentration activate the channel, increases in intracellular ATP concentration inhibit KATP channel function. KATP channels can also be regulated by ROS signaling and PKC/PKA-mediated pathways [195]. Therefore, KATP channels can couple vascular smooth muscle metabolic state to electrical activity to regulate cell excitability, myogenic tone and vascular reactivity [14, 212, 213]. Given that hyperglycemia is likely to alter the cellular metabolic state of vascular smooth muscle, it is then reasonable to speculate that KATP channel activity can be modulated by acute and short-term elevations in extracellular glucose. Surprisingly, however, a study using rat aorta smooth muscle cells found that 24 h elevations in extracellular glucose from 5.5 to 23 mM d-glucose had no effect on KATP channel activity as measured using single-channel recordings (Table 4) [214]. This study also reported that 23 mM d-glucose impaired the activation of KATP channels by the anesthetic isoflurane [214], which is known to induce dilation of blood vessels, at least in part, by activating these channels [215, 216]. Although this work did not examine the mechanisms by which elevated glucose impaired the isoflurane effects on KATP channels, subsequent studies using short-term incubation of human omental artery preparations in elevated glucose suggest that enhanced ROS signaling may be involved [217,218,219,220]. Accordingly, incubating endothelium-denuded human omental arteries for at least 60 min in 20 mM d-glucose was sufficient to increase the production of superoxide, likely via a mechanism mediated by activation of PKC and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling [219, 220]. This excessive superoxide production prevents/reduces endothelium-denuded human omental artery relaxation (as measured using isometric force transducers) in response to application of the KATP agonist levcromakalim [217,218,219,220]. Consistent with these observations, inhibition of PKC and PI3K/Akt signaling or treatment with a synthetic peroxisome proliferator-activated receptor γ (PPARγ) agonist that may act as an antioxidant, reduced glucose-induced superoxide production and prevented the impairment in human omental artery relaxation in response to levcromakalim [217,218,219,220]. These results suggest, at least indirectly, that elevations in extracellular glucose could impair agonist-mediated activation of KATP channels in vascular smooth muscle and vasorelaxation. Whether acute and/or short-term elevations in glucose alter basal KATP channel activity and how this may impact myogenic tone remains unclear. The consequences of glucose-induced alterations in KATP channel activity are likely to be tissue-specific, as KATP channels contribute to the regulation of basal myogenic tone in coronary and skeletal muscle arteries but not in other vascular beds [14]. Further studies revealing the effects of acute and short-term glucose exposure on KATP channel function should attempt to directly measure the activity of KATP channels and consequences on myogenic tone in different vascular beds.
Effects of hyperglycemia on sarcoplasmic reticulum ion channels
Vascular smooth muscle cells express a number of sarcoplasmic reticulum (SR) Ca2+ release ion channels, including RyR and IP3R [14, 105]. These ion channels are instrumental in regulating vascular smooth muscle function.
In vascular smooth muscle, RyRs mediate the localized release of Ca2+ from the SR in the form of a Ca2+ spark [14, 105, 208]. RyRs in the SR are in close spatial proximity to BKCa channels located at the surface membrane [14, 105, 208]. This is crucial as RyR-mediated Ca2+ sparks are critical for activation of BKCa channels to produce spontaneous transient outward currents (STOCs), which hyperpolarize vascular smooth muscle leading to relaxation [207, 221]. Thus, changes in the activity of RyRs in response to acute and/or short-term hyperglycemia may have significant consequences on vascular smooth muscle function. Surprisingly, how acute and/or short-term elevations in extracellular glucose impact RyR function, including its coupling to BKCa channels, in vascular smooth muscle has not been extensively and rigorously examined. A single study has reported that RyR expression may be elevated in cultured A7r5 cells exposed for a minimum of 10 h in 75 mM d-glucose, compared to 25 mM d-glucose [222]. On the other hand, IP3Rs mediate localized (e.g. Ca2+ puffs) but also cell-wide (e.g. Ca2+ waves) Ca2+ signals that help regulate vascular smooth muscle function [14, 105, 223, 224]. Like RyRs, changes in IP3R function due to acute and/or short-term hyperglycemia may alter vascular smooth muscle function, but studies looking at this are also limited. In a comparative study examining IP3R expression in cultured A7r5 cells exposed to 25 or 75 mM d-glucose for at least 10 h, downregulation of IP3R protein levels was observed in the higher glucose concentration condition [222]. Conversely, another independent study found no changes in IP3R protein expression in cultured A7r5 cells incubated between 3 and 28 days in 5.5 mM and 25 mM d-glucose [225]. The reasons for the disparities between these studies are unclear. Nevertheless, the studies described above for both RyR and IP3R highlight major limitations with the use of culturing cells and supraphysiological glucose conditions that may confound interpretation of results. Moreover, given that the mechanisms underlying the changes in RyR and IP3R functional expression have not been described, there is a clear opportunity for additional research in this area where physiological conditions are utilized.
Further considerations
Vascular smooth muscle cells expresses a number of other ion channels [14]. Inward-rectifying K+ (Kir) channels, various members of the TRP channel family and two members of the T-type Ca2+ channel family have all been demonstrated to play essential roles in the regulation of vascular smooth muscle function, myogenic tone and vascular reactivity [14]. Yet, whether the function of these ion channels is altered by acute and short-term elevations in extracellular glucose is unknown. Addressing this issue offers unique opportunities to provide new insights into the regulation of different ion channels, how they are regulated by a highly relevant pathophysiological stimulus and to uncover new mechanisms underlying vascular complications during diabetic hyperglycemia.
Another area that should be considered in future studies relates to mechanisms underlying glucose uptake in vascular smooth muscle. Available studies have shown the expression of several glucose transporters, including insulin-independent Glut1 and insulin-dependent Glut4 in vascular smooth muscle cells [127, 226, 227]. The expression of these glucose transporters can be influenced by glucose concentration both in the intracellular and extracellular milieu. Accordingly, one study found decreased expression of Glut1 upon incubation of cultured rat thoracic aortic smooth muscle in 20 mM d-glucose for 24 h [227]. Interestingly, intracellular glucose levels remained elevated despite the downregulation in Glut1 expression in these cells [227], suggesting that other glucose transporters may also contribute to facilitate glucose uptake in vascular smooth muscle cells. Indeed, in native cerebral vascular smooth muscle cells pre-treated with indinavir, which is considered a selective Glut4 inhibitor [228, 229], elevated glucose-induced potentiation of L-type Ca2+ channels was completely blocked [31]. Moreover, Glut4 activity was found to account for 50% of the total glucose uptake in mouse aortic vascular smooth muscle and to be necessary for agonist-induced contraction of mouse thoracic aortic rings [127]. These results suggest a potential role for Glut4 in glucose metabolism in vascular smooth muscle cells. Further studies are necessary to comprehensively understand how glucose enters vascular smooth muscle cells and how this process is regulated.
Mechanisms impairing vascular ion channels activity during diabetes
Much like in acute and chronic hyperglycemia, vascular smooth muscle function is altered in diabetes, and changes in vascular ion channels expression and/or activity can contribute to this outcome. Below we discuss our current knowledge of the mechanisms altering L-type Ca2+ channel, KV, BKCa and KATP channel activity in vascular smooth muscle during diabetes. RyRs and IP3Rs will be briefly discussed given the limited data on the subject.
Effects of diabetes on L-type Ca2+ channels
L-type Ca2+ channel activity is distinctly altered in vascular smooth muscle during diabetes (Table 3). Decreased L-type Ca2+ channel activity in vascular smooth muscle has been observed in kidney afferent arterioles, aorta and tail arteries from rat STZ models [106,107,108]. Mechanisms for this reduction in L-type Ca2+ channel activity are unclear. Yet, physiological implications of this observation have been suggested to involve impaired responses to vasoconstrictor stimuli that may contribute to altering glomerular filtration, blood flow regulation and even proliferation of vascular smooth muscle [106,107,108, 230].
Most studies indicate that L-type Ca2+ channel activity is increased in vascular smooth muscle from diverse animal models of diabetes [15, 31, 32, 48, 59, 111,112,113,114,115, 231, 232]. Moreover, a recent report showed that the activity of L-type Ca2+ channels is also elevated in vascular smooth muscle from diabetic patients [31], suggesting similarities in how diabetes alters the function of this ion channel in animal models and humans. Physiologically, changes in L-type Ca2+ channel activity in vascular smooth muscle during diabetes may contribute to altering myogenic tone and vascular reactivity, impairing tissue perfusion and blood pressure, and even triggering the modulation of the contractile and proliferative gene program [14, 15, 51, 230].
The mechanisms underlying the increase in L-type Ca2+ channel activity during diabetes are less well understood. However, they do not seem to involve changes in pore-forming subunit expression, as similar protein abundance for CaV1.2 was reported in cerebral/mesenteric and adipose arteries from high fat diet (HFD, type-2 diabetic model) mice and diabetic patients [31, 210]. Rather, current evidence suggests that the effects of diabetes on L-type Ca2+ channel function in vascular smooth muscle may be mainly due to post-translational modifications. Accordingly, an initial study examining this issue found that an increase in the expression of the phosphatidylinositol 3-kinase δ isoform (PI3Kδ) underlie the potentiation of L-type Ca2+ currents in aortic vascular smooth muscle from a mouse model of type-1 diabetes (STZ) [113]. How PI3Kδ increased L-type Ca2+ channel activity during diabetes was not examined. Other studies have suggested that PI3K may act through Akt and protein kinase B (PKB) to increase phosphorylation of the accessory CaV β subunit that then promotes channel trafficking to the membrane and the stimulation in channel activity [233]. It is thus tempting to speculate that a similar mechanism may be engaged to potentiate L-type Ca2+ channel activity in vascular smooth muscle from type-1 diabetic mice. Another study using cerebral vascular smooth muscle from rats in HFD found that increased L-type Ca2+ channel activity correlates with elevations in plasma fatty acids, although the specific mechanisms for this were not described [114]. In skeletal artery vascular smooth muscle from STZ rat, increased L-type Ca2+ channel activity was associated with enhanced PKC function, but mechanism remains unclear [48].
In dB/dB and HFD models of diabetes, the increase in L-type Ca2+ channel activity in cerebral vascular smooth muscle cells was attributed to activation of an unexpected AKAP5-anchored PKA signaling pathway leading to phosphorylation of the CaV1.2 subunit at S1928 (Fig. 2) [31, 112]. Similar results were observed in adipose artery vascular smooth muscle from diabetic patients [31], thus bestowing translational relevance to the aforementioned observations in animal models. Accordingly, genetic depletion of AKAP5, inhibition of PKA, disruption of the interaction between AKAP5 and PKA, and preventing the phosphorylation of the S1928 site halted the increase in L-type Ca2+ channel activity in vascular smooth muscle from HFD mice [31]. More recently, the activity of the AC5 isoform was found to be essential for generating local cAMP signaling leading to PKA-dependent potentiation of L-type Ca2+ channels in vascular smooth muscle from mice in HFD and STZ [32]. Intriguingly, an independent microarray analysis found that AC5 expression was upregulated in vascular smooth muscle upon chronic hyperglycemic conditions [51]. Augmented AC5 expression may contribute to the formation of additional local complexes between AKAP5, PKA and CaV1.2 that further stimulate L-type Ca2+ channel activity during diabetic hyperglycemia. Consistent with this possibility, increased nanometer AC5 and CaV1.2 proximity and localized Ca2+ influx frequency (e.g. persistent Ca2+ sparklets) were found in vascular smooth muscle from different animal models of diabetes (dB/dB, HFD and STZ) [32, 112]. These results are important as they suggest the engagement of similar signaling pathways in the mechanism altering L-type Ca2+ channel activity in different models of diabetes. Moreover, these results are starting to uncover a novel signaling nanocomplex involving AKAP5, AC5, PKA and CaV1.2. This nanocomplex affords a level of compartmentalization that can be selectively engaged to promote cAMP/PKA-dependent phosphorylation of S1928 to stimulate L-type Ca2+ channel activity and alter myogenic tone and vascular reactivity during diabetes. Altogether, results suggest altered L-type Ca2+ channel activity via multiple mechanisms in vascular smooth muscle that may contribute to modulate myogenic tone and vascular reactivity during diabetes. Results may identify potential new targets (e.g. AKAP5, AC5, S1928) for drug development to treat vascular complications during diabetes.
