
Abstract
While scientific advances have led to large-scale production and widespread distribution of vaccines and antiviral drugs, viruses still remain a major cause of human diseases today. The ever-increasing reports of viral resistance and the emergence and re-emergence of viral epidemics pressure the health and scientific community to constantly find novel molecules with antiviral potential. This search involves numerous different approaches, and the use of antimicrobial peptides has presented itself as an interesting alternative. Even though the number of antimicrobial peptides with antiviral activity is still low, they already show immense potential to become pharmaceutically available antiviral drugs. Such peptides can originate from natural sources, such as those isolated from mammals and from animal venoms, or from artificial sources, when bioinformatics tools are used. This review aims to shed some light on antimicrobial peptides with antiviral activities against human viruses and update the data about the already well-known peptides that are still undergoing studies, emphasizing the most promising ones that may become medicines for clinical use.
Similar content being viewed by others

Introduction
Even though viral infections have been reported since ancient times, it was only during the nineteenth century that scientists were able to isolate “the filterable particles”, later called viruses. Since then, major breakthroughs regarding the control of viral reproduction, infection, and the production of vaccines have led to remarkable advances in the human–virus interaction, such as smallpox eradication and the control of measles and poliomyelitis transmission. Nevertheless, viruses still remain one of the main causes of human diseases, mainly because the discovery and development of new vaccines is usually challenging and time consuming [1]. For this reason, the most commonly utilized alternative available for viral control is treatment with antiviral drugs [2, 3]. In an overall view, the most common mechanisms of action for antiviral drugs are virus-targeting antivirals and host-targeting antivirals [3]. Virus-targeting antivirals focus on the inhibition of important transcription and replication enzymes, such as proteases and polymerases [4, 5], or the direct inactivation of viral structural proteins [6]. In contrast, host-targeting antivirals focus on: the inhibition cyclophilins, known to be important cellular factors that are hijacked by some viruses during the replication cycle [3]; the use of immunomodulators such as interferons [7, 8]; and gamma globulins [9].
First-generation antiviral molecules (described in the 60 s and early 70 s) had serious side effects on humans due to their poor specificity. For example, vidarabine, an adenosine analog used as a replication inhibitor, can affect not only viral DNA polymerase, but also the eukaryotic analog [10]. The advance of research in the area led to the description of better molecules, such as acyclovir, the first nucleoside analog, and antiviral drug to be considered successful for the treatment of herpes simplex virus (HSV) and varicella zoster virus (VZV) infections. Because of its specificity—it requires a phosphorylation step mediated by a viral protein—this molecule causes lower toxicity for the host when compared with previously used treatments [10, 11]. Unfortunately, the low efficacy of antiviral treatments is still evidenced by the ever-increasing reports of viral resistance [12,13,14], concomitant viral infections [15], and the emergence and re-emergence of viral epidemics in relatively short periods of time, as observed for H1N1, Ebola and zika virus (ZIKV) only in the first 5 years of the present decade [16,17,18,19,20]. Therefore, the demand for production of new antiviral drugs is higher than ever, with increased preference for molecules capable of presenting broad-spectrum activity [21]. The search for these new molecules involves different approaches such as bioinformatics-assisted predictions based on molecule interaction with important viral structures or enzymes [22,23,24] and the isolation of new compounds obtained from natural sources [25,26,27]. Using such techniques, many new molecules have been described so far, and, most recently, the description of antimicrobial peptides has been gaining attention [28,29,30,31,32].
Recent evidence highlights the function of antiviral proteinaceous compounds as a defensive barrier, and it is being demonstrated that some antimicrobial peptides may also present activity against a broad range of viruses, thus being called antiviral peptides (AVPs) [33,34,35,36]. These molecules can also be obtained through the utilization of bioinformatic tools, and are then called designed or artificial AVPs. They can derive from bait studies where an artificial peptide is tested for interaction against a specific target, such as a surface glycoprotein or an important viral enzyme [37]; or obtained in silico using specific software designed for the prediction of peptides [38, 39]. In both cases, many settings are taken into consideration, such as the topology, amino acid composition, charge, and many other chemical and structural characteristics that may influence the antiviral activity of a peptide [40, 41].
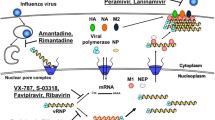
The study of AVPs has been the focus of numerous research projects in recent years, and the structures and mechanisms of action of such molecules have been previously reviewed and even compiled in online databases such as the antiviral peptide database (AVPdb—http://crdd.osdd.net/servers/avpdb/) with 2683 entries of experimentally tested peptides [42,43,44,45]. Regarding their mechanism of action (Fig. 1), these are mostly called virucidal when they act directly by inhibiting the viral particle; or by competing for the protein link site in the host cell membrane, interfering in their interaction and consequent adsorption [46]. However, they may also act in other stages of the viral cycle, causing, for example, the suppression of viral gene expression [47, 48].

Antiviral peptide inhibition sites on viral replication cycle. The antiviral peptides with a described mechanism of action were placed in their inhibition sites as follows: 1, virion inhibition; 2, adsorption; 3, viral penetration; 4, endosomal escape; 5, viral uncoating; 6, viral genome replication and 7, release of mature virions
Due to the increasing number of studies demonstrating the antiviral activity of peptides and the pressing need for new antiviral drugs, this article aimed to compile relevant information from the most promising antiviral peptides that may become effective drugs and that are still undergoing studies and/or in clinical trials.
Natural AMPS with antiviral activities: the AVPs
Peptides have been studied for at least 40 decades, and a broad spectrum of biological activities has been described so far. In some cases, a given peptide shows more than one activity and is, therefore, called a promiscuous peptide. The overall biochemical features of AVPs are cationic and amphipathic characteristics and positive net charges, all of which are essential for these peptides to work as antimicrobials [49]. Moreover, there is no evidence of great physicochemical differences between AMPs and other AVPs, as Wang et al., showed in a large-scale analysis correlating antimicrobial activities with amphipathicity and charge. However, data show that hydrophobicity seems to be a very important property for those peptides with activity against enveloped viruses [50, 51]. Among the advantages of natural AVPs, one can cite the possibility of high specificity and effectiveness, low toxicity and peptidase biodegradability (which limits the accumulation in tissues), and low molecular weight. Otherwise, short half-life, immunogenic potential, high cost of production, and low oral absorption are some of the limitations in the use of those compounds as antivirals [46]. In the following sections, we provide a description of naturally derived AVPs, and their mode of action and potential as a pharmaceutical molecule. Moreover, some examples of antiviral peptide structures and alignments are shown in Figs. 2 and 3, respectively.

Structure of some AVPs already described. Magainin 1 and 2, melittin, lactarcin, clavanin, dermaseptin S4, lactoferricin, HNP1 and 4, HBD 2 and 3, protegrin, and temporin B

Antimicrobial peptides aligned per family
AVPs derived from plants
Plants utilize a diverse array of small, cationic, cysteine-rich proteins as toxic weapons to fend off pest and pathogen attack. These peptides are well known for their capacity to obstruct a wide array of virus infections that cause economically important diseases in crops [43, 52]. In recent years, plant-derived defensive peptides have become the focus of numerous studies for their potential use as novel molecules in the treatment of human viral diseases. Cyclotides are a large family of plant-derived peptides characterized by a cyclic backbone and three conserved disulfide bonds that form a knot-like rigid structure [53]. They have a broad range of biological roles, including antimicrobial, anthelminthic, nematocidal, and insecticidal activities [53, 54]. Cyclotides from different plant species have been significantly investigated for their ability to hinder the growth of viruses involved in human diseases, such as human immunodeficiency virus (HIV), influenza H1N1, and dengue (DENV) [55,56,57,58,59]. One example is kalata B1, a well-studied cyclotide found in the leaves of the African plant Oldenlandia affinis. Kalata B1 shows strong anti-HIV activity, being capable of destroying the viral particles prior to cell entry as well as inhibiting fusion of the virus to the host membrane [57, 60]. The antiviral activity of kalata B1 and other plant cyclotides seems to rely on the same mechanism utilized by these peptides to hinder the growth of microbial pathogens: its ability to bind and disrupt the pathogen cell membrane. Due to their cationic and amphipathic nature, as well as their small size, cyclotides can easily bind and aggregate within lipids of bacterial and fungal membranes. This interaction disrupts the membrane organization, leading to the formation of pores that cause leakage of internal cell components and, consequently, cell death [52, 54]. Enveloped viruses such as HIV use the host cell membrane to form an outer wrapping composed of specific lipid microdomains that are necessary for the virus replication cycle [61]. Thus, cyclotides are also capable of disturbing the virus’ lipid envelope, leading to inhibition of viral fusion to the target cells and to destruction of the viral particles [57, 62, 63]. The pharmaceutical potential of cyclotides has been highlighted by their aforementioned antiviral properties and also by their capacity to tolerate a high number of substitutions in their amino acid backbone while maintaining their biological activities. For this reason, cyclotides are constantly used as scaffolds for novel drug design [64]. As an example, amino acid modifications performed on kalata B1 allowed the design of a novel “kalata B1-inspired peptide” that presents activity against the dengue virus [58].