Effects of diabetes on K+ channels
Most studies report a reduction in KV channel activity in vascular smooth muscle from different vascular beds and animal models of diabetes (Table 4) [199, 200, 204, 234,235,236,237]. In coronary vascular smooth muscle from a rat model of type-1 diabetes (STZ), reduced KV function was associated with a decreased expression and increased nitration state of KV1.2 but not KV1.5 subunits due to enhanced peroxynitrite production [234, 236]. A decrease in Kv7 function was also reported as a potential mechanism altering vascular reactivity in coronary arteries from STZ mice [204]. However, in coronary vascular smooth muscle from HFD mice, decreased KV channel activity was correlated with AGEs-mediated downregulation of KV1.2 and KV1.5 subunits’ transcript and protein levels [199]. In cerebral and mesenteric vascular smooth muscle from HFD mice, decreased KV channel activity was linked to selective transcriptional suppression of KV2.1 subunits via activation of the AKAP5-anchored calcineurin/NFATc3 signaling pathway (Fig. 2) [200]. This change in KV2.1 functional expression was correlated with enhanced myogenic tone in HFD cerebral and mesenteric arteries [200]. The studies described above reveal the engagement of distinct mechanisms that could synergize to impair KV channel function and vascular smooth muscle function that may alter myogenic tone and vascular reactivity during diabetes. Future studies should examine whether these mechanisms also alter KV function in vascular smooth muscle from diabetic patients.
BKCa channel activity is also impaired in vascular smooth muscle cells from different vascular beds, animal models of diabetes, and diabetic patients (Table 4) [42, 210, 211, 238,239,240,241,242,243,244,245]. Indeed, there seems to be consensus among all these studies of a reduction in vascular smooth muscle BKCa channel activity during diabetes. Moreover, impaired BKCa channel activity has been associated with impaired vasodilation, increased basal myogenic tone and/or enhanced hyper-reactivity to vasoactive agonists [42, 210, 211, 238,239,240,241,242,243,244,245]. However, in cerebral vascular smooth muscle from dB/dB mice, changes in BKCa channel activity during diabetes seems to be dependent on biological sex, with females being protected from diabetes-mediated changes in channel function [246]. These results contrast with observations of decreased BKCa channel activity in adipose artery vascular smooth muscle from both female and male diabetic patients [42], thus suggesting species and/or vessel-specific differences. The general mechanism for reduced BKCa channel activity has been linked to a downregulation in the functional expression of the BKCa β1 subunit with no change in expression or function of the pore-forming BKCa α subunit, or reduced BKCa β1/ BKCa α expression ratio [42, 210, 211, 238,239,240,241,242,243,244,245]. Accordingly, reduced BKCa β1, but not BKCa α, subunit expression was found in cerebral arteries from a Swiss Webster STZ mouse model [238], aorta from rats and mice in STZ as well as Zucker rat model [211, 239, 241], and aorta, coronary, cerebral, mesenteric arteries from HFD mouse model [210, 245]. In aorta from dB/dB mice, a concomitant increase in BKCa α subunit and decrease in BKCa β1 subunit expression was observed [242].
Several mechanisms have been proposed to account for the reduction in BKCa β1 subunit expression during diabetes. For instance, NFATc3 activation, via an AKAP5-anchored calcineurin-dependent pathway, was found to mediate downregulation in the expression and function of BKCa β1 subunit, contributing to enhanced myogenic tone of cerebral arteries in a HFD model of diabetes (Fig. 2) [210]. Other studies reported accelerated BKCa β1 subunit degradation that is mediated by ROS-dependent activation of a FOXO-3a/FBXO and a NFκB-mediated MuRF1 pathway in mouse aorta and coronary cells (Fig. 4a) [211, 241, 245, 247]. Activation of the NFκB pathway could also lead to suppression of the nuclear factor erythroid-2-related factor 2 (Nrf2) activity and subsequent impairment of Nrf2-mediated transcriptional regulation of the BKCa β1 subunit expression [245, 247]. Intriguingly, two recent studies reported decreased BKCa channel activity in vascular smooth muscle from diabetic patients that was mediated by a reduction in the expression and/or function of the BKCa β1 subunit (Table 4) [42, 243]. One study using adipose artery vascular smooth muscle from diabetic patients showed that impaired BKCa channel activity was likely due to reduced functional coupling of BKCa α and β1 subunits rather than changes in the cellular abundance of each subunit or altered Ca2+ sparks properties [42]. The other study using coronary artery vascular smooth muscle from diabetic patients reported that altered BKCa channel function was correlated with downregulation in the protein levels of both BKCa α and β1 subunits [243]. The differences between these studies and in the mechanisms underlying the alteration in BKCa channel activity could be due to the use of cells from different vascular beds. Nonetheless, results using human samples are essential to pinpoint similarities and differences with animal models that could help define mechanisms involved in diabetic vasculopathy.

Schematic of models for regulation f gene expression and excitability in vascular smooth muscle during diabetic hyperglycemia. a Nrf2 model based on work from Refs. [245, 247]. In this model, diabetic hyperglycemia induces downregulation of Nrf2 via mechanisms that require further examination but that may include NFκB regulation. Decreased expression of Nrf2-regulated genes contributes to ROS production and NF-κB activation. NFκB then inhibits Nrf2-mediated transcriptional induction of BKCa β1 subunit expression and stimulates MuRF1-mediated degradation of BKCa β1 protein leading to a reduction in of BKCa channel activity. Impairment of BKCa channel function may contribute to altered vascular smooth muscle excitability leading to impaired vasodilation and/or hypercontractility. Key open questions (double question marks) are: (1) how elevations in extracellular glucose and/or diabetes reduce Nrf2 expression, (2) what are the mechanisms regulating the crosstalk between Nrf2 and NFκB? (3) can ROS signaling regulate the functional expression of Nrf2? b Proposed model for glucose-induced regulation of vascular smooth muscle contractile and cytoskeletal gene profile based on data from Refs. [51, 263, 265]. Arrows = activation, lines with bars = inhibition, dotted lines = reduced effects.
There is substantial evidence suggesting reduced KATP channel function in vascular smooth muscle from different vascular beds during diabetes that may contribute to impaired cell excitability. Much of this evidence comes from indirect observations showing impaired vasodilation to KATP openers during diabetes. Accordingly, reduced sensitivity to KATP openers (e.g. levcromakalim, aprokalin, lemakalim, cromakalim) has been reported in STZ-treated rat aorta [248], cerebral [50], and coronary arteries [249], as well as human coronary arterioles from type-1 and type-2 diabetic patients [36]. Intriguingly, a single study concomitantly examining vascular reactivity in multiple vascular beds in response to the KATP opener lemakalim found reduced vasodilation in coronary rings, but not in aortic and mesenteric rings from rats in STZ [249], suggesting differential regulation in vascular beds even from the same animal model.
Other studies have correlated the impaired vasodilation to KATP openers during diabetes to a reduction in KATP currents (Table 4) [250,251,252]. In mesenteric vascular smooth muscle from STZ rats, KATP currents were reported to decrease in a time-dependent manner, reaching a plateau after 14 days of STZ treatment [250]. Although the mechanisms for diabetes-induced alterations in KATP currents were not examined in this study, it was found that the xanthine derivative KMUP-1 prevented impaired KATP channel function [250]. Because KMUP-1 can stimulate guanylate cyclase signaling and eNOS activity as well as inhibit phosphodiesterases [253, 254], it is tempting to speculate that this xanthine can simultaneously increase PKB, nitric oxide (NO) and PKA signaling, which converge to improve KATP channel activity in vascular smooth muscle and rescue vascular function during diabetes. In another study using aorta from STZ rats, decreased KATP currents in vascular smooth muscle were correlated with a reduction in total protein levels and increase in S-glutathionation of the Kir6.X subunit [251]. Enhanced S-glutathionation of Kir6.X was suggested to underlie oxidative stress inhibition of KATP currents [251]. In human umbilical smooth muscle obtained from patients with gestational diabetes, a reduction in KATP currents was associated with decreased expression of the KATP channel subunits Kir6.X and SUR2B [252]. Interestingly, KATP currents in diabetic cells were restored to levels observed in control cells by activation of the PKA signaling pathway with forskolin [252]. These results suggest that, in addition to downregulation of KATP subunits expression, other mechanisms may also contribute to impairing KATP channel function and KATP-dependent vasodilation during gestational diabetes. Altogether, results suggest that changes in K+ channels functional expression due to the engagement of several mechanisms in vascular smooth muscle cells may contribute to the altered myogenic tone and vascular reactivity during diabetes.
Further considerations
RyR and IP3R play a key role in regulating vascular smooth muscle Ca2+ dynamics and excitability. However, little is known about how their function is altered in the context of diabetes. A reduction in RyR expression was found in aortic lysates from dB/dB mice [242]. This was linked to a decrease in Ca2+ sparks amplitude with no change in frequency in dB/dB cerebral vascular smooth muscle [242]. This study also found that SR Ca2+ content was impaired in dB/dB cerebral vascular smooth muscle [242], perhaps indicating modifications in the expression and/or function of the SR Ca2+ ATPase SERCA. Indeed, an independent study using rat aorta from the Diabetes Resistant Bio-Breeding (DR-BB) and STZ models of type-1 diabetes found a significant reduction in SERCA2 expression [222]. This alteration may contribute to impair Ca2+ spark amplitude in dB/dB cerebral vascular smooth muscle. Ultimately, reduced Ca2+ sparks amplitude may alter the RyR-BKCa channel coupling to impair the negative tonic feedback regulation of contraction mediated by the activation of BKCa channels. Intriguingly, larger Ca2+ spark amplitude was observed in vascular smooth muscle from retinal arterioles of rats in STZ [244]. This is a potential compensatory mechanism to stimulate BKCa channel activity, which is also impaired due to downregulation of BKCa β1 in these cells [244]. Whether RyR expression is increased in vascular smooth muscle from STZ-treated rat retinal arterioles is unknown, but a significant elevation in RyR proteins levels has been reported in aorta from STZ rats [222].
The role of IP3R in the regulation of vascular smooth muscle excitability during diabetes is less clear. Different reports suggest that intracellular Ca2+ transients mediated by IP3R are either increased or decreased depending on the species and diabetic model [222, 255]. A decrease and impairment in Ca2+ transient dynamics were observed in aortic smooth muscle from DR-BB and STZ rats [222]. These observations were correlated with a decrease in IP3R type-1 expression in diabetic cells [222], but the mechanisms were not explored. In stark contrast, an increase in IP3-evoked [Ca2+]i was found in aortic smooth muscle from dB/dB mice [255]. This effect was directly related to altered stimulation of IP3R activity in dB/dB smooth muscle as SR Ca2+ content and IP3R expression were similar in non-diabetic and diabetic cells [255]. Rather, an increase in the association of IP3Rs with the anti-apoptotic protein Bcl-2 was suggested as a potential culprit in enhancing IP3-evoked [Ca2+]i during diabetes [255], as this protein is known to “sensitize” IP3Rs [256]. Indeed, pharmacological inhibition of Bcl-2 restored IP3-evoked [Ca2+]i transients, but surprisingly did not prevented the agonist-induced hypercontractility in diabetic cells [255]. Thus, the Bcl-2-IP3R axis is likely to modulate [Ca2+]i to activate other signaling pathways, but play no role in regulation of vascular smooth muscle contraction during diabetes. Taken together, results suggest that diabetes impinges on vascular smooth muscle RyRs and IP3Rs via different mechanisms. These data further highlight the need for additional studies examining the functional role of these intracellular ion channels in diabetic vascular complications.
Vascular smooth muscle, gene expression and diabetic hyperglycemia
Hyperglycemia may also alter vascular smooth muscle excitability by influencing the contractile gene program. For instance, it has been shown that acute elevation in extracellular glucose can stimulate the activation and nuclear accumulation of the transcription factor nuclear factor of activated t cells cytoplasmic 3 isoform (NFATc3) in aorta and cerebral vascular smooth muscle within just 30 min [181, 200]. This time scale for NFATc3 nuclear accumulation seems sufficient to trigger changes in gene expression as altered expression of NFATc3-responsive genes can be observed as soon as 1 h after exposure to elevated glucose [200, 257, 258]. The mechanisms underlying glucose-induced NFATc3 activation seem to involve the stimulation of L-type Ca2+ channels to increase Ca2+ influx (Fig. 2) [210, 259]. This elevation in [Ca2+]i is then “sensed” by calmodulin (CaM), which forms a Ca2+/CaM complex that activates the AKAP5-anchored phosphatase calcineurin [134, 210, 260]. AKAP5-anchored calcineurin can then dephosphorylate multiple targets, including NFATc3. Phosphorylated NFATc3 remains inactive in the cytosol, whereas dephosphorylated NFATc3 can translocate into the nucleus where it can alter gene expression, including genes involved in regulation of vascular smooth muscle excitability, such as those coding for K+ channels [15, 16, 134]. The reduction of K+ channel expression results in membrane potential depolarization, activation of L-type Ca2+ channels and alterations in vascular smooth muscle function (see Fig. 2).
Glucose can also regulate the transcriptional activity of Nrf2 in vascular smooth muscle. Nrf2 is a basic leucine zipper transcription factor that plays a significant role in redox homeostasis via modulation of the expression of antioxidant genes [261, 262]. Much like NFATc3, Nrf2 remains in the cytosol where it is found in complex with the E3 ubiquitin ligase, which promotes its degradation in the proteasome to maintain low cellular levels of this transcription factor [261, 262]. In response to changes in cellular oxidative stress, Nrf2 is released from the E3 ubiquitin ligase complex [261, 262]. Free Nrf2 can then translocate to the nucleus where it modulates the expression of genes containing antioxidant responsive elements [261, 262]. Thus, changes in the functional expression of Nrf2 during diabetic hyperglycemia may have profound effects on vascular smooth muscle function, including the regulation of cell excitability. Accordingly, a reduction in Nrf2 protein levels was observed in mesenteric arteriole and aorta lysates from hyperglycemic dB/dB and HFD mice [37, 245, 247], as well as cultured human coronary vascular smooth muscle cells incubated in 22 mM d-glucose (control was cultured cells in 5 mM d-glucose) [247]. In one study, this observation was correlated with downregulation of Nrf2-regulated genes, including many genes coding for antioxidant molecules, and increased production of vascular smooth muscle ROS [37]. Enhanced ROS production in dB/dB cells, perhaps via potentiation of L-type Ca2+ channels [124,125,126], was then suggested to underlie the increased myogenic response in mesenteric arterioles [37]. In a subsequent comprehensive series of experiments using aorta preparations from dB/dB and HFD mice and cultured human coronary vascular smooth muscle exposed to elevated glucose, the decreased Nrf2 expression was correlated with ROS-mediated activation of the transcription factor NFκB and the E3 ubiquitin ligase MuRF1 (Fig. 4a) [245, 247]. Activation of NFκB and MuRF1 during diabetic hyperglycemia were correlated with an Nrf2-dependent reduction in the functional expression of the BKCa β1 subunit and impaired vasodilation [245, 247]. Intriguingly, these studies show a direct relationship between Nrf2 and BKCa β1 subunit expression and an inverse relationship between Nrf2 and NFκB/MuRF1 in vascular smooth muscle [245, 247], but the mechanisms for this remains unclear. The schematic in Fig. 4a provides a suggested model integrating observations from the Nrf2 studies in vascular smooth muscle described above during diabetic hyperglycemia.
A recent study used microarray analysis to examine the effects of elevated extracellular glucose, both in vitro and in vivo, on vascular smooth muscle gene expression programming (Fig. 4b) [51]. It was found that chronic glucose stimulation of vascular smooth muscle for as short as 1 week promoted the activation of a contractile and cytoskeletal gene program that could contribute to vascular complications in diabetes [51]. The glucose-induced activation of the contractile/cytoskeletal gene program was mediated by stimulation of myocardin-related transcription factors (MRTF) that translocate to the nucleus to activate serum response factors (SRF) [51]. Repression of the zinc-finger transcription factor Krüppel-like factor 4 (Klf4) was also observed and further hypothesized to contribute to SRF regulation of the contractile gene program [51]. This elegant study found that increased expression of contractile/cytoskeletal markers due to diabetic hyperglycemia was dependent on AGEs, PKC/Rho activation, the expression of the miR143-145 cluster, which can regulate CaV1.2 expression [263, 264], and the activity of L-type Ca2+ channels (Fig. 4b) [51]. It was subsequently found that repression of Klf4 expression during chronic hyperglycemia was mediated by miR-29 [265], thus uncovering another layer of regulation. It is intriguing to speculate whether disruption of CaV1.2 S1928 phosphorylation (as in S1928A cells) to prevent potentiation of L-type Ca2+ channel activity upon elevated glucose in vascular smooth muscle will normalize the gene expression profile. Nonetheless, glucose-induced activation of contractile/cytoskeletal markers seems to be a general phenomenon of pathophysiological relevance as it was observed in cells from animal models of diabetes and diabetic patients [51]. Overall, these studies highlight mechanisms by which elevated glucose can contribute to regulating vascular smooth muscle function by triggering changes in gene programs. Moreover, this is an exciting area of research that may reveal new targets to treat vascular dysfunction during diabetic hyperglycemia.
Further considerations
Although outside the scope of this work, hyperglycemia and diabetes may also trigger the activation of a gene program that promotes differentiation of vascular smooth muscle into a synthetic, non-contractile phenotype. The synthetic phenotype supports a proliferative state with altered migratory capacity resulting in the thickening of the intima-media arterial wall [266]. Intriguingly, L-type Ca2+ channel activity is essential for activation of both a contractile and synthetic phenotype [51, 230]. The synthetic phenotype has been suggested to contribute to vascular complications, such as atherosclerosis, calcification and inflammation during diabetes [267,268,269,270,271]. However, a role for hyperglycemia in the phenotypic switch is unclear [272]. Nonetheless, activation of both the contractile and synthetic gene program could occur concurrently or via graded activation and/or regulation of different signaling pathways during diabetic hyperglycemia. Accordingly, the synthetic phenotype may be hyperactivated in vascular beds disposed to atherosclerosis, which could also contribute to less responsiveness to vasoactive substances. On the other hand, the contractile phenotype may dominate in blood vessels in which maintaining a level of contraction is essential for proper blood flow and blood pressure regulation (i.e. resistance arteries). Future studies should define whether hyperglycemia itself contributes to the phenotypic switch, especially in an in vivo setting. Moreover, given the "omics era", comparative studies examining how diabetic hyperglycemia regulates contractile and synthetic gene programs in conduit and resistance arteries, are a logical step for future research.
Therapeutic considerations
The insight gained about the biochemical pathways, signaling generators and ion channels involved in altering vascular smooth muscle function in response to hyperglycemic conditions and diabetes may identify new targets for therapeutic intervention [273]. Indeed, potential treatment options have considered inhibiting (1) the activity of PKC, (2) the generation of AGE and/or activation of RAGE and (3) the generation of ROS [273]. These therapeutic interventions may act by restoring ion channels activity and/or the contractile/synthetic phenotype to basal conditions in vascular smooth muscle (but also in other cell types, such as endothelial cells).
The identification of the P2Y11 receptor as a signaling generator promoting glucose-induced PKA-dependent potentiation of L-type Ca2+ channels offers an attractive candidate for therapeutic targeting. Inhibition of P2Y11 receptors may not only prevent hyperactivity of L-type Ca2+ channels during hyperglycemia and diabetes, but also the activation of additional pathological signaling pathways. Indeed, activation of P2Y11 receptors have been associated with inflammation, and cardiac and vascular metabolic complications [274], further highlighting the potential for this purinergic receptor as a therapeutic target. Findings indicating that hyperphosphorylation of CaV1.2 at S1928 underlies L-type Ca2+ channel potentiation during hyperglycemia and diabetes also open new possibilities to develop improved Ca2+ channel blockers that correct impaired channel function (e.g. prevent S1928 hyperphosphorylation), as opposed to established therapies that aim to reduce Ca2+ influx. Indeed, despite calcium channel blocker being used to treat cardiovascular complications in diabetic patients [2], clinical trials have shown a modest effect of this drug class on renal and cardiovascular outcomes when compared to other drugs, such as blockers of the renin-angiotensin system [275,276,277]. Perhaps this new approach of correcting channel function could improve renal and cardiovascular outcome (likely in combination with other drugs, such as renin-angiotensin system inhibitors) [277], and reduce unwanted side effects associated with existing Ca2+ channel blockers [278,279,280].
Targeting K+ channels is also an attractive possibility that has been proposed to correct/ameliorate vascular smooth muscle dysfunction in different pathologies, including diabetic hyperglycemia [281]. For example, treatment options that increase functional expression of BKCa β1 subunit may contribute to rescue vascular smooth muscle function. Accordingly, the use of dimethyl fumarate (DMF), a Food and Drug Administration (FDA) approved Nrf2 activator, has been shown to rescue BKCa β1 subunit, BKCa channel activity and sheer-stress induced coronary vasodilation in dB/dB and HFD mice [245, 247]. Inhibition of NFATc3 has also been shown to prevent BKCa β1 and KV2.1 subunits downregulation in vascular smooth muscle and rescue vascular function in HFD mice [200, 210]. It is also intriguing to speculate that AGE/RAGE inhibitors could contribute to improve vascular smooth muscle function by preventing or restoring KV subunit expression [199]. The use of K+ channel openers is also a therapeutic option that has been proposed for many years due to their antihypertensive properties [282, 283]. However, as discussed above, the use of a given KATP opener itself does not rescue vascular reactivity, at least in some vascular beds. The use of compounds, such as KMUP-1 [250], may rescue other signals (e.g. NO, PKA) to restore KATP activity and vascular function. Consistent with this, perhaps a pharmacological approach targeting many of these ion channels, signaling generators and signaling pathways will be essential for the long-term success in the treatment of vascular smooth muscle dysfunction and vascular complications during diabetic hyperglycemia.
Conclusion
Diabetes is a multifactorial disorder and a major risk factor for cardiovascular diseases. The vasculature is particularly sensitive to the detrimental effects of diabetes. Hyperglycemia plays a central role in mechanisms underlying both vascular endothelial and smooth muscle dysfunction. Whereas the molecular and cellular mechanisms underlying endothelial dysfunction during hyperglycemia and diabetes have been extensively studied [3, 7, 21,22,23,24], much less is known about how vascular smooth muscle is affected. Here we chose to focus on how vascular smooth muscle ion channels are affected by hyperglycemia and diabetes. We did so because ion channels are (1) essential for the regulation of vascular smooth muscle function, and (2) heavily impacted by hyperglycemia and diabetes. In our narrative, we deemed relevant to briefly discuss the information available on how hyperglycemia and diabetes may influence the functional expression of ion channels in endothelial cells as well as the underlying consequences in myogenic tone and vascular reactivity. We then dove into examining the growing body of literature describing how glucose and diabetes can promote changes in the activity of ion channels (particularly L-type Ca2+ channels, KV/BKCa/KATP K+ channels, RyRs and IP3R) in vascular smooth muscle. We summarized mechanisms underlying changes in their function, when possible attempted to reconcile differences, integrate observations and identify knowledge gaps, described how these changes could be related to alterations in myogenic tone and vascular reactivity, and highlighted potential therapeutic considerations. A key take-home message is how one can leverage glucose as a stimulatory molecule to uncover and characterize novel signaling pathways that play a central role in controlling vascular smooth muscle excitability (and perhaps the phenotypic profile). It is clear that mechanisms underlying ion channels dysfunction during hyperglycemia and diabetes are variable, but not necessarily mutually exclusive. It is also clear that vascular smooth muscle ion channels function in response to hyperglycemia and diabetes can be distinctively modulated depending on species, vascular bed and general experimental conditions, such as hyperglycemic and diabetic state. Moreover, the single mechanism approach that is often employed taking advantage of in vitro tools, has to be corroborated in vivo to account for a complex integration of many factors at the organism level. Indeed, implementation of computational modeling could provide an advantage in this regard by allowing the integration of multiple factors that interact non-linearly to affect outcomes and inform experimental designs to be tested in vivo. As our knowledge of the functional expression of other ion channels in vascular smooth muscle expands, it is likely that those newly discovered channels also contribute to vascular smooth muscle dysfunction, thus opening additional research and therapeutic opportunities.