Other plant defense-related peptides may curtail human virus infections by interaction and interference with proteins that are fundamental for the viral replication cycle. For example, phaseococcin and sesquin, two antimicrobial peptides isolated, respectively, from the seeds of runner beans (Phaseolus coccineus) and ground beans (Vigna sesquipedalis) are capable of inhibiting the reverse transcriptase activity of the HIV virus, thus hindering viral replication [65, 66]. HSV-1 virucidal activity observed in a 2 kDa antiviral peptide isolated from Sorghum bicolor seeds may be explained by the capacity of the peptide to bind and mask essential viral envelope proteins [67]. In fact, modulation in the activity of enzymes involved in viral infection and replication appears to be one of the most prominent mechanisms utilized by plant peptides to inhibit viral proliferation [68].
Arthropod-derived AVPs
Arthropods are a rich source of compounds with diverse activities, including several antimicrobial peptides. Surprisingly, until recently, there were not many arthropod-derived molecules with described antiviral activity [69]. This situation appears to be changing, as numerous novel molecules with antiviral properties have now been isolated from these organisms. For example, cecropin A, a 37-amino acid peptide derived from the moth Hyalophora cecropia [70], showed inhibitory activity against HIV through a mechanism that seems to suppress viral gene expression [71]. In 2004, another assay showed that cecropin A also has inhibitory activity against herpes simplex virus 1 and 2 (HSV) and against Junin virus (JV). The replication inhibition of JV has reached 90% at 40 µM [72]. Bee (Apis mellifera) venom is also an interesting source of AVP. One example of bee venom-derived AVP is melittin, which was tested against HIV-1 in an assay with infected T lymphoma cells. Interestingly, the cell culture treated with melittin showed an almost total absence of viral particles [73]. This peptide was also tested against HSV-1, 2, and JV, showing high inhibition of viral replication for all tested viruses at relatively low concentrations (3 µM) [72]. In a previous study carried out by Wachinger et al. [73], the action of the antiviral melittin against HIV-1 had already been proven. It is believed that peptide activity against enveloped viruses relies on viral envelope lysis [46]. Recently, Hood et al. [74] have shown that nanoparticles incorporated with melittin were able to prevent in vitro HIV infection, and were not toxic for the vaginal epithelium cells used in the assay.
Arthropod venoms are a recurring theme in studies searching for new AVPs. Recently, researchers used mastoparan, a peptide derived from wasp venom (Vespula lewisii), to develop a new peptide, mastoparan 7, which showed broad antiviral activity against enveloped viruses. From the ten viruses tested, this AVP reached 99% of replication inhibition in eight of them. In pre-incubation, in vivo assays performed with Indian vesicular stomatitis virus (VSV) and MP7-NH2; the peptide made the virus non-infectious and unable to replicate in vivo. In a transmission electron microscopy (TEM) assay, it was observed that VSV virions treated with mastoparan 7 had a significant envelope disruption, appearing to be separated from the capsid [75].
Lactarcin 1, derived from the venom of Lachesana tarabaeve, a spider from central Asia, demonstrated inhibitory activity against DENV2. A protein–protein docking analysis suggested that lactarcin 1 could bind to the viral protease NS2B-NS3 (NS2B-NS3Bpro) near its active site, which was verified with ELISA and western blot assays, and antiviral activity confirmed in cell culture assays, reaching viral inhibition at 12.68 µM at 37 °C and 6.58 µM at 40 °C. This viral protease seems to be vital for viral replication, and its blocking leads to consequent dengue inhibition [27].
Venoms from scorpion are another rich source of arthropod venom-derived AVPs [76]. The venomous secretion of the Asian forest scorpion (Heterometrus petersii) contains the peptides Hp1090, Hp1239, and Hp1036, which are capable of inhibiting the replication of hepatitis C virus (HCV), preventing infection installation [77]. Further assays using Hp1239 and Hp1036 against HSV-1 showed that these AVPs were capable of blocking the virus attachment to the target cell and of inactivating viral particles that had already attached but not yet penetrated [78]. Mucroporin-M1, an artificially designed peptide derived from the Chinese swimming scorpion (Lychas mucronatus), presented outstanding inhibitory activity against the measles virus, influenza H5N1, and severe acute respiratory syndrome coronavirus (SARS-CoV). The difference between mucroporin-M1 and the original molecule is the exchange of all glycine and proline residues by arginine or lysine (G3R, P6K, G10K, and G11R), which gives the variant a greater positive net charge on the peptide hydrophilic side, improving interaction with the virus envelopes. For this reason, the authors suggested a virucidal mechanism of action [79]. In a further study, mucroporin-M1 showed activity against the hepatitis B virus (HBV) both in vitro and in vivo, inhibiting viral replication by activating the MAP kinase route and decreasing the expression of HNF4α, an important factor for HBV replication [80]. In 2012, Chen et al. [81] synthesized a 13-amino acid residue peptide named Kn2-7 from the improvement of the Bmkn1 peptide, derived from Mesobuthus martensii scorpion venom. Kn2-7 showed low cytotoxicity and antiviral activity against 13 variants of the HIV-1 subtype B, reaching almost 99% of viral inhibition at 16 µg/mL, by direct inactivation of the viral particle, presenting itself as a promising antiviral drug candidate. Recently, Zeng et al. [82] screened venom peptides derived from Euscorpiops validus scorpion and identified Eva1418 as an antiviral peptide against HSV-1. In this study, the authors tried to improve this peptide’s cellular uptake and intracellular distribution, by introducing histidine residues that would enhance helicity and amphiphilicity. The results showed that modified peptide Eva1418-FH5 had the lowest cytotoxicity, highest antiviral activity against HSV-1, enhanced cellular uptake, and better cellular distribution.
Finally, the peptides alloferon 1 and 2, derived from the hemolymph of blowfly (Calliphora vicina), showed antitumor activity as well as antiviral activity against two influenza variants. These peptides were also tested regarding their immunomodulatory activity, and it was demonstrated that alloferon 1 and 2 have the capacity to influence the activation of natural killer cells and host release of interferon, an important cytokine involved in the immune response process [83]. In more recent studies, Kuczer et al. [69, 84, 85] tested alloferon 1 and some analogs against HSV-1 and against coxsackievirus B2 (CBV-2) in three different types of cell culture. The results showed inhibitory activity of the original peptide and analogs 1 and 2, which considerably diminished viral replication at 24 h of contact in the in vitro assay, and of compounds 2, 4, 5, and 9, all against HSV-1. The analogs that showed the best inhibitory activity against CBV-2 were analogs 1 and 4, both with modifications in their N-terminal portions. Alloferon 1 is a promising candidate for the design of new AVPs, due to its optimal antiviral activity and absence of toxicity towards mammalian cells. However, its mechanism of action remains to be fully elucidated [85].