References
World Health Organization (2016) Global report on diabetes. World Health Organization, Switzerland, pp 88. ISBN: 978 92 4 156525 7
Association AD (2017) Standards of medical care in diabetes—2017. Diabetes Care 40:S1–S135
Creager MA, Luscher TF, Cosentino F, Beckman JA (2003) Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Circulation 108:1527–1532
Grundy SM (2012) Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol 59:635–643
Raghavan S, Vassy JL, Ho YL, Song RJ, Gagnon DR, Cho K, Wilson PWF, Phillips LS (2019) Diabetes mellitus-related all-cause and cardiovascular mortality in a national cohort of adults. J Am Heart Assoc 8:e011295
Brown A, Reynolds LR, Bruemmer D (2010) Intensive glycemic control and cardiovascular disease: an update. Nat Rev Cardiol 7:369–375
Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, Stefanadis C (2013) Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol 62:667–676
Cooper ME, Bonnet F, Oldfield M, Jandeleit-Dahm K (2001) Mechanisms of diabetic vasculopathy: an overview. Am J Hypertens 14:475–486
Stehouwer CDA (2018) Microvascular dysfunction and hyperglycemia: a vicious cycle with widespread consequences. Diabetes 67:1729–1741
Serne EH, de Jongh RT, Eringa EC, Stehouwer CD (2007) Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension 50:204–211
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The diabetes control and complications trial research group. N Engl J Med 329:977–986
Bayliss WM (1902) On the local reaction of the arterial wall to changes in internal pressure. J Physiol 28:220–231
Davis MJ, Hill MA (1999) Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79:387–423
Tykocki NR, Boerman EM, Jackson WF (2017) Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr Physiol 7:485–581
Navedo MF, Amberg GC (2013) Local regulation of l-type Ca(2)(+) channel sparklets in arterial smooth muscle. Microcirculation 20:290–298
Navedo MF, Santana LF (2013) Cav1.2 sparklets in heart and vascular smooth muscle. J Mol Cell Cardiol 58:67–76
Somlyo AP, Somlyo AV (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by g proteins, kinases, and myosin phosphatase. Physiol Rev 83:1325–1358
El-Yazbi AF, Abd-Elrahman KS (2017) Rok and arteriolar myogenic tone generation: molecular evidence in health and disease. Front Pharmacol 8:87
Knot HJ, Nelson MT (1998) Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508(Pt 1):199–209
Abd-Elrahman KS, Walsh MP, Cole WC (2015) Abnormal rho-associated kinase activity contributes to the dysfunctional myogenic response of cerebral arteries in type 2 diabetes. Can J Physiol Pharmacol 93:177–184
Zhang HN, Xu QQ, Thakur A, Alfred MO, Chakraborty M, Ghosh A, Yu XB (2018) Endothelial dysfunction in diabetes and hypertension: role of micrornas and long non-coding rnas. Life Sci 213:258–268
Meza CA, La Favor JD, Kim DH, Hickner RC (2019) Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int J Mol Sci 20(15):3775. https://doi.org/10.3390/ijms20153775
Hadi HA, Suwaidi JA (2007) Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 3:853–876
Sena CM, Pereira AM, Seica R (2013) Endothelial dysfunction—a major mediator of diabetic vascular disease. Biochem Biophys Acta 1832:2216–2231
Montero D, Walther G, Perez-Martin A, Vicente-Salar N, Roche E, Vinet A (2013) Vascular smooth muscle function in type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia 56:2122–2133
Aronson D (2008) Hyperglycemia and the pathobiology of diabetic complications. Adv Cardiol 45:1–16
Fleischhacker E, Esenabhalu VE, Spitaler M, Holzmann S, Skrabal F, Koidl B, Kostner GM, Graier WF (1999) Human diabetes is associated with hyperreactivity of vascular smooth muscle cells due to altered subcellular Ca2+ distribution. Diabetes 48:1323–1330
Sonoyama K, Greenstein A, Price A, Khavandi K, Heagerty T (2007) Vascular remodeling: implications for small artery function and target organ damage. Ther Adv Cardiovasc Dis 1:129–137
Schofield I, Malik R, Izzard A, Austin C, Heagerty A (2002) Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation 106:3037–3043
Nacci C, Tarquinio M, De Benedictis L, Mauro A, Zigrino A, Carratu MR, Quon MJ, Montagnani M (2009) Endothelial dysfunction in mice with streptozotocin-induced type 1 diabetes is opposed by compensatory overexpression of cyclooxygenase-2 in the vasculature. Endocrinology 150:849–861
Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F et al (2017) Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal 10(463):eaaf9647. https://doi.org/10.1126/scisignal.aaf9647
Syed AU, Reddy GR, Ghosh D, Prada MP, Nystoriak MA, Morotti S, Grandi E, Sirish P, Chiamvimonvat N, Hell JW, Santana LF, Xiang YK, Nieves-Cintron M, Navedo MF (2019) Adenylyl cyclase 5-generated camp controls cerebral vascular reactivity during diabetic hyperglycemia. J Clin Investig 129:3140–3152
Bjarnegard N, Arnqvist HJ, Lindstrom T, Jonasson L, Jonsson A, Lanne T (2009) Long-term hyperglycaemia impairs vascular smooth muscle cell function in women with type 1 diabetes mellitus. Diab Vasc Dis Res 6:25–31
Cipolla MJ (1999) Elevated glucose potentiates contraction of isolated rat resistance arteries and augments protein kinase c-induced intracellular calcium release. Metab Clin Exp 48:1015–1022
McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, Andrews JW, Hayes JR (1992) Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 35:771–776
Miura H, Wachtel RE, Loberiza FR Jr, Saito T, Miura M, Nicolosi AC, Gutterman DD (2003) Diabetes mellitus impairs vasodilation to hypoxia in human coronary arterioles: reduced activity of atp-sensitive potassium channels. Circ Res 92:151–158
Velmurugan GV, Sundaresan NR, Gupta MP, White C (2013) Defective nrf2-dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes. Cardiovasc Res 100:143–150
Okon EB, Chung AW, Rauniyar P, Padilla E, Tejerina T, McManus BM, Luo H, van Breemen C (2005) Compromised arterial function in human type 2 diabetic patients. Diabetes 54:2415–2423
Heinonen SE, Genove G, Bengtsson E, Hubschle T, Akesson L, Hiss K, Benardeau A, Yla-Herttuala S, Jonsson-Rylander AC, Gomez MF (2015) Animal models of diabetic macrovascular complications: key players in the development of new therapeutic approaches. J Diabetes Res 2015:404085
Islam MS, du Loots T (2009) Experimental rodent models of type 2 diabetes: a review. Methods Find Exp Clin Pharmacol 31:249–261
Bagi Z, Koller A, Kaley G (2003) Superoxide-no interaction decreases flow- and agonist-induced dilations of coronary arterioles in type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol 285:H1404–1410
Nieves-Cintron M, Syed AU, Buonarati OR, Rigor RR, Nystoriak MA, Ghosh D, Sasse KC, Ward SM, Santana LF, Hell JW, Navedo MF (2017) Impaired bkca channel function in native vascular smooth muscle from humans with type 2 diabetes. Scientific reports 7:14058
Ito I, Jarajapu YP, Guberski DL, Grant MB, Knot HJ (2006) Myogenic tone and reactivity of rat ophthalmic artery in acute exposure to high glucose and in a type ii diabetic model. Investig Ophthalmol Vis Sci 47:683–692
Kold-Petersen H, Brondum E, Nilsson H, Flyvbjerg A, Aalkjaer C (2012) Impaired myogenic tone in isolated cerebral and coronary resistance arteries from the goto-kakizaki rat model of type 2 diabetes. J Vasc Res 49:267–278
Abd-Elrahman KS, Colinas O, Walsh EJ, Zhu HL, Campbell CM, Walsh MP, Cole WC (2017) Abnormal myosin phosphatase targeting subunit 1 phosphorylation and actin polymerization contribute to impaired myogenic regulation of cerebral arterial diameter in the type 2 diabetic goto-kakizaki rat. J Cereb Blood Flow Metab 37:227–240
Bagi Z, Erdei N, Toth A, Li W, Hintze TH, Koller A, Kaley G (2005) Type 2 diabetic mice have increased arteriolar tone and blood pressure: enhanced release of cox-2-derived constrictor prostaglandins. Arterioscler Thromb Vasc Biol 25:1610–1616
Jarajapu YP, Guberski DL, Grant MB, Knot HJ (2008) Myogenic tone and reactivity of cerebral arteries in type ii diabetic bbzdr/wor rat. Eur J Pharmacol 579:298–307
Ungvari Z, Pacher P, Kecskemeti V, Papp G, Szollar L, Koller A (1999) Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase c. Cardiovasc Res 43:1018–1028
Sauve M, Hui SK, Dinh DD, Foltz WD, Momen A, Nedospasov SA, Offermanns S, Husain M, Kroetsch JT, Lidington D, Bolz SS (2016) Tumor necrosis factor/sphingosine-1-phosphate signaling augments resistance artery myogenic tone in diabetes. Diabetes 65:1916–1928
Zimmermann PA, Knot HJ, Stevenson AS, Nelson MT (1997) Increased myogenic tone and diminished responsiveness to atp-sensitive k+ channel openers in cerebral arteries from diabetic rats. Circ Res 81:996–1004
Hien TT, Turczynska KM, Dahan D, Ekman M, Grossi M, Sjogren J, Nilsson J, Braun T, Boettger T, Garcia-Vaz E, Stenkula K, Sward K, Gomez MF, Albinsson S (2016) Elevated glucose levels promote contractile and cytoskeletal gene expression in vascular smooth muscle via rho/protein kinase c and actin polymerization. J Biol Chem 291:3552–3568
Xie Z, Su W, Guo Z, Pang H, Post SR, Gong MC (2006) Up-regulation of cpi-17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells. Cardiovasc Res 69:491–501
Kizub IV, Pavlova OO, Johnson CD, Soloviev AI, Zholos AV (2010) Rho kinase and protein kinase c involvement in vascular smooth muscle myofilament calcium sensitization in arteries from diabetic rats. Br J Pharmacol 159:1724–1731
Lagaud GJ, Masih-Khan E, Kai S, van Breemen C, Dube GP (2001) Influence of type ii diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res 38:578–589
Kautzky-Willer A, Harreiter J, Pacini G (2016) Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev 37:278–316
Mauvais-Jarvis F (2018) Gender differences in glucose homeostasis and diabetes. Physiol Behav 187:20–23
Zhang R, Thor D, Han X, Anderson L, Rahimian R (2012) Sex differences in mesenteric endothelial function of streptozotocin-induced diabetic rats: a shift in the relative importance of edrfs. Am J Physiol Heart Circ Physiol 303:H1183–1198
Cipolla MJ, Porter JM, Osol G (1997) High glucose concentrations dilate cerebral arteries and diminish myogenic tone through an endothelial mechanism. Stroke 28:405–410
Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, Ghosh D, Simo S, Sato D, Sasse KC, Ward SM et al (2019) A Gs-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. eLife 8:e42214. https://doi.org/10.7554/eLife.42214
Straub SV, Girouard H, Doetsch PE, Hannah RM, Wilkerson MK, Nelson MT (2009) Regulation of intracerebral arteriolar tone by k(v) channels: effects of glucose and pkc. Am J Physiol Cell Physiol 297:C788–796
Ottolini M, Hong K, Sonkusare SK (2019) Calcium signals that determine vascular resistance. Wiley Interdiscip Rev Syst Biol Med 11:e1448
Kaiser N, Sasson S, Feener EP, Boukobza-Vardi N, Higashi S, Moller DE, Davidheiser S, Przybylski RJ, King GL (1993) Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes 42:80–89
Artwohl M, Brunmair B, Furnsinn C, Holzenbein T, Rainer G, Freudenthaler A, Porod EM, Huttary N, Baumgartner-Parzer SM (2007) Insulin does not regulate glucose transport and metabolism in human endothelium. Eur J Clin Investig 37:643–650
Wu N, Shen H, Liu H, Wang Y, Bai Y, Han P (2016) Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc Diabetol 15:109
Maeda M, Hayashi T, Mizuno N, Hattori Y, Kuzuya M (2015) Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: role of superoxide production by nadph oxidase. PLoS ONE 10:e0123169
Murata M, Adachi H, Oshima S, Kurabayashi M (2017) Glucose fluctuation and the resultant endothelial injury are correlated with pancreatic beta cell dysfunction in patients with coronary artery disease. Diabetes Res Clin Pract 131:107–115
Feletou M (2016) Endothelium-dependent hyperpolarization and endothelial dysfunction. J Cardiovasc Pharmacol 67:373–387
Welsh DG, Tran CHT, Hald BO, Sancho M (2017) The conducted vasomotor response: function, biophysical basis, and pharmacological control. Annu Rev Pharmacol Toxicol. https://doi.org/10.1146/annurev-pharmtox-010617-052623
Earley S, Brayden JE (2015) Transient receptor potential channels in the vasculature. Physiol Rev 95:645–690
Kwan HY, Huang Y, Yao X (2007) Trp channels in endothelial function and dysfunction. Biochem Biophys Acta 1772:907–914
Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT (2012) Elementary Ca2+ signals through endothelial trpv4 channels regulate vascular function. Science 336:597–601
Straub AC, Zeigler AC, Isakson BE (2014) The Bmyoendothelial junction: connections that deliver the message. Physiology (Bethesda, Md) 29:242–249
Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA (2012) Low intravascular pressure activates endothelial cell trpv4 channels, local Ca2+ events, and ikca channels, reducing arteriolar tone. Proc Natl Acad Sci USA 109:18174–18179
Ma X, Du J, Zhang P, Deng J, Liu J, Lam FF, Li RA, Huang Y, Jin J, Yao X (2013) Functional role of trpv4-kca2.3 signaling in vascular endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension 62:134–139
Bagher P, Segal SS (2011) Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol 202:271–284
Socha MJ, Behringer EJ, Segal SS (2012) Calcium and electrical signalling along endothelium of the resistance vasculature. Basic Clin Pharmacol Toxicol 110:80–86
Marziano C, Hong K, Cope EL, Kotlikoff MI, Isakson BE, Sonkusare SK (2017) Nitric oxide-dependent feedback loop regulates transient receptor potential vanilloid 4 (trpv4) channel cooperativity and endothelial function in small pulmonary arteries. J Am Heart Assoc 6(12):e007157. https://doi.org/10.1161/JAHA.117.007157
Monaghan K, McNaughten J, McGahon MK, Kelly C, Kyle D, Yong PH, McGeown JG, Curtis TM (2015) Hyperglycemia and diabetes downregulate the functional expression of trpv4 channels in retinal microvascular endothelium. PLoS ONE 10:e0128359
He D, Pan Q, Chen Z, Sun C, Zhang P, Mao A, Zhu Y, Li H, Lu C, Xie M, Zhou Y, Shen D, Tang C, Yang Z, Jin J, Yao X, Nilius B, Ma X (2017) Treatment of hypertension by increasing impaired endothelial trpv4-kca2.3 interaction. EMBO Mol Med 9:1491–1503
Sun H, Leng T, Zeng Z, Gao X, Inoue K, Xiong ZG (2013) Role of trpm7 channels in hyperglycemia-mediated injury of vascular endothelial cells. PLoS ONE 8:e79540
Bishara NB, Ding H (2010) Glucose enhances expression of trpc1 and calcium entry in endothelial cells. Am J Physiol Heart Circ Physiol 298:H171–178
Sun J, Pu Y, Wang P, Chen S, Zhao Y, Liu C, Shang Q, Zhu Z, Liu D (2013) Trpv1-mediated ucp2 upregulation ameliorates hyperglycemia-induced endothelial dysfunction. Cardiovasc Diabetol 12:69
Tian XY, Ma S, Tse G, Wong WT, Huang Y (2018) Uncoupling protein 2 in cardiovascular health and disease. Front Physiol 9:1060
Mishra J, Jhun BS, Hurst S, Csordas G, Sheu SS (2017) The mitochondrial Ca(2+) uniporter: structure, function, and pharmacology. Handb Exp Pharmacol. 240:129–156
Chen W, Yang J, Chen S, Xiang H, Liu H, Lin D, Zhao S, Peng H, Chen P, Chen AF, Lu H (2017) Importance of mitochondrial calcium uniporter in high glucose-induced endothelial cell dysfunction. Diab Vasc Dis Res 14:494–501
Putney JW Jr (1986) A model for receptor-regulated calcium entry. Cell Calcium 7:1–12
Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA (2005) Stim1, an essential and conserved component of store-operated ca2+ channel function. The Journal of cell biology 169:435–445
Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD (2005) Stim1 is a ca2+ sensor that activates crac channels and migrates from the ca2+ store to the plasma membrane. Nature 437:902–905
Ruhle B, Trebak M (2013) Emerging roles for native orai ca2+ channels in cardiovascular disease. Curr Top Membr 71:209–235
Tanwar J, Trebak M, Motiani RK (2017) Cardiovascular and hemostatic disorders: role of stim and orai proteins in vascular disorders. Adv Exp Med Biol 993:425–452
Tamareille S, Mignen O, Capiod T, Rucker-Martin C, Feuvray D (2006) High glucose-induced apoptosis through store-operated calcium entry and calcineurin in human umbilical vein endothelial cells. Cell Calcium 39:47–55
Daskoulidou N, Zeng B, Berglund LM, Jiang H, Chen GL, Kotova O, Bhandari S, Ayoola J, Griffin S, Atkin SL, Gomez MF, Xu SZ. High glucose enhances store-operated calcium entry by upregulating orai/stim via calcineurin-nfat signalling. Journal of molecular medicine. 2014
Garcia-Vaz E, McNeilly AD, Berglund LM, Ahmad A, Gallagher JR, Dutius Andersson AM, McCrimmon RJ, Zetterqvist AV, Gomez MF, Khan F (2020) Inhibition of nfat signaling restores microvascular endothelial function in diabetic mice. Diabetes 69:424–435
Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT (2008) Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A 105:9627–9632
Sonkusare SK, Dalsgaard T, Bonev AD, Nelson MT. Inward rectifier potassium (kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J Physiol. 2016;594:3271–3285
Sancho M, Samson NC, Hald BO, Hashad AM, Marrelli SP, Brett SE, Welsh DG (2017) Kir channels tune electrical communication in cerebral arteries. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 37:2171–2184
Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, Li W, Oulidi A, Boop FA, Feng Y, Jaggar JH, Welsh DG, Earley S. Localized trpa1 channel ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Science signaling. 2015;8:ra2
Su J, Lucchesi PA, Gonzalez-Villalobos RA, Palen DI, Rezk BM, Suzuki Y, Boulares HA, Matrougui K (2008) Role of advanced glycation end products with oxidative stress in resistance artery dysfunction in type 2 diabetic mice. Arterioscler Thromb Vasc Biol 28:1432–1438
Ren X, Ren L, Wei Q, Shao H, Chen L, Liu N (2017) Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovascular diabetology 16:52
Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A (2014) Role of advanced glycation end products in cellular signaling. Redox Biol 2:411–429
Naser N, Januszewski AS, Brown BE, Jenkins AJ, Hill MA, Murphy TV (2013) Advanced glycation end products acutely impair ca(2+) signaling in bovine aortic endothelial cells. Frontiers in physiology 4:38
Amberg GC, Navedo MF (2013) Calcium dynamics in vascular smooth muscle. Microcirculation 20:281–289
Barbagallo M, Shan J, Pang PK, Resnick LM (1995) Glucose-induced alterations of cytosolic free calcium in cultured rat tail artery vascular smooth muscle cells. J Clin Invest 95:763–767
Kharade SV, Sonkusare SK, Srivastava AK, Thakali KM, Fletcher TW, Rhee SW, Rusch NJ (2013) The beta3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin ii-infused c57bl/6 mice. Hypertension 61:137–142
Ghosh D, Syed AU, Prada MP, Nystoriak MA, Santana LF, Nieves-Cintron M, Navedo MF (2017) Calcium channels in vascular smooth muscle. Adv Pharmacol 78:49–87
Fulton DJ, Hodgson WC, Sikorski BW, King RG (1991) Attenuated responses to endothelin-1, kcl and cacl2, but not noradrenaline, of aortae from rats with streptozotocin-induced diabetes mellitus. Br J Pharmacol 104:928–932
Wang R, Wu Y, Tang G, Wu L, Hanna ST (2000) Altered l-type ca(2+) channel currents in vascular smooth muscle cells from experimental diabetic rats. Am J Physiol Heart Circ Physiol 278:H714–722
Carmines PK, Ohishi K, Ikenaga H (1996) Functional impairment of renal afferent arteriolar voltage-gated calcium channels in rats with diabetes mellitus. J Clin Invest 98:2564–2571
Abebe W, MacLeod KM (1990) Protein kinase c-mediated contractile responses of arteries from diabetic rats. Br J Pharmacol 101:465–471
Mulhern M, Docherty JR (1989) Effects of experimental diabetes on the responsiveness of rat aorta. Br J Pharmacol 97:1007–1012
White RE, Carrier GO (1990) Vascular contraction induced by activation of membrane calcium ion channels is enhanced in streptozotocin-diabetes. J Pharmacol Exp Ther 253:1057–1062
Navedo MF, Takeda Y, Nieves-Cintron M, Molkentin JD, Santana LF (2010) Elevated ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am J Physiol Cell Physiol 298:C211–220
Pinho JF, Medeiros MA, Capettini LS, Rezende BA, Campos PP, Andrade SP, Cortes SF, Cruz JS, Lemos VS (2010) Phosphatidylinositol 3-kinase-delta up-regulates l-type ca2+ currents and increases vascular contractility in a mouse model of type 1 diabetes. Br J Pharmacol 161:1458–1471
Wilde DW, Massey KD, Walker GK, Vollmer A, Grekin RJ (2000) High-fat diet elevates blood pressure and cerebrovascular muscle ca(2+) current. Hypertension 35:832–837
Ma YG, Wang JW, Bai YG, Liu M, Xie MJ, Dai ZJ (2017) Salidroside contributes to reducing blood pressure and alleviating cerebrovascular contractile activity in diabetic goto-kakizaki rats by inhibition of l-type calcium channel in smooth muscle cells. BMC Pharmacol Toxicol 18:30
Clunn GF, Wijetunge S, Hughes AD (1999) Effect of hypercholesterolaemia on voltage-operated calcium channel currents in rabbit arterial smooth muscle cells. J Hum Hypertens 13:849–853
Bowles DK, Heaps CL, Turk JR, Maddali KK, Price EM (2004) Hypercholesterolemia inhibits l-type calcium current in coronary macro-, not microcirculation. J Appl Physiol 96:2240–2248
McCarty MF (2006) Pkc-mediated modulation of l-type calcium channels may contribute to fat-induced insulin resistance. Med Hypotheses 66:824–831
Navedo MF, Amberg GC, Votaw VS, Santana LF (2005) Constitutively active l-type ca2+ channels. Proc Natl Acad Sci U S A 102:11112–11117
Dixon RE, Moreno CM, Yuan C, Opitz-Araya X, Binder MD, Navedo MF, Santana LF. Graded ca(2)(+)/calmodulin-dependent coupling of voltage-gated cav1.2 channels. eLife. 2015;4
Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. Ca2+ signaling amplification by oligomerization of l-type cav1.2 channels. Proc Natl Acad Sci U S A. 2012;109:1749–1754
Zhong Z, Han J, Zhang J, Xiao Q, Hu J, Chen L (2018) Pharmacological activities, mechanisms of action, and safety of salidroside in the central nervous system. Drug Des Devel Ther 12:1479–1489
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H (2000) High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase c–dependent activation of nad(p)h oxidase in cultured vascular cells. Diabetes 49:1939–1945