AVPs derived from amphibians
Frog skin is considered an abundant source of antimicrobial peptides; however, there are still not many frog-derived peptides with antiviral activities described in the literature. They are produced in dermal glands, deployed in events of stress and, generally, present a cationic, amphipathic α-helical secondary structure, with 10–50 amino acids [86, 87]. Examples are magainin 1 and 2, derived from the frog Xenopus laevis, with 23 amino acid residues each. These AVPs were tested against HSV-1 and -2, and showed efficient inhibition of both viruses [72]. Previously, some magainin variants were also tested against HSV-1, and the ones presenting lysine-rich regions or many lysine residues in their structure showed the best results in inhibiting the virus. The authors suggested that cationic charge associated with an amphipathic structure may enable these peptides to interact with the viral envelope anionic phospholipids, consequently disrupting its structure by some unknown mechanism and exerting virucidal activity [88]. In 2010, Dean et al. [89] tested an alanine-substituted magainin-2 amide along with three other peptides against vaccinia virus to evaluate their virucidal activity, and the magainin-2 variant showed satisfactory activity against the virus, attacking its envelope.
Another peptide capable of targeting viral envelopes is temporin B, derived from the frog species Rana temporaria. This AVP showed high antiviral activity against HSV-1 in in vitro plaque assays. The authors showed that temporin B inhibited HSV-1 in a dose-dependent manner (reaching IC50 at 2.507 µg/ml); interfered in cell-to-cell spread of the virus, suggesting that the peptide may target other cellular mechanisms used by the virus; and had virucidal action reaching 99.99% of inhibition, suggesting that the peptide acts on the viral particle. To verify the hypothesis, a TEM study was conducted and showed that virions treated with temporin B had a loss of envelope integrity [86].
Dermaseptins are a large family of antimicrobial peptides produced by frogs from the Phyllomedusa genus. The antiviral activity of these peptides had already been described against HSV-1, -2, and HIV-1 [22, 90,91,92], where viral envelopes appear to be the preferential target of dermaseptin S4 and its derivatives. Most recently, derivatives of dermaseptins S3, S4, and S4 derivatives have been tested against rabies virus in both in vitro and in vivo assays. S4 and an S4 derivative, where methionine 4 was exchanged for a lysine (S4M4K) derivative, showed the strongest antiviral activity, reaching over 85% of inhibition from 7.5 µM to 10 µM in the cell culture assay. Moreover, when tested in mice, S4M4K showed a protective effect by increasing the survival rate of treated mice, in relation to control mice, by 62.5% at 100 µg and 75% at 200 µg. Thus, the authors suggested that there was a specific exchange of a Met4 by a Lys4 in dermaseptin S4, therefore, creating S4M4K and decreasing cytotoxicity because of the substitution of a hydrophobic with a positively net charged amino acid. Given these results, not only do these dermaseptins have a virucidal mechanism of action, but they also affect early stages of the intracellular infection of the rabies virus [93].
Moreover, HS-1, an AVP derived from anuran Hypsiboas semilineatus skin, showed remarkable antiviral activity against DENV2 and 3. More specifically, in the pre-treatment and inhibition of viral adsorption assays, the percentage of inhibition reached almost 100% for both viruses, but no antiviral activity was observed in other stages of the viral cycle, which led the authors to infer that HS-1 acts upon the early stages of the infection. To corroborate these results, a qPCR assay and atomic force microscopy analysis were conducted, showing not only that the number of viral genomes of both viruses was reduced but also that the viral envelope appeared disturbed by some invaginations when in contact with the peptide [94].
A last example of virucidal frog peptide is urumin, an AVP derived from the Indian frog Hydrophylax bahuvistara. Urumin showed strong inhibitory activity against influenza virus in both in vitro and in vivo tests. In the in vitro assays, urumin showed inhibition against all strains of H1N1 and H1N2, but H3N1 and H3N2 were unaffected. This fact led the authors to suggest that urumin was able to interact with hemagglutinin 1 (H1); a TEM assay was, therefore, conducted, and this showed that influenza virions were destroyed. In the in vivo assays, BALB/C mice were treated with urumin, administered intranasally, and then inoculated with influenza. The results showed that urumin-treated mice had less morbidity than the non-treated [95].
AVPs derived from marine organisms
Another rich source of AVPs is found among aquatic living beings. The most studied of them are possibly the clavanins, a class of peptides derived from a tunicate called Styela clava. In a study performed by Yasin et al., clavanin AK showed inhibition against HSV-1 (percentage of inhibition of 70.4%) [96]. Clavanin A had been tested against rotavirus and adenovirus, both non-enveloped, and in vitro, it was possible to verify the viral inactivation before inoculation in cell culture, in a pre-treatment step with the peptide, and after inoculation in the cell culture—called a simultaneous test—clavanin A showed better inhibition of rotavirus during the pre-treatment, reaching 95% of inhibition at 50 µM. However, the peptide’s best activity against adenovirus happened in the simultaneous test, reaching 94% of inhibition at 25 µM [97]. Clavanin B also showed inhibitory activity against HIV [55].
In addition, the four cyclic depsipeptides, mirabamides E, F, G, and H, isolated from the marine sponge Stelletta clavosa, had their chemical properties and structures defined and were tested against an HIV strain showing strong inhibitory activity. In the in vitro assays, the peptide mirabamide H showed the best antiviral activity with an IC50 value around 40 µM, followed by F and G, both with 65 µM, and mirabamide E with 120 µM [98]. Most recently, in a study carried out by Migliolo et al. (2012), the peptide Pa-MAP 1, derived from the polar fish called Pleunorectus americanus, had its structural, physicochemical, and antimicrobial properties extensively studied and showed not only antibacterial and antifungal activity, but also anti-tumoral and finally antiviral activity against HSV-1 (94%) and -2 (97%) [99]. In later assays, it was found that Pa-MAP 1 has a virucidal mechanism of action, which means that this peptide possibly interacts with some viral surface glycoprotein [100].
AVPs derived from mammals
Among the antimicrobial peptides derived from mammals, two main families are frequently studied for their antiviral properties: the defensins and the cathelicidins. Both have cationic charges and amphipathic properties, but defensins have a predominance of β sheets stabilized by three disulfide bonds and range from 18 to 45 amino acid residues [101], whereas cathelicidins have an N-terminal signal sequence, a conserved cathelin-like domain and a variable C-terminal domain that needs proteolytic cleavage to be matured and range from 12 to 88 amino acid residues [44]. The defensins are produced by keratinocytes upon a skin lesion and are classified in three groups; α-defensins, also known as human neutrophil peptide (HNP), β-defensins (HBD), and θ-defensins, which are not produced by higher primates such as human beings [101]. Regarding their antiviral activity, the defensin HNPs 1, 2, and 4 showed antiviral activity against HIV, among which HNP-4 presented the best results [102, 103]. In another study, Salvatore et al. [104] also showed the activity of HNP-1 against the influenza A virus and suggested that this peptide inhibits the virus in its most initial stages of the cycle. To establish the infection, influenza uses the PKC signaling pathway of the cell, and the data showed that PKC phosphorylation was decreased in infected cells treated with HNP-1, showing that it interfered with the viral cycle [104].
On the other hand, from the many human β-defensins already described, two stand out for their potential antiviral activity, HBD-2 and HBD-3, as both have been described as having their production induced by viral presence in the organism. Meyer-Hoffert et al. [105] investigated the presence of these peptides in lesions of verrucae vulgaris and condylomata acuminata, both cutaneous lesions caused by different subtypes of HPV. The results showed an increased expression of HDB-2 and HDB-3 during viral infection. However, the authors were unable to ensure if the expression of those peptides is, in fact, induced by HPV infection, since it can also be induced by pro-inflammatory cytokines, and they suggested further studies to address the matter. Moreover, HBD-3 also demonstrates the capability of inhibiting the vaccinia virus, demonstrating that the presence of the virus induces the production of this peptide in keratinocytes [106]. In addition, another study showed VZV inhibition in keratinocytes by HBD-2 [35]. In 2003, Quiñones-Mateu et al. [107] showed the antiviral activity of HBD-2 and HBD-3 against HIV, and Zapata et al. [48] recently showed HDB2 and HBD-3 mRNA expression in HIV-1-exposed seronegative (HESN) individuals and in seropositive patients. The results suggested that HBDs can inhibit the virus not only in the early steps of the cycle, but also the late reverse transcripts and nuclear import of HIV; and indicated that HBD-3 had the best results. Finally, the authors proposed these peptides could be used in retroviral therapy in the future.