Chaplin NL, Amberg GC (2012) Hydrogen peroxide mediates oxidant-dependent stimulation of arterial smooth muscle l-type calcium channels. Am J Physiol Cell Physiol 302:C1382–1393
Chaplin NL, Nieves-Cintron M, Fresquez AM, Navedo MF, Amberg GC (2015) Arterial smooth muscle mitochondria amplify hydrogen peroxide microdomains functionally coupled to l-type calcium channels. Circ Res 117:1013–1023
Amberg GC, Earley S, Glapa SA (2010) Local regulation of arterial l-type calcium channels by reactive oxygen species. Circ Res 107:1002–1010
Park JL, Loberg RD, Duquaine D, Zhang H, Deo BK, Ardanaz N, Coyle J, Atkins KB, Schin M, Charron MJ, Kumagai AK, Pagano PJ, Brosius FC 3rd (2005) Glut4 facilitative glucose transporter specifically and differentially contributes to agonist-induced vascular reactivity in mouse aorta. Arterioscler Thromb Vasc Biol 25:1596–1602
Jackson R, Brennan S, Fielding P, Sims MW, Challiss RA, Adlam D, Squire IB, Rainbow RD (2016) Distinct and complementary roles for alpha and beta isoenzymes of pkc in mediating vasoconstrictor responses to acutely elevated glucose. Br J Pharmacol 173:870–887
Hashim S, Li Y, Anand-Srivastava MB (2006) G protein-linked cell signaling and cardiovascular functions in diabetes/hyperglycemia. Cell Biochem Biophys 44:51–64
Patel NA, Chalfant CE, Yamamoto M, Watson JE, Eichler DC, Cooper DR (1999) Acute hyperglycemia regulates transcription and posttranscriptional stability of pkcbetaii mrna in vascular smooth muscle cells. FASEB J 13:103–113
Williams B, Gallacher B, Patel H, Orme C (1997) Glucose-induced protein kinase c activation regulates vascular permeability factor mrna expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes 46:1497–1503
Williams B, Schrier RW (1992) Characterization of glucose-induced in situ protein kinase c activity in cultured vascular smooth muscle cells. Diabetes 41:1464–1472
Navedo MF, Amberg GC, Nieves M, Molkentin JD, Santana LF (2006) Mechanisms underlying heterogeneous ca2+ sparklet activity in arterial smooth muscle. J Gen Physiol 127:611–622
Nieves-Cintron M, Amberg GC, Navedo MF, Molkentin JD, Santana LF (2008) The control of ca2+ influx and nfatc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci U S A 105:15623–15628
Payne GW, Smeda JS (2002) Cerebrovascular alterations in pressure and protein kinase c-mediated constriction in dahl salt-sensitive rats. J Hypertens 20:1355–1363
Ruan X, Arendshorst WJ. Role of protein kinase c in angiotensin ii-induced renal vasoconstriction in genetically hypertensive rats. Am J Physiol. 1996;270
Salamanca DA, Khalil RA (2005) Protein kinase c isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol 70:1537–1547
Keef KD, Hume JR, Zhong J. Regulation of cardiac and smooth muscle ca(2+) channels (ca(v)1.2a,b) by protein kinases. Am J Physiol Cell Physiol. 2001;281:C1743–1756
Porter VA, Bonev AD, Knot HJ, Heppner TJ, Stevenson AS, Kleppisch T, Lederer WJ, Nelson MT (1998) Frequency modulation of ca2+ sparks is involved in regulation of arterial diameter by cyclic nucleotides. Am J Physiol 274:C1346–1355
Wellman GC, Santana LF, Bonev AD, Nelson MT (2001) Role of phospholamban in the modulation of arterial ca2+ sparks and ca2+-activated k+ channels by camp. Am J Physiol Cell Physiol 281:C1029–1037
Wellman GC, Bonev AD, Nelson MT, Brayden JE (1996) Gender differences in coronary artery diameter involve estrogen, nitric oxide, and ca2+-dependent k+ channels. Circ Res 79:1024–1030
Aiello EA, Walsh MP, Cole WC (1995) Phosphorylation by protein kinase a enhances delayed rectifier k+ current in rabbit vascular smooth muscle cells. Am J Physiol 268:H926–934
Moore CL, Nelson PL, Parelkar NK, Rusch NJ, Rhee SW (2014) Protein kinase a-phosphorylated kv1 channels in psd95 signaling complex contribute to the resting membrane potential and diameter of cerebral arteries. Circ Res 114:1258–1267
Langeberg LK, Scott JD (2015) Signalling scaffolds and local organization of cellular behaviour. Nat Rev Mol Cell Biol 16:232–244
Johnstone TB, Agarwal SR, Harvey RD, Ostrom RS (2018) Camp signaling compartmentation: adenylyl cyclases as anchors of dynamic signaling complexes. Mol Pharmacol 93:270–276
Man KNM, Navedo MF, Horne MC, Hell JW. Beta2 adrenergic receptor complexes with the l-type ca(2+) channel cav1.2 and ampa-type glutamate receptors: paradigms for pharmacological targeting of protein interactions. Annual review of pharmacology and toxicology. 2019
Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM (1997) Camp-dependent regulation of cardiac l-type ca2+ channels requires membrane targeting of pka and phosphorylation of channel subunits. Neuron 19:185–196
Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD (2006) Dynamic regulation of camp synthesis through anchored pka-adenylyl cyclase v/vi complexes. Mol Cell 23:925–931
Navedo MF, Nieves-Cintron M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF (2008) Akap150 is required for stuttering persistent ca2+ sparklets and angiotensin ii-induced hypertension. Circ Res 102:e1–e11
Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, Dessauer CW (2010) Akap79 interacts with multiple adenylyl cyclase (ac) isoforms and scaffolds ac5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (ampa) receptors. J Biol Chem 285:14450–14458
Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell'Acqua ML (2014) Akap-anchored pka maintains neuronal l-type calcium channel activity and nfat transcriptional signaling. Cell reports 7:1577–1588
Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, Xiang YK, Hell JW (2013) Adenylyl cyclase anchoring by a kinase anchor protein akap5 (akap79/150) is important for postsynaptic beta-adrenergic signaling. J Biol Chem 288:17918–17931
Dai S, Hall DD, Hell JW (2009) Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev 89:411–452
Davare MA, Hell JW. Increased phosphorylation of the neuronal l-type ca(2+) channel ca(v)1.2 during aging. Proc Natl Acad Sci U S A. 2003;100:16018–16023
De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA (1996) Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac l-type calcium channel by adenosine 3',5'- cyclic monophosphate-dependent protein kinase. Biochemistry 35:10392–10402
Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal c-terminal domain of cardiac cav1.2 channels during beta1-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:16574–16579
Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves-Cintron M, Buonarati OR, Chowdhury D, Nanou E, Nystoriak MA, Catterall WA, Poomvanicha M, Hofmann F, Navedo MF, Hell JW. Phosphorylation of ser1928 mediates the enhanced activity of the l-type ca2+ channel cav1.2 by the beta2-adrenergic receptor in neurons. Science signaling. 2017;10:eaaf9659
Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, Hofmann F, Moosmang S. Unchanged beta-adrenergic stimulation of cardiac l-type calcium channels in ca v 1.2 phosphorylation site s1928a mutant mice. J Biol Chem. 2008;283:34738–34744
Katchman A, Yang L, Zakharov SI, Kushner J, Abrams J, Chen BX, Liu G, Pitt GS, Colecraft HM, Marx SO. Proteolytic cleavage and pka phosphorylation of alpha1c subunit are not required for adrenergic regulation of cav1.2 in the heart. Proc Natl Acad Sci U S A. 2017;114:9194–9199
Ganesan AN, Maack C, Johns DC, Sidor A, O'Rourke B (2006) Beta-adrenergic stimulation of l-type ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of alpha1c but not serine 1928. Circ Res 98:e11–18
Liu Y, Terata K, Rusch NJ, Gutterman DD (2001) High glucose impairs voltage-gated k+ channel current in rat small coronary arteries. Circ Res 89:146–152
Rainbow RD, Hardy ME, Standen NB, Davies NW (2006) Glucose reduces endothelin inhibition of voltage-gated potassium channels in rat arterial smooth muscle cells. J Physiol 575:833–844
Knot HJ, Nelson MT (1995) Regulation of membrane potential and diameter by voltage-dependent k+ channels in rabbit myogenic cerebral arteries. Am J Physiol 269:H348–355
Kapela A, Bezerianos A, Tsoukias NM (2008) A mathematical model of ca2+ dynamics in rat mesenteric smooth muscle cell: agonist and no stimulation. J Theor Biol 253:238–260
Morotti S, Nieves-Cintron M, Nystoriak MA, Navedo MF, Grandi E. Predominant contribution of l-type cav1.2 channel stimulation to impaired intracellular calcium and cerebral artery vasoconstriction in diabetic hyperglycemia. Channels (Austin). 2017;11:340–346
Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A, Dangas K, Gygi SP, Pitt GS, Colecraft HM, Ben-Johny M, Kalocsay M, Marx SO. Mechanism of adrenergic cav1.2 stimulation revealed by proximity proteomics. Nature. 2020
Munder T, Kuntzel H (1989) Glucose-induced camp signaling in saccharomyces cerevisiae is mediated by the cdc25 protein. FEBS Lett 242:341–345
Dyachok O, Idevall-Hagren O, Sagetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjarvi G, Gylfe E, Tengholm A (2008) Glucose-induced cyclic amp oscillations regulate pulsatile insulin secretion. Cell Metab 8:26–37
Glorian M, Limon I (2013) The role of cyclic 3’-5’ adenosine monophosphate (camp) in differentiated and trans-differentiated vascular smooth muscle cells. In: Rezzani R (ed) Current trends in atherogenesis. IntechOpen, London, UK, pp 121–145
Schleicher K, Zaccolo M. Using camp sensors to study cardiac nanodomains. J Cardiovasc Dev Dis. 2018;5
Zhang L, Bouadjel K, Manoury B, Vandecasteele G, Fischmeister R, Leblais V (2019) Cyclic nucleotide signalling compartmentation by pdes in cultured vascular smooth muscle cells. Br J Pharmacol 176:1780–1792
Nelson CP, Rainbow RD, Brignell JL, Perry MD, Willets JM, Davies NW, Standen NB, Challiss RA (2011) Principal role of adenylyl cyclase 6 in k(+) channel regulation and vasodilator signalling in vascular smooth muscle cells. Cardiovasc Res 91:694–702
Liu S, Zhang J, Xiang YK (2011) Fret-based direct detection of dynamic protein kinase a activity on the sarcoplasmic reticulum in cardiomyocytes. Biochem Biophys Res Commun 404:581–586
Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, Seifert R. International union of basic and clinical pharmacology. Ci. Structures and small molecule modulators of mammalian adenylyl cyclases. Pharmacological reviews. 2017;69:93–139
Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, Insel PA (2002) Localization of adenylyl cyclase isoforms and g protein-coupled receptors in vascular smooth muscle cells: expression in caveolin-rich and noncaveolin domains. Mol Pharmacol 62:983–992
Webb JG, Yates PW, Yang Q, Mukhin YV, Lanier SM (2001) Adenylyl cyclase isoforms and signal integration in models of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 281:H1545–1552
Hashim S, Li Y, Nagakura A, Takeo S, Anand-Srivastava MB (2004) Modulation of g-protein expression and adenylyl cyclase signaling by high glucose in vascular smooth muscle. Cardiovasc Res 63:709–718
Li Y, Descorbeth M, Anand-Srivastava MB (2008) Role of oxidative stress in high glucose-induced decreased expression of gialpha proteins and adenylyl cyclase signaling in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 294:H2845–2854
Matsumoto T, Wakabayashi K, Kobayashi T, Kamata K (2005) Functional changes in adenylyl cyclases and associated decreases in relaxation responses in mesenteric arteries from diabetic rats. Am J Physiol Heart Circ Physiol 289:H2234–2243
Parodi J, Flores C, Aguayo C, Rudolph MI, Casanello P, Sobrevia L (2002) Inhibition of nitrobenzylthioinosine-sensitive adenosine transport by elevated d-glucose involves activation of p2y2 purinoceptors in human umbilical vein endothelial cells. Circ Res 90:570–577
Nilsson J, Nilsson LM, Chen YW, Molkentin JD, Erlinge D, Gomez MF (2006) High glucose activates nuclear factor of activated t cells in native vascular smooth muscle. Arterioscler Thromb Vasc Biol 26:794–800
Erlinge D. Extracellular atp: a central player in the regulation of vascular smooth muscle phenotype. Focus on "dual role of pka in phenotype modulation of vascular smooth muscle cells by extracellular atp". Am J Physiol Cell Physiol. 2004;287:C260–262
Brayden JE, Li Y, Tavares MJ (2013) Purinergic receptors regulate myogenic tone in cerebral parenchymal arterioles. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 33:293–299
Burnstock G, Ralevic V (2014) Purinergic signaling and blood vessels in health and disease. Pharmacol Rev 66:102–192
von Kugelgen I, Harden TK (2011) Molecular pharmacology, physiology, and structure of the p2y receptors. Adv Pharmacol 61:373–415
Communi D, Govaerts C, Parmentier M, Boeynaems JM (1997) Cloning of a human purinergic p2y receptor coupled to phospholipase c and adenylyl cyclase. J Biol Chem 272:31969–31973
Morita H, Sharada T, Takewaki T, Ito Y, Inoue R (2002) Multiple regulation by external atp of nifedipine-insensitive, high voltage-activated ca(2+) current in guinea-pig mesenteric terminal arteriole. J Physiol 539:805–816
Hoffmann C, Ziegler N, Reiner S, Krasel C, Lohse MJ (2008) Agonist-selective, receptor-specific interaction of human p2y receptors with beta-arrestin-1 and -2. J Biol Chem 283:30933–30941
Barragan-Iglesias P, Mendoza-Garces L, Pineda-Farias JB, Solano-Olivares V, Rodriguez-Silverio J, Flores-Murrieta FJ, Granados-Soto V, Rocha-Gonzalez HI (2015) Participation of peripheral p2y1, p2y6 and p2y11 receptors in formalin-induced inflammatory pain in rats. Pharmacol Biochem Behav 128:23–32
Barragan-Iglesias P, Pineda-Farias JB, Cervantes-Duran C, Bravo-Hernandez M, Rocha-Gonzalez HI, Murbartian J, Granados-Soto V (2014) Role of spinal p2y6 and p2y11 receptors in neuropathic pain in rats: possible involvement of glial cells. Molecular pain 10:29
King BF, Townsend-Nicholson A (2008) Involvement of p2y1 and p2y11 purinoceptors in parasympathetic inhibition of colonic smooth muscle. J Pharmacol Exp Ther 324:1055–1063
Ecke D, Hanck T, Tulapurkar ME, Schafer R, Kassack M, Stricker R, Reiser G (2008) Hetero-oligomerization of the p2y11 receptor with the p2y1 receptor controls the internalization and ligand selectivity of the p2y11 receptor. Biochem J 409:107–116
Kennedy C (2017) P2y11 receptors: properties, distribution and functions. Adv Exp Med Biol 1051:107–122
Nelson MT, Quayle JM (1995) Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol 268:C799–822
Cheong A, Dedman AM, Xu SZ, Beech DJ (2001) K(v)alpha1 channels in murine arterioles: differential cellular expression and regulation of diameter. Am J Physiol Heart Circ Physiol 281:H1057–1065
Amberg GC, Santana LF (2006) Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol 291:C348–356
Albarwani S, Nemetz LT, Madden JA, Tobin AA, England SK, Pratt PF, Rusch NJ. Voltage-gated k+ channels in rat small cerebral arteries: molecular identity of the functional channels. J Physiol. 2003
Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, Greenwood IA (2010) Participation of kcnq (kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol 588:3277–3293
Su W, Li W, Chen H, Liu H, Huang H, Li H (2015) Advanced glycation end products impair voltage-gated k+ channels-mediated coronary vasodilation in diabetic rats. PLoS ONE 10:e0142865
Nieves-Cintron M, Nystoriak MA, Prada MP, Johnson K, Fayer W, Dell'Acqua ML, Scott JD, Navedo MF. Selective downregulation of kv2.1 function contributes to enhanced arterial tone during diabetes. Journal of Biological Chemistry. 2015;290:7918–7929
Li H, Chai Q, Gutterman DD, Liu Y (2003) Elevated glucose impairs camp-mediated dilation by reducing kv channel activity in rat small coronary smooth muscle cells. Am J Physiol Heart Circ Physiol 285:H1213–1219
Li H, Gutterman DD, Rusch NJ, Bubolz A, Liu Y (2004) Nitration and functional loss of voltage-gated k+ channels in rat coronary microvessels exposed to high glucose. Diabetes 53:2436–2442
Iadecola C (2017) The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96:17–42
Morales-Cano D, Moreno L, Barreira B, Pandolfi R, Chamorro V, Jimenez R, Villamor E, Duarte J, Perez-Vizcaino F, Cogolludo A (2015) Kv7 channels critically determine coronary artery reactivity: left-right differences and down-regulation by hyperglycaemia. Cardiovasc Res 106:98–108
Thallas-Bonke V, Lindschau C, Rizkalla B, Bach LA, Boner G, Meier M, Haller H, Cooper ME, Forbes JM (2004) Attenuation of extracellular matrix accumulation in diabetic nephropathy by the advanced glycation end product cross-link breaker alt-711 via a protein kinase c-alpha-dependent pathway. Diabetes 53:2921–2930
Bu DX, Rai V, Shen X, Rosario R, Lu Y, D'Agati V, Yan SF, Friedman RA, Nuglozeh E, Schmidt AM (2010) Activation of the rock1 branch of the transforming growth factor-beta pathway contributes to rage-dependent acceleration of atherosclerosis in diabetic apoe-null mice. Circ Res 106:1040–1051
Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ (1995) Relaxation of arterial smooth muscle by calcium sparks. Science 270:633–637
Jaggar JH, Porter VA, Lederer WJ, Nelson MT (2000) Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278:C235–256
Lu T, He T, Katusic ZS, Lee HC (2006) Molecular mechanisms mediating inhibition of human large conductance ca2+-activated k+ channels by high glucose. Circ Res 99:607–616
Nystoriak MA, Nieves-Cintron M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell'acqua ML, Scott JD, Santana LF, Navedo MF (2014) Akap150 contributes to enhanced vascular tone by facilitating large-conductance ca2+-activated k+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res 114:607–615
Lu T, Chai Q, Yu L, d'Uscio LV, Katusic ZS, He T, Lee HC (2012) Reactive oxygen species signaling facilitates foxo-3a/fbxo-dependent vascular bk channel beta1 subunit degradation in diabetic mice. Diabetes 61:1860–1868
Flagg TP, Enkvetchakul D, Koster JC, Nichols CG (2010) Muscle katp channels: recent insights to energy sensing and myoprotection. Physiol Rev 90:799–829
Quayle JM, Nelson MT, Standen NB (1997) Atp-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev 77:1165–1232
Kawano T, Tanaka K, Mawatari K, Oshita S, Takahashi A, Nakaya Y. Hyperglycemia impairs isoflurane-induced adenosine triphosphate-sensitive potassium channel activation in vascular smooth muscle cells. Anesth Analg. 2008;106:858–864, table of contents
Zhou X, Abboud W, Manabat NC, Salem MR, Crystal GJ (1998) Isoflurane-induced dilation of porcine coronary arterioles is mediated by atp-sensitive potassium channels. Anesthesiology 89:182–189
Tanaka K, Kawano T, Nakamura A, Nazari H, Kawahito S, Oshita S, Takahashi A, Nakaya Y (2007) Isoflurane activates sarcolemmal adenosine triphosphate-sensitive potassium channels in vascular smooth muscle cells: a role for protein kinase a. Anesthesiology 106:984–991
Kinoshita H, Matsuda N, Iranami H, Ogawa K, Hatakeyama N, Azma T, Kawahito S, Yamazaki M (2012) Isoflurane pretreatment preserves adenosine triphosphate-sensitive k(+) channel function in the human artery exposed to oxidative stress caused by high glucose levels. Anesth Analg 115:54–61
Kinoshita H, Azma T, Iranami H, Nakahata K, Kimoto Y, Dojo M, Yuge O, Hatano Y (2006) Synthetic peroxisome proliferator-activated receptor-gamma agonists restore impaired vasorelaxation via atp-sensitive k+ channels by high glucose. J Pharmacol Exp Ther 318:312–318
Kinoshita H, Azma T, Nakahata K, Iranami H, Kimoto Y, Dojo M, Yuge O, Hatano Y (2004) Inhibitory effect of high concentration of glucose on relaxations to activation of atp-sensitive k+ channels in human omental artery. Arterioscler Thromb Vasc Biol 24:2290–2295
Kinoshita H, Matsuda N, Kaba H, Hatakeyama N, Azma T, Nakahata K, Kuroda Y, Tange K, Iranami H, Hatano Y (2008) Roles of phosphatidylinositol 3-kinase-akt and nadph oxidase in adenosine 5'-triphosphate-sensitive k+ channel function impaired by high glucose in the human artery. Hypertension 52:507–513
Perez GJ, Bonev AD, Patlak JB, Nelson MT (1999) Functional coupling of ryanodine receptors to kca channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol 113:229–238
Searls YM, Loganathan R, Smirnova IV, Stehno-Bittel L (2010) Intracellular ca2+ regulating proteins in vascular smooth muscle cells are altered with type 1 diabetes due to the direct effects of hyperglycemia. Cardiovascular diabetology 9:8
Gonzales AL, Amberg GC, Earley S (2010) Ca2+ release from the sarcoplasmic reticulum is required for sustained trpm4 activity in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 299:C279–288
Gonzales AL, Earley S (2012) Endogenous cytosolic ca(2+) buffering is necessary for trpm4 activity in cerebral artery smooth muscle cells. Cell Calcium 51:82–93
El-Najjar N, Kulkarni RP, Nader N, Hodeify R, Machaca K (2017) Effects of hyperglycemia on vascular smooth muscle ca(2+) signaling. Biomed Res Int 2017:3691349
Pyla R, Poulose N, Jun JY, Segar L (2013) Expression of conventional and novel glucose transporters, glut1, -9, -10, and -12, in vascular smooth muscle cells. Am J Physiol Cell Physiol 304:C574–589
Howard RL (1996) Down-regulation of glucose transport by elevated extracellular glucose concentrations in cultured rat aortic smooth muscle cells does not normalize intracellular glucose concentrations. J Lab Clin Med 127:504–515
Murata H, Hruz PW, Mueckler M (2000) The mechanism of insulin resistance caused by hiv protease inhibitor therapy. J Biol Chem 275:20251–20254
Murata H, Hruz PW, Mueckler M (2002) Indinavir inhibits the glucose transporter isoform glut4 at physiologic concentrations. AIDS 16:859–863
Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK (2004) L-type voltage-gated ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/srf-dependent mechanism. Circ Res 95:406–414
Park SH, Choi HJ, Lee JH, Yang IS, Han HJ (2001) High glucose stimulates ca2+ uptake via camp and plc/pkc pathways in primary cultured renal proximal tubule cells. Kidney Blood Press Res 24:10–17
Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF (2010) Increased coupled gating of l-type ca2+ channels during hypertension and timothy syndrome. Circ Res 106:748–756