Furthermore, cathelicidin LL-37 [108] demonstrates inhibitory activity toward several enveloped viruses, including: VZV; the vaccinia virus (VV), HSV-1; HIV; the syncytial respiratory virus (RSV); influenza A virus; HCV; dengue virus serotype 2; Zika virus; and, more recently, Venezuelan equine encephalitis virus (VEEV), which can infect both equines and humans [35, 45, 109,110,111,112,113,114]. The proposed mechanism of action against enveloped viruses is described as damaging the envelope or protecting the target cells against infection. The modulation of viral components necessary for replication and infection (such as HIV reverse transcriptase) has also been described [44]. Moreover, LL-37 showed antiviral activity against non-enveloped virus such as adenovirus, Aichi virus, and rhinovirus [100, 115, 116]. However, it is suggested that this peptide uses a different mechanism to inhibit non-enveloped viruses such as the adenovirus strains. Therefore, more studies are needed for elucidation of this matter [44].
Besides defensins and cathelicidins, there are other peptides found in mammals, such as lactoferrin, a peptide derived from mammals’ milk and studied for the past 30 years, which possesses various antimicrobial and immunomodulatory properties. Lactoferrin’s antiviral activity has already been described against many viruses such as CMV, HSV-1, and -2, adenovirus, rotavirus, poliovirus, RSV, HIV, influenza, HCV, HBV, and, recently, bovine lactoferrin showed activity against dengue, chikungunya, and Zika viruses, all three transmitted by the Aedes aegypti mosquito [117,118,119,120]. Lactoferricin, a smaller peptide derived from the N-terminal region of lactoferrin, has also been described as an antiviral peptide. Its inhibitory activity has been showed against various viruses such as CMV, in which a cyclic form of lactoferricin was able to prevent viral entry into fibroblasts [121]. Variants of this peptide were also tested against HPV, demonstrating different percentages of inhibition in in vitro tests [122]. Lactoferricin was further tested against HSV-1 in an in vitro assay, where the results showed that intracellular HSV-1 trafficking is delayed in the presence of this peptide. The molecular mechanism of action proposed possibly involves an interference with the host cell microtubules, which are needed for successful viral replication [123]. Moreover, when this peptide was tested against HSV-2 in a well-established in vivo assay, female C57/BL6 mice were inoculated with the virus and the peptide, and showed no signs of disease [124]. Finally, Wang et al. [125] showed that when in the presence of lactoferricin, there is a reduction of the HIV-1 integrase nuclear distribution, the mechanism used by the virus to integrate its genetic material within the cell's, mediated by its own enzyme, called integrase.
In addition, protegrin-1, a cyclical cationic peptide with 18 amino acid residues, derived from swine white blood cells, showed activity against dengue virus. This peptide was able to inhibit the specific viral protease important for its replication, named NS2B-NS3pro [28]. Interestingly, a protegrin-1 enantiomer, composed entirely of amino acids in their D form, showed activity against HSV-1 (82%) and HSV-2 (75%) [96]. Finally, there is CYVIP derived from human hemofiltrate, which showed efficient inhibition of CMV and HSV-1 in vitro. Its mechanism of action resides in the peptide’s interaction with the heparan sulfate receptors in the host cell surfaces, which are also these viruses’ target. These receptors are used by the viral surface glycoproteins for its adsorption, and, therefore, if its binding sites are not available, adsorption would be compromised [126].
Finally, Sala et al. [127] evaluated several synthetic peptides derived from different human serum proteins against four viruses. Among them, the peptide called KP showed remarkable antiviral activity against HSV-1 (reaching 99% of inhibition), in a virucidal manner. Against VSV, another enveloped virus, the inhibition rate reached only 78%. The authors suggested that KP shows hydrophobic amino acids in its sequence that may interact with viral envelope lipids and/or glycoproteins. Nevertheless, the different composition and organization of HSV-1 and VSV enveloped may have influenced the different results.
Rationally designed AVPs
Besides the description of natural peptides, another way of discovering new antiviral drugs is the prediction and rational design of novel molecules. Basically, there are three major methods used for rational design: template-based design, physicochemical, and de novo methods, all of which aim to create novel peptides and/or improve already existing ones. Template-based design aims to add selectivity and/or increase activity of a known peptide sequence, by including an amino acid or by changing its position, therefore, reducing the peptide sizes. These modifications can lead to the creation of a novel AVP even from inactive peptides. The physicochemical design also uses a known sequence to generate analogs with different physicochemical properties. Finally, the de novo method creates new peptides using amino acid patterns or frequencies [128]. Recently, a new algorithm was created specifically to predict antiviral peptides, called AntiVPP 1.0 (available at https://github.com/bio-coding/AntiVPP). The authors suggest that this algorithm is a fast, accurate, and intuitive tool, and stated that the number of hydrogen-bond donors is an important feature to be considered in the development of AVP prediction algorithms [129].
When these methods are applied to AVP design, researchers usually base their new molecules on specific viral structures such as surface glycoproteins [37], viral proteases, and other important enzymes brought within the viral particles. Therefore, the knowledge of the viral genome, protein structure, replication cycle, and host cell targets is of great importance for the description of efficient inhibitors [23]. Interestingly, in some cases, a peptide designed to target a specific virus can be used as a template for the design of a new peptide against a different virus [130]. Viral surface glycoproteins are often a target, because they are essential in both the entry and penetration process, and require conformational changes given the protein–protein interaction which they undergo [23]. In addition, their sequence has conserved regions that are used as models for new molecules [131]. Given that the rational design of AVPs tends to be virus-specific, in the following section of the review, we have listed the most recent AVPs separated by the virus which they target.
Dengue virus as a scaffold for peptide design
Dengue virus is a mosquito-borne Flavivirus. Its genome encodes three structural proteins: capsid (C), pre-membrane (prM) and envelope (E), and seven non-structural proteins (NSs). The E protein is glycosylated and has an important role in viral adsorption and entry, with 500 amino acids; a C-terminal that consist of a highly conserved stem region, formed by two helices (H1 and H2) and partially inserted in the lipid envelope; and finally, the N-terminal ectodomain formed by three domains (EDI, EDII, and EDII) [132]. DN59, a peptide mimetic to the E protein stem region, was tested against DENV2 and WNV, showing inhibition of both viruses at a concentration lower than 25 µM [133], and in a more recent assay, when tested against all four dengue serotypes, DN59 showed inhibition of infectivity by the release of the viral genome through holes formed in the envelope. Moreover, in in vitro assays with mammalian epithelial and mosquito cells, no toxicity was found. Inhibition was neither seen in pre-incubation assays with the peptide nor in post-infection assays—only in co-incubation—proving that DN59 clearly attacks the viral particle directly [134].
Costin et al. [135] designed various peptides that mimic sequences from EDII, near the EDI/EDII hinge region, and from an extended beta sheet region comprising the first connection between EDI and EDII. After computational work aiming to optimize their structure, seven artificial AVPs were selected for synthesis and in vitro challenges against DENV2. The results showed that the peptides DN57opt, DN81opt and 1OAN1 were able to inhibit the virus at concentrations of 50 µM (97, 57, and 99%, respectively). Furthermore, DN57opt and 1OAN1 were chosen for cryoEM assay, and the results showed that the viral particles treated with these peptides lost icosahedral symmetry, leading to inhibition of viral entry [135].
Recently, Cui et al. [131] tested the hypothesis that dengue E protein could interact with β3 integrin (a host cell surface receptor). After some specific assays, the occurrence of this interaction was proven. Therefore, eight peptides were synthesized based on the structure of the E protein. The results showed that the two peptides designed based on the EDIII region, named P4 and P7, were able to inhibit the interaction between the DENV 2 E protein and β3 integrin, and only P7 against DENV 1. The authors also mentioned that the mechanism of inhibition of these two peptides is by occupying the binding site in β3 integrin and, consequently, preventing viral entry in the host cell [131].
AVPs designed from herpes simplex virus
Glycoprotein B is a well-conserved and important surface protein present in the HSV-1 and 2 envelopes, which, along with 10 other surface glycoproteins (gC-gM), is involved in virus attachment and penetration in the host cells [136]. Its structure has been the target of assays in search of an HSV entry inhibitor, such as the one conducted by Akkarawongsa et al. [137], in which multiple peptides homologous to gB were synthesized and tested against HSV-1. The results showed that, from the 138 molecules synthesized, gB94, gB122, and gB131 had the best inhibitory activities at concentrations lower than 20 µM. Further assays performed to infer the mechanism of action showed that gB122 and gB131 were able to interfere in the HSV-1 entry step with an EC50 of 12 and 18 µM, respectively; however, gB94 could only inhibit direct viral particles at higher concentrations (125 µM). Finally, VP16 translocation and ICP0 expression, a viral tegument protein and an immediate early protein, respectively, were measured to confirm gB122 activity at an early step of infection, showing a reduction in nuclear VP16 and ICP0 expression in the presence of the peptide [137].
In another study, peptides were synthesized based on a highly conserved amino acid sequence from both gB and gH for inhibition assays against HSV-1 and -2 and, given the established parameters, only four peptides (two from each glycoprotein) were selected for virucidal and antiviral in vitro assays. Interestingly, both peptides derived from gB (U-1 and U-2) showed virucidal activity against both viruses, reaching 80% of inhibition at 100 µM, whereas CB-2, derived from gH, was able to inhibit HSV-1 infectivity particles in more than 90%, but with higher concentrations, and it also presented limited activity against HSV-2. Almost the same was observed in the antiviral assay: while gB peptides showed more than 80% of inhibition against both viruses (U-1 at 6.25 µM, and U-2 at 50 µM), gH peptides CB-1 showed better protection against HSV-2 (90%) and CB-2 against HSV-1, again in much higher concentrations, 250 and 500 µM, respectively. Further assays led the authors to suggest that U-1 and U-2 may act in a post-binding step, possibly in upstream components of the entry machinery, and that CB-1 and CB-2 may interact with gC, the least herpes-conserved glycoprotein. In conclusion, further improvement of gH derived peptides are needed to diminish the concentration necessary to achieve satisfactory viral inhibition [138].
Human immunodeficiency virus
Among all the viruses causing human diseases, none has ever caused more concern worldwide than HIV, with more than 36 million people infected [139]. The currently available antiviral treatment targets four steps of the viral cycle: viral entry, reverse transcription, integration, and virion maturation [140]. However, since HIV-1 and HIV-2 have different evolutionary histories, sharing only 50% of genetic similarity, most antivirals are capable of inhibiting only HIV-1, and a few inhibit HIV-2 [141, 142]. Recently, many studies have focused on entry/fusion inhibitor molecules, mainly the ones targeting the gp41 of the HIV envelope, which is an important glycoprotein for viral fusion and entry into the host cells [6]. The gp41 has 345 amino acid residues, which form a cytoplasmic domain, a transmembrane domain, and an ectodomain. The ectodomain has three important regions: a fusion peptide region, an N-terminal helical heptad repeats region (NHR), and a C-terminal helical heptad repeat region (CHR); when, in the fusion process, the NHR and CHR form a six-helix bundle (6-HB) core. Moreover, the hydrophobic pocket in the NHR trimer is very important to stabilize the gp41 6-HB, being, thus, an interesting target for small molecule design. [6, 143]. Briefly, there are two major classes of HIV entry inhibitors: those targeting the CHR and those targeting the NHR of gp41 [144].
Based on the gp41 native CHR sequence, peptide T-20 (generic name “Efurtivide”, brand name “Fuzeon”) is the only example of an antiviral peptide already approved for clinical use in HIV patients. It represents the first generation of entry inhibitor, basically used for treatment of patients who show evidence of viral replication despite ongoing antiretroviral therapy [145]. Despite its efficacy, T-20 resistance occurs due to extended exposure, poor bioavailability, large-dose requirements, and cross reaction with preexisting antibodies in the patient. Furthermore, it has been shown that activity against HIV-2 is decreased [142, 146,147,148]. In addition, a C-peptide-based molecule, Sifuvirtide, showed longer half-life, higher potency and a lower threshold for resistance than T-20. Besides, in the phase IIb clinical trial in China, it showed improved efficacy and a better rate of undetectable viral loads, recently being approved for phase III clinical trials [144, 149]. Another promising C-peptide is V2o, which was designed with an amino acid profile that minimized antigenicity and immunogenicity, while preserving and enhancing antiviral activity from its mother molecule C46-EHO. The decrease in antigenicity and immunogenicity is a strategy used in several studies to avoid cross reactions with the patient’s preexisting antibodies, which can potentially impair treatment. Furthermore, the authors tested whether V2o would be recognized by HIV-1-infected patient’s serum when already immunized for C46-EHO, and no recognition was observed, thus, suggesting that V2o could be a safer drug [150].
The strategy mentioned above was also used with another peptide called P3, an N-peptide. Borrego et al. [147] studied ancestral sequences from gp41 derived from HIV-2 and simian immunodeficiency virus (SIV) and designed the peptide P3, which overlaps the N-terminal pocket-binding region and heptad repeat core of the HR2 region. In the in vitro assay, P3 inhibited both HIV-1 and HIV-2 infection, being significantly more active against the former than the latter. Moreover, when, in the presence of a patient infected with HIV-1 plasma, a poor reaction of peptide P3 and the patients’ antibodies was seen in comparison with the T-20 reaction. This result was expected, since the HR2 region of HIV-1 and HIV-2 differs significantly; therefore, antibodies generated against this region in HIV-1 are unlikely to bind to a peptide derived from HIV-2/SIV. Finally, the authors suggest that P3 could be used as an alternative treatment for T-20-resistant HIV-1-infected patients. In a further assay, Bártolo et al. [151] formulated P3 in a gel of hydroxyethyl cellulose (HEC) and performed in vivo tests with Balb/c mice. The idea was to create a microbicide that could be used intravaginally to prevent HIV infection. The authors described high stability and bioactivity in genital human fluids even a month after exposure, in different pH values and temperatures, and in the presence of hydrogen peroxide. More importantly, no bacterial toxicity of vaginal microbiota was observed, making P3/HEC gel a promising candidate for the development of a vaginal microbicide gel to be used as prophylaxis in women.
The Met115–Thre116 (or simply M–T) hook structure is another approach used to improve entry inhibitor peptides for HIV treatment, which is formed by two residues that precede the pocket-binding domain (PBD) of CHR peptides. Such a modification confers extensive hydrophobic interactions with the PBD, thus improving binding affinity and antiviral activity, suggesting a highly conserved pocket region between HIV-1, -2 and SIV. Therefore, Xiong et al. [152] generated 2P23, a fusion inhibitor peptide that had the pocket site of gp41 as the target. In comparison with peptides used as the control, like T-20, 2P23 showed increased inhibitory activity against HIV-1, HIV-2, and SIV. It was also effective against subtypes of T-20-resistant HIV mutants and primary HIV-2 isolates. A further assay from the same group led to the improvement of 2P23 which, by adding a fatty acid group (C16) to its C terminus, produced LP-19 [16]. The results showed that LP-19 could inhibit HIV-1, -2 and SIV in much lower concentrations than 2P23, especially in the assay for inhibition of virus entry. The lipid conjugation strategy improved the peptide’s binding stability and antiviral activity in both in vitro and ex vivo assays, providing a good candidate for drug development [140].
Influenza virus inspired peptides
The influenza virus presents a major threat to human health as it has been responsible for many epidemics over the years [16]. Given their segmented single-stranded RNA genome, influenza viruses have a high degree of genomic variation, which is caused by point mutation changes or large sequence reassortment [153], triggered when an animal is infected by different strains of influenza that can exchange genome segments, thus resulting in a new strain [154]. Despite frequent vaccination policies to minimize viral transmission, events of antigenic mismatch between the viruses used in the vaccine and the ones circulating in the community still make this measure ineffective. In addition, the only two classes of antiviral drugs against influenza are the adamantane and neuraminidase inhibitors, for which there are already described cases of resistance [155]. Thus, new molecules are needed, and current studies target one of the viral spikes called hemagglutinin (HA); this and neuraminidase (NA) are the principal glycoproteins involved in the entry and release process [156]. Influenza HA is formed by two subunits (HA1 and HA2), and it is considered a homotrimeric type 1 membrane glycoprotein. The subunits are connected by one disulfide bond, forming a globular head domain, which contains the receptor-binding site (RBS), while HA stem structure is responsible for intraendosomal membrane fusion [156, 157].
Based on a conserved region of the HA from influenza A, several peptides were designed by López-Martinéz et al. [158]. From the nine peptides designed, three derived from the N-terminal region of HA1, three derived from the C-terminal of the HA1, and another three derived from HA2. All of them were tested in vitro against four different strains of influenza from human, swine and avian origin, with HA subtype H1 or H5. The results showed that all nine peptides were able to inhibit the four influenza strains in concentrations ranging from 20 to 74 µM. Furthermore, docking analysis with the subtype H1 suggested that the antiviral activity could be related with multiple interactions between each AVP and relevant regions of HA, which could impair the conformational changes needed for the membrane fusion process.
A recent strategy is the design of peptides based on broad neutralizing monoclonal antibodies (bnAbs) that bind to the conserved HA stem region. Koday et al. [159] described a peptide named HB36.5, whose structure mimics the stem-binding region of bnAbs for HA in vitro. The results showed that HB36.5 has activity against multiple HA subtypes, and neutralized distinct human avian influenza viruses such as H5N1. Furthermore, when tested in mice, a pre-exposure treatment not only prevented infection but also did not induce an inflammatory response. Hence, as a post-exposure treatment HB36.5 blocked and interfered with the viral spread. Altogether, these results suggest the peptide could be used both to prevent infection and to treat the disease without exacerbating the inflammatory response.
AVPs against coronavirus
Human coronaviruses are positive-sense RNA enveloped viruses that belong to the Coronaviridae family. So far, six coronaviruses (CoV) have been reported to infect humans: HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, severe acute respiratory syndrome coronavirus (SARS-CoV), and the Middle East respiratory syndrome coronavirus (MERS-CoV) [160]. While HCoV-229E and HCoV-OC43 are associated with upper and mild respiratory tract infections, SARS-CoV and MERS-CoV cause a variety of severe flu-like symptoms and were responsible for recent epidemics (in 2002/3 and 2015, respectively) [161, 162].
Basically, the viral particle is formed by spike glycoprotein (S), the envelope (E), the membrane (M), and the nucleocapsid (N). The spike is a type 1 transmembrane protein and is formed by two subunits (S1 and S2) which are involved in the fusion/entry process. While S1 has the receptor-binding domain (RBD) and is responsible for binding to the cellular receptor, S2 has the fusion peptide (FP), the heptad repeat 1 (HR1), the heptad repeat 2 (HR2), a transmembrane domain (TM), and a cytoplasmic domain peptide (CP), and acts in the viral fusion and entry into the cell [163, 164]. Both HR1 and HR2 have three segments each that, altogether, form a six-helix bundle (6-HB) fusion core. Disruption of this core formation leads to viral fusion inhibition, therefore, preventing infection [165].
For this reason, Gao et al. [165] designed two AVPs (P1 and P2) based on the HR2 sequence and challenged its effects against a pseudotyped MERS-CoV system. The peptide P1 showed good inhibitory effect with an EC50 ~ 3.013 µM, while P2 showed no activity at all even at the highest concentration. In another assay, Lu et al. [166] also designed peptides based on the HR2 and the HR1 regions of MERS-CoV. Among the tests performed, they analyzed whether these peptides interact with each other to form the 6-HB core, which was proved by an SDS-PAGE gel, suggesting that this interaction mimics the fusion core structure in vitro for academic purposes. Furthermore, cell–cell fusion inhibition assays showed that HR2P was more efficient at inhibiting both MERS-CoV spread and syncytium formation. The mechanism described by the authors was that HR2P could interact with the viral HR1 domain, blocking viral fusion core formation. In addition, an antiviral assay performed in Vero cells expressing the DPP4 receptor, which is used by MERS-CoV, showed only that HRP2 inhibited the viral replication in a dose-dependent manner. Finally, the authors suggest that this peptide should be used as a template for the design of analogs with enhanced activity against MERS-CoV infection and possibly used in clinical patients. As another example, Sun et al. [167], inspired by the same fusion core proteins, designed a peptide named MERS-5HB, which contains three copies of HR1 and two copies of HR2. Besides the tests describing the peptide’s structure and possible interaction with another MERS inspired peptide, a pseudotyped inhibition assay and a cell–cell spread assay were carried out. The results showed that MERS-5HB inhibited both the MERS-pseudovirus and syncytia formation. Finally, the author suggested that this peptide may prevent the interaction of the HR1 and HR2 regions, consequently blocking the formation of 6HB fusion core.
Most recently, the same group designed peptides for the HR1 and HR2 regions from the HCoV-229E and tested them against HCoV-229E S protein-mediated cell–cell fusion, and both pseudotyped and live HCoV-229E. The peptides named 229E-HR1P and 229E-HR2P both showed inhibition of cell–cell spread, and inhibition of the pseudovirus infection, but 229E-HR2P was much more effective. Besides, in vivo assays showed that 229E-HR2P could retain its antiviral activity in both upper and lower respiratory tracts when administered intranasally. In the end, the authors suggested that 229E-HR2P could become an antiviral drug to be used along with different antiviral molecules with a different mechanism of action, possibly exerting synergistic activity [168].
Final considerations
Since the majority of viral infections still have no available treatment, and due to the emergence/re-emergence of some virus strains, chances of viral pandemics that pose real threats to the worldwide population are still a real risk. In a highly globalized world, an infected individual can spread diseases much faster than centuries ago. Such an example is the Ebola virus outbreak from 2013 to 2016, which spread from West Africa, where it is endemic, to other places in Europe and the United States by infected health care agents. The rise of the disease in mid-2014 led the World Health Organization (WHO) to consider it a public health event of international concern [18].
Therefore, novel antiviral molecules for clinical treatments are indispensable. Current approaches have proved to be insufficient in some cases, such as nucleoside analogs that were considered satisfactory in the past, but their first generation showed many side effects, and later generations struggled with viral resistance. As indicated at the beginning of this review, many alternatives are proposed nowadays, and antiviral peptides are among them, here, as summarized in Table 1. However, even if there are several peptides described as antivirals, why do so few of these molecules actually reach the clinical trial phase?
Despite these molecules’ advantages, some potential problems still need to be addressed. First, the production cost is high, due to current solid-phase techniques that require coupling reagents, resin, and, in some cases, protective amino acids. The process can become quite challenging depending on the peptide composition; for instance, the presence of disulfide bridges and the repetition of a single amino acid residue [169]. Cost reduction could be achieved by rethinking both synthesis and purification methods, like the use of recombinant peptide expression [170]. Second, short half-life and poor oral absorption are common challenges, since peptides are highly susceptible to degradation by proteases and peptidases [171]. This issue could be solved by the use of d-enantiomers, which allow increased stability and decreased substrate recognition and binding affinity to proteolytic enzymes; or post-translational modifications such as amidation and acetylation, also to improve peptide stability; or even by the addition of fatty acid chains to improve membrane permeability [87, 172]. Finally, there is the challenge of delivery systems. Thus, it will depend on the viral target, if it is the viral envelope or intracellular replication steps; and drug administration via oral or parenteral; solutions may be the conjugation with nanocarriers, antibodies, carbohydrates, and lipids [173, 174].
With the development of techniques to produce and to improve both pharmacodynamics and pharmacokinetics of AVPs, such problems will be overcome one day, since interest in peptide-based drugs is rising. Large-scale production and screening are speeding up the drug discovery phase, and it is expected that more antiviral peptides will enter the phase of clinical trials. Even so, for recently discovered viruses, there is little information, and the current methods for antiviral peptide design seem to work not only for drug description but also to help understand viral structure.
In addition, some authors suggest the use of peptide-based drugs as adjuvants or in combination therapy with other antivirals with different mechanisms of action, thus diminishing drug resistance establishment and producing fewer side effects. Finally, the description of new antiviral drugs supplements the existing therapies and provides alternatives to treat viral diseases that cause serious pandemics, reducing the mortality/morbidity associated with them.
References
Mahmoud A (2016) New vaccines: challenges of discovery. Microb Biotechnol 9:549–552. https://doi.org/10.1111/1751-7915.12397
Enquist LW (2009) Virology in the 21st Century. J Virol 83:5296–5308. https://doi.org/10.1128/jvi.00151-09
Lou Z, Sun Y, Rao Z (2014) Current progress in antiviral strategies. Trends Pharmacol Sci 35:86–102. https://doi.org/10.1016/j.tips.2013.11.006
McDonald CK, Kuritzkes DR (1997) Human immunodeficiency virus type 1 protease inhibitors. Arch Intern Med 157:951. https://doi.org/10.1001/archinte.1997.00440300037003
Kiser JJ, Flexner C (2013) Direct-acting antiviral agents for hepatitis C virus infection. Annu Rev Pharmacol Toxicol 53:427–449. https://doi.org/10.1146/annurev-pharmtox-011112-140254
Yu F, Lu L, Du L et al (2013) Approaches for identification of HIV-1 entry inhibitors targeting gp41 pocket. Viruses 5:127–149
El Raziky M, Fathalah WF, El-Akel WA et al (2013) The effect of peginterferon Alpha-2a vs. Peginterferon Alpha-2b in treatment of naive chronic HCV genotype-4 patients: a single centre Egyptian study. Hepat Mon 13:10069. https://doi.org/10.5812/hepatmon.10069
Fching Lin, Young HA (2014) Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev 25:369–376
Buttinelli G, Donati V, Fiore S et al (2003) Nucleotide variation in Sabin type 2 poliovirus from an immunodeficient patient with poliomyelitis. J Gen Virol 84:1215–1221. https://doi.org/10.1099/vir.0.18974-0
Thompson C, Whitley R (2011) Neonatal herpes simplex virus infections: where are we now? Adv Exp Med Biol 697:221–230. https://doi.org/10.1007/978-1-4419-7185-2_15
Elion GB (1982) Mechanism of action and selectivity of acyclovir. Am J Med 73:7–13. https://doi.org/10.1016/0002-9343(82)90055-9
Duraffour S, Andrei G, Topalis D et al (2012) Mutations conferring resistance to viral DNA polymerase inhibitors in camelpox virus give different drug-susceptibility profiles in vaccinia virus. J Virol 86:7310–7325. https://doi.org/10.1128/jvi.00355-12
Le Page AK, Jager MM, Iwasenko JM et al (2013) Clinical aspects of cytomegalovirus antiviral resistance in solid organ transplant recipients. Clin Infect Dis 56:1018–1029. https://doi.org/10.1093/cid/cis1035
Musiime V, Kaudha E, Kayiwa J et al (2013) Antiretroviral drug resistance profiles and response to second-line therapy among HIV type 1-infected ugandan children. AIDS Res Hum Retroviruses 29:449–455. https://doi.org/10.1089/aid.2012.0283
Deming P, McNicholl IR (2011) Coinfection with human immunodeficiency virus and hepatitis C virus: challenges and therapeutic advances—insights from the society of infectious diseases pharmacists. Pharmacotherapy 4:357–368. https://doi.org/10.1592/phco.31.4.357
Hui DSC, Lee N, Chan PKS (2017) A clinical approach to the threat of emerging influenza viruses in the Asia–Pacific region. Respirology 22:1300–1312. https://doi.org/10.1111/resp.13114
Marston BJ, Dokubo EK, van Steelandt A et al (2017) Ebola response impact on public health programs, West Africa, 2014–2017. Emerg Infect Dis 23:S25–S32. https://doi.org/10.3201/eid2313.170727
Suijkerbuijk AWM, Swaan CM, Mangen M-JJ et al (2017) Ebola in the Netherlands, 2014–2015: costs of preparedness and response. Eur J Heal Econ 19:1–9. https://doi.org/10.1007/s10198-017-0940-4
De Souza WV, De Albuquerque MDFPM, Vazquez E et al (2018) Microcephaly epidemic related to the Zika virus and living conditions in Recife, Northeast Brazil. BMC Public Health 18:1–7. https://doi.org/10.1186/s12889-018-5039-z
Lowe R, Barcellos C, Brasil P et al (2018) The zika virus epidemic in brazil: from discovery to future implications. Int J Environ Res Public Health 15:1–18. https://doi.org/10.3390/ijerph15010096
Da ZhuJ, Meng W, Wang XJ, Wang HCR (2015) Broad-spectrum antiviral agents. Front Microbiol 6:1–15. https://doi.org/10.3389/fmicb.2015.00517
Wang G, Watson KM, Peterkofsky A, Buckheit RW (2010) Identification of novel human immunodeficiency virus type 1-inhibitory peptides based on the antimicrobial peptide database. Antimicrob Agents Chemother 54:1343–1346. https://doi.org/10.1128/aac.01448-09
Jesus T, Rogelio L, Abraham C et al (2012) Prediction of antiviral peptides derived from viral fusion proteins potentially active against herpes simplex and influenza A viruses. Bioinformation 8:870–874. https://doi.org/10.6026/97320630008870
Elshabrawy HA, Fan J, Haddad CS et al (2014) Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and ebola, hendra, and nipah viruses by using a novel high-throughput screening assay. J Virol 88:4353–4365. https://doi.org/10.1128/jvi.03050-13
Martins FO, da Rocha Gomes MM, Pereira Nogueira FL et al (2009) In vitro inhibitory effect of Urera baccifera (L.) Gaudich. extracts against herpes simplex. African J Pharm Pharmacol 3:581–584
Cantatore A, Randall SD, Traum D, Adams SD (2013) Effect of black tea extract on herpes simplex virus-1 infection of cultured cells. BMC Compl Altern Med 13:1–10. https://doi.org/10.1186/1472-6882-13-139
Rothan HA, Bahrani H, Rahman NA, Yusof R (2014) Identification of natural antimicrobial agents to treat dengue infection: in vitro analysis of latarcin peptide activity against dengue virus. BMC Microbiol 14:1–10. https://doi.org/10.1186/1471-2180-14-140
Rothan HA, Abdulrahman AY, Sasikumer PG et al (2012) Protegrin-1 inhibits dengue NS2B-NS3 serine protease and viral replication in MK2 cells. J Biomed Biotechnol 2012:1–6. https://doi.org/10.1155/2012/251482
Rothan HA, Han HC, Ramasamy TS et al (2012) Inhibition of dengue NS2B-NS3 protease and viral replication in Vero cells by recombinant retrocyclin-1. BMC Infect Dis 12:1–9. https://doi.org/10.1186/1471-2334-12-314
Hakim A, Nguyen JB, Basu K et al (2013) Crystal structure of an insect antifreeze protein and its implications for ice binding. J Biol Chem 288:12295–12304. https://doi.org/10.1074/jbc.m113.450973
Ishag HZA, Li C, Huang L et al (2013) Inhibition of Japanese encephalitis virus infection in vitro and in vivo by pokeweed antiviral protein. Virus Res 171:89–96. https://doi.org/10.1016/j.virusres.2012.10.032
Chen L, Liu Y, Wang S et al (2017) Antiviral activity of peptide inhibitors derived from the protein E stem against Japanese encephalitis and Zika viruses. Antiviral Res 141:140–149. https://doi.org/10.1016/j.antiviral.2017.02.009
Chinchar VG, Bryan L, Silphadaung U et al (2004) Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology 323:268–275. https://doi.org/10.1016/j.virol.2004.02.029
Falco A, Mas V, Tafalla C et al (2007) Dual antiviral activity of human alpha-defensin-1 against viral haemorrhagic septicaemia rhabdovirus (VHSV): inactivation of virus particles and induction of a type I interferon-related response. Antiviral Res 76:111–123. https://doi.org/10.1016/j.antiviral.2007.06.006
Crack LR, Jones L, Malavige GN et al (2012) Human antimicrobial peptides LL-37 and human β-defensin-2 reduce viral replication in keratinocytes infected with varicella zoster virus. Clin Exp Dermatol 37:534–543. https://doi.org/10.1111/j.1365-2230.2012.04305.x
Altmann SE, Brandt CR, Jahrling PB, Blaney JE (2012) Antiviral activity of the EB peptide against zoonotic poxviruses. Virol J 6:1–6. https://doi.org/10.1186/1743-422x-9-6
Okazaki K, Kida H (2004) A synthetic peptide from a heptad repeat region of herpesvirus glycoprotein B inhibits virus replication. J Gen Virol 85:2131–2137. https://doi.org/10.1099/vir.0.80051-0
Tiwari V, Liu J, Valyi-Nagy T, Shukla D (2011) Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J Biol Chem 286:25406–25415. https://doi.org/10.1074/jbc.m110.201103
Mooney C, Haslam NJ, Pollastri G, Shields DC (2012) Towards the improved discovery and design of functional peptides: common features of diverse classes permit generalized prediction of bioactivity. PLoS One 7:1–12. https://doi.org/10.1371/journal.pone.0045012
Maccari G, Di Luca M, Nifosí R et al (2013) Antimicrobial peptides design by evolutionary multiobjective optimization. PLoS Comput Biol 9:1–12. https://doi.org/10.1371/journal.pcbi.1003212
Sharma A, Singla D, Rashid M, Raghava GPS (2014) Designing of peptides with desired half-life in intestine-like environment. BMC Bioinf 15:1–8. https://doi.org/10.1186/1471-2105-15-282
Qureshi A, Thakur N, Tandon H, Kumar M (2014) AVPdb: a database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res 42:1147–1153. https://doi.org/10.1093/nar/gkt1191
Mulder KCL, Lima LA, Miranda VJ et al (2013) Current scenario of peptide-based drugs: the key roles of cationic antitumor and antiviral peptides. Front Microbiol 4:1–23. https://doi.org/10.3389/fmicb.2013.00321
Barlow PG, Findlay EG, Currie SM, Davidson DJ (2014) Antiviral potential of cathelicidins. Future Microbiol 9:55–73. https://doi.org/10.2217/fmb.13.135
Gwyer Findlay E, Currie SM, Davidson DJ (2013) Cationic host defence peptides: potential as antiviral therapeutics. BioDrugs 27:479–493. https://doi.org/10.1007/s40259-013-0039-0
Galdiero S, Falanga A, Tarallo R et al (2013) Peptide inhibitors against herpes simplex virus infections. J Pept Sci 19:148–158. https://doi.org/10.1002/psc.2489
Qureshi A, Thakur N, Kumar M (2013) HIPdb: a database of experimentally validated hiv inhibiting peptides. PLoS One 8:1–5. https://doi.org/10.1371/journal.pone.0054908
Zapata W, Aguilar-Jiménez W, Feng Z et al (2016) Identification of innate immune antiretroviral factors during in vivo and in vitro exposure to HIV-1. Microbes Infect 18:211–219. https://doi.org/10.1016/j.micinf.2015.10.009
Bulet P, Stöcklin R, Menin L (2004) Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev 198:169–184
Badani H, Garry RF, Wimley WC (2014) Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim Biophys Acta Biomembr 1838:2180–2197. https://doi.org/10.1016/j.bbamem.2014.04.015
Wang C-K, Shih L-Y, Chang K (2017) Large-scale analysis of antimicrobial activities in relation to amphipathicity and charge reveals novel characterization of antimicrobial peptides. Molecules 22:2037. https://doi.org/10.3390/molecules22112037
Tam JP, Wang S, Wong KH, Tan WL (2015) Antimicrobial peptides from plants. Pharmaceuticals 8:711–757. https://doi.org/10.3390/ph8040711
Burman R, Yeshak MY, Larsson S et al (2015) Distribution of circular proteins in plants: large-scale mapping of cyclotides in the Violaceae. Front Plant Sci 6:1–13. https://doi.org/10.3389/fpls.2015.00855
Weidmann J, Craik DJ (2016) Discovery, structure, function, and applications of cyclotides: circular proteins from plants. J Exp Bot 67:4801–4812. https://doi.org/10.1093/jxb/erw210
Ireland DC, Wang CKL, Wilson JA et al (2008) Cyclotides as natural anti-HIV agents. Biopolym - Pept Sci Sect 90:51–60. https://doi.org/10.1002/bip.20886
Wang CKL, Colgrave ML, Gustafson KR et al (2008) Anti-HIV cyclotides from the Chinese medicinal herb Viola yedoensis. J Nat Prod 71:47–52. https://doi.org/10.1021/np070393g
Henriques ST, Craik DJ (2010) Cyclotides as templates in drug design. Drug Discov Today 7:179–194. https://doi.org/10.1016/j.drudis.2009.10.007
Gao Y, Cui T, Lam Y (2010) Synthesis and disulfide bond connectivity-activity studies of a kalata B1-inspired cyclopeptide against dengue NS2B-NS3 protease. Bioorganic Med Chem 18:1331–1336. https://doi.org/10.1016/j.bmc.2009.12.026
Sencanski M, Radosevic D, Perovic V et al (2015) Natural products as promising therapeutics for treatment of influenza disease. Curr Pharm Des 21:5573–5588. https://doi.org/10.2174/1381612821666151002113426
Henriques ST, Huang YH, Rosengren KJ et al (2011) Decoding the membrane activity of the cyclotide kalata B1: the importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti-HIV activities. J Biol Chem 286:1–24. https://doi.org/10.1074/jbc.m111.253393
Waheed AA, Freed EO (2009) Lipids and membrane microdomains in HIV-1 replication. Virus Res 143:162–176. https://doi.org/10.1016/j.virusres.2009.04.007
Daly NL, Gustafson KR, Craik DJ (2004) The role of the cyclic peptide backbone in the anti-HIV activity of the cyclotide kalata B1. FEBS Lett 574:69–72. https://doi.org/10.1016/j.febslet.2004.08.007
Nawae W, Hannongbua S, Ruengjitchatchawalya M (2017) Molecular dynamics exploration of poration and leaking caused by Kalata B1 in HIV-infected cell membrane compared to host and HIV membranes. Sci Rep 7:1–13. https://doi.org/10.1038/s41598-017-03745-2
Craik DJ, Du J (2017) Cyclotides as drug design scaffolds. Curr Opin Chem Biol 38:8–16. https://doi.org/10.1016/j.cbpa.2017.01.018
Ngai PH, Ng TB (2005) Phaseococcin, an antifungal protein with antiproliferative and anti-HIV-1 reverse transcriptase activities from small scarlet runner beans. Biochem Cell Biol 83:212–220. https://doi.org/10.1139/o05-037
Jack HW, Tzi BN (2005) Sesquin, a potent defensin-like antimicrobial peptide from ground beans with inhibitory activities toward tumor cells and HIV-1 reverse transcriptase. Peptides 26:1120–1126. https://doi.org/10.1016/j.peptides.2005.01.003
Camargo Filho I, Cortez DAG, Ueda-Nakamura T et al (2008) Antiviral activity and mode of action of a peptide isolated from Sorghum bicolor. Phytomedicine 15:202–208. https://doi.org/10.1016/j.phymed.2007.07.059
Salas CE, Badillo-Corona JA, Ramírez-Sotelo G, Oliver-Salvador C (2015) Biologically active and antimicrobial peptides from plants. Biomed Res Int 2015:1–11. https://doi.org/10.1155/2015/102129
Kuczer M, Dziubasik K, Midak-Siewirska A et al (2010) Studies of insect peptides alloferon, Any-GS and their analogues. Synthesis and antiherpes activity. J Pept Sci 16:186–189. https://doi.org/10.1002/psc.1219
Hultmark D, Steiner H, Rasmuson T, Boman HG (2005) Insect immunity. Purification and properties of three inducible bactericidal proteins from hemolymph of immunized pupae of Hyalophora cecropia. Eur J Biochem 106:7–16. https://doi.org/10.1111/j.1432-1033.1980.tb05991.x
Wachinger M, Kleinschmidt A, Winder D et al (1998) Antimicrobial peptides melittin and cecropin inhibit replication of human immunodeficiency virus 1 by suppressing viral gene expression. J Gen Virol 79:731–740. https://doi.org/10.1099/0022-1317-79-4-731
Albiol Matanic VC, Castilla V (2004) Antiviral activity of antimicrobial cationic peptides against Junin virus and herpes simplex virus. Int J Antimicrob Agents 23:382–389. https://doi.org/10.1016/j.ijantimicag