Viard P, Butcher AJ, Halet G, Davies A, Nurnberg B, Heblich F, Dolphin AC (2004) Pi3k promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat Neurosci 7:939–946
Bubolz AH, Wu Q, Larsen BT, Gutterman DD, Liu Y (2007) Ebselen reduces nitration and restores voltage-gated potassium channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol 293:H2231–2237
Chai Q, Liu Z, Chen L (2005) Effects of streptozotocin-induced diabetes on kv channels in rat small coronary smooth muscle cells. The Chinese journal of physiology 48:57–63
Bubolz AH, Li H, Wu Q, Liu Y (2005) Enhanced oxidative stress impairs camp-mediated dilation by reducing kv channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol 289:H1873–1880
Nieves-Cintron M, Syed AU, Nystoriak MA, Navedo MF. Regulation of voltage-gated potassium channels in vascular smooth muscle during hypertension and metabolic disorders. Microcirculation. 2018;25
Dong L, Zheng YM, Van Riper D, Rathore R, Liu QH, Singer HA, Wang YX (2008) Functional and molecular evidence for impairment of calcium-activated potassium channels in type-1 diabetic cerebral artery smooth muscle cells. Journal of cerebral blood flow and metabolism : Official journal of the International Society of Cerebral Blood Flow and Metabolism 28:377–386
Lu T, Ye D, He T, Wang XL, Wang HL, Lee HC (2008) Impaired ca2+-dependent activation of large-conductance ca2+-activated k+ channels in the coronary artery smooth muscle cells of zucker diabetic fatty rats. Biophys J 95:5165–5177
Lu T, Zhang DM, Wang XL, He T, Wang RX, Chai Q, Katusic ZS, Lee HC (2010) Regulation of coronary arterial bk channels by caveolae-mediated angiotensin ii signaling in diabetes mellitus. Circ Res 106:1164–1173
Zhang DM, He T, Katusic ZS, Lee HC, Lu T (2010) Muscle-specific f-box only proteins facilitate bk channel beta(1) subunit downregulation in vascular smooth muscle cells of diabetes mellitus. Circ Res 107:1454–1459
Rueda A, Fernandez-Velasco M, Benitah JP, Gomez AM (2013) Abnormal ca2+ spark/stoc coupling in cerebral artery smooth muscle cells of obese type 2 diabetic mice. PLoS ONE 8:e53321
Lu T, Chai Q, Jiao G, Wang XL, Sun X, Furuseth JD, Stulak JM, Daly RC, Greason KL, Cha YM, Lee HC (2019) Downregulation of bk channel function and protein expression in coronary arteriolar smooth muscle cells of type 2 diabetic patients. Cardiovasc Res 115:145–153
McGahon MK, Dash DP, Arora A, Wall N, Dawicki J, Simpson DA, Scholfield CN, McGeown JG, Curtis TM (2007) Diabetes downregulates large-conductance ca2+-activated potassium beta 1 channel subunit in retinal arteriolar smooth muscle. Circ Res 100:703–711
Lu T, Sun X, Li Y, Chai Q, Wang XL, Lee HC (2017) Role of nrf2 signaling in the regulation of vascular bk channel beta1 subunit expression and bk channel function in high-fat diet-induced diabetic mice. Diabetes 66:2681–2690
Fernandez-Velasco M, Ruiz-Hurtado G, Gomez AM, Rueda A (2014) Ca(2+) handling alterations and vascular dysfunction in diabetes. Cell Calcium 56:397–407
Li Y, Wang XL, Sun X, Chai Q, Li J, Thompson B, Shen WK, Lu T, Lee HC (2017) Regulation of vascular large-conductance calcium-activated potassium channels by nrf2 signalling. Diab Vasc Dis Res 14:353–362
Kamata K, Miyata N, Kasuya Y (1989) Functional changes in potassium channels in aortas from rats with streptozotocin-induced diabetes. Eur J Pharmacol 166:319–323
Bouchard JF, Dumont EC, Lamontagne D (1999) Modification of vasodilator response in streptozotocin-induced diabetic rat. Can J Physiol Pharmacol 77:980–985
Lee CH, Dai ZK, Yen CT, Hsieh SL, Wu BN (2018) Kmup-1 protects against streptozotocin-induced mesenteric artery dysfunction via activation of atp-sensitive potassium channels. Pharmacol Rep 70:746–752
Li SS, Cui N, Yang Y, Trower TC, Wei YM, Wu Y, Zhang S, Jin X, Jiang C (2015) Impairment of the vascular katp channel imposes fatal susceptibility to experimental diabetes due to multi-organ injuries. J Cell Physiol 230:2915–2926
Li H, Shin SE, Seo MS, An JR, Ha KS, Han ET, Hong SH, Kim J, Yim MJ, Lee JM, An TG, Jeon J, Lee SJ, Na SH, Park WS (2018) Alterations of atp-sensitive k(+) channels in human umbilical arterial smooth muscle during gestational diabetes mellitus. Pflugers Arch 470:1325–1333
Wu BN, Lin RJ, Lin CY, Shen KP, Chiang LC, Chen IJ (2001) A xanthine-based kmup-1 with cyclic gmp enhancing and k(+) channels opening activities in rat aortic smooth muscle. Br J Pharmacol 134:265–274
Chung HH, Dai ZK, Wu BN, Yeh JL, Chai CY, Chu KS, Liu CP, Chen IJ (2010) The xanthine derivative kmup-1 inhibits models of pulmonary artery hypertension via increased no and cgmp-dependent inhibition of rhoa/rho kinase. Br J Pharmacol 160:971–986
Velmurugan GV, White C (2012) Calcium homeostasis in vascular smooth muscle cells is altered in type 2 diabetes by bcl-2 protein modulation of insp3r calcium release channels. Am J Physiol Heart Circ Physiol 302:H124–134
Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C (2007) Apoptosis regulation by bcl-x(l) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci U S A 104:12565–12570
Rossow CF, Dilly KW, Santana LF (2006) Differential calcineurin/nfatc3 activity contributes to the ito transmural gradient in the mouse heart. Circ Res 98:1306–1313
Nieves-Cintrón M, Amberg GC, Nichols CB, Molkentin JD, Santana LF (2007) Activation of nfatc3 down-regulates the β1 subunit of large conductance, calcium-activated k+ channels in arterial smooth muscle and contributes to hypertension. J Biol Chem 282:3231–3240
Oliveria SF, Dell'Acqua ML, Sather WA (2007) Akap79/150 anchoring of calcineurin controls neuronal l-type ca2+ channel activity and nuclear signaling. Neuron 55:261–275
Nieves-Cintron M, Hirenallur-Shanthappa D, Nygren PJ, Hinke SA, Dell'Acqua ML, Langeberg LK, Navedo M, Santana LF, Scott JD (2016) Akap150 participates in calcineurin/nfat activation during the down-regulation of voltage-gated k(+) currents in ventricular myocytes following myocardial infarction. Cell Signal 28:733–740
Smolkova K, Miko E, Kovacs T, Leguina-Ruzzi A, Sipos A, Bai P. Nuclear factor erythroid 2-related factor 2 in regulating cancer metabolism. Antioxid Redox Signal. 2020
Ramprasath T, Freddy AJ, Velmurugan G, Tomar D, Rekha B, Suvekbala V, Ramasamy S. Context-dependent regulation of nrf2/are axis on vascular cell function during hyperglycemic condition. Curr Diabetes Rev. 2020
Turczynska KM, Hellstrand P, Sward K, Albinsson S (2013) Regulation of vascular smooth muscle mechanotransduction by micrornas and l-type calcium channels. Commun Integr Biol 6:e22278
Turczynska KM, Sadegh MK, Hellstrand P, Sward K, Albinsson S (2012) Micrornas are essential for stretch-induced vascular smooth muscle contractile differentiation via microrna (mir)-145-dependent expression of l-type calcium channels. J Biol Chem 287:19199–19206
Hien TT, Garcia-Vaz E, Stenkula KG, Sjogren J, Nilsson J, Gomez MF, Albinsson S (2018) Microrna-dependent regulation of klf4 by glucose in vascular smooth muscle. J Cell Physiol 233:7195–7205
Gomez D, Swiatlowska P, Owens GK (2015) Epigenetic control of smooth muscle cell identity and lineage memory. Arterioscler Thromb Vasc Biol 35:2508–2516
Sward K, Stenkula KG, Rippe C, Alajbegovic A, Gomez MF, Albinsson S (2016) Emerging roles of the myocardin family of proteins in lipid and glucose metabolism. J Physiol 594:4741–4752
Xi G, Shen X, Wai C, White MF, Clemmons DR (2019) Hyperglycemia induces vascular smooth muscle cell dedifferentiation by suppressing insulin receptor substrate-1-mediated p53/klf4 complex stabilization. J Biol Chem 294:2407–2421
Chen Y, Zhao X, Wu H. Arterial stiffness: a focus on vascular calcification and its link to bone mineralization. Arteriosclerosis, thrombosis, and vascular biology. 2020:ATVBAHA120313131
Wang K, Deng X, Shen Z, Jia Y, Ding R, Li R, Liao X, Wang S, Ha Y, Kong Y, Wu Y, Guo J, Jie W (2017) High glucose promotes vascular smooth muscle cell proliferation by upregulating proto-oncogene serine/threonine-protein kinase pim-1 expression. Oncotarget 8:88320–88331
Casella S, Bielli A, Mauriello A, Orlandi A (2015) Molecular pathways regulating macrovascular pathology and vascular smooth muscle cells phenotype in type 2 diabetes. Int J Mol Sci 16:24353–24368
Chait A, Bornfeldt KE (2009) Diabetes and atherosclerosis: is there a role for hyperglycemia? J Lipid Res 50(Suppl):S335–339
Calcutt NA, Cooper ME, Kern TS, Schmidt AM (2009) Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials. Nat Rev Drug Discov 8:417–429
Danila MD, Piollet M, Aburel OM, Angoulvant D, Lefort C, Chadet S, Roger S, Muntean MD, Ivanes F (2020) Modulation of p2y11-related purinergic signaling in inflammation and cardio-metabolic diseases. Eur J Pharmacol 876:173060
Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I, Collaborative Study G (2001) Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345:851–860
Moser M (2002) Current recommendations for the treatment of hypertension: are they still valid? J Hypertens Suppl 20:S3–10
Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr, Williamson JD, Wright JT Jr (2018) 2017 acc/aha/aapa/abc/acpm/ags/apha/ash/aspc/nma/pcna guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Hypertension 71:e13–e115
Estacio RO, Jeffers BW, Hiatt WR, Biggerstaff SL, Gifford N, Schrier RW (1998) The effect of nisoldipine as compared with enalapril on cardiovascular outcomes in patients with non-insulin-dependent diabetes and hypertension. N Engl J Med 338:645–652
Muntwyler J, Follath F (2001) Calcium channel blockers in treatment of hypertension. Prog Cardiovasc Dis 44:207–216
Zakhari S (1986) Mechanism of action of calcium antagonists on myocardial and smooth muscle membranes. Drugs Exp Clin Res 12:817–829
Rusch NJ (1996) Potassium channels: new targets for drug treatment. Wis Med J 95:713–716
Lawson K (2000) Potassium channel openers as potential therapeutic weapons in ion channel disease. Kidney Int 57:838–845
Rubaiy HN (2016) The therapeutic agents that target atp-sensitive potassium channels. Acta Pharm 66:23–34
Acknowledgements
We thank members of the Navedo and Nieves-Cintron Labs for critically reading early versions of the manuscript. This work was supported by NIH grants R01HL098200, R01HL121059 and R01HL149127 (to MFN), UC MEXUS-CONACYT CN-19-147 (to MFN) and a UC Davis Academic Federation Innovative Development Award (to MN-C).
Author information
Authors and Affiliations
Contributions
MN-C and MFN provided overall direction and supervision of the work. VAF-T generated figures, wrote and revised the manuscript. MN-C, TL and MAM-AB wrote and revised the manuscript. MFN generated tables, wrote and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Nothing to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nieves-Cintrón, M., Flores-Tamez, V.A., Le, T. et al. Cellular and molecular effects of hyperglycemia on ion channels in vascular smooth muscle. Cell. Mol. Life Sci. 78, 31–61 (2021). https://doi.org/10.1007/s00018-020-03582-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03582-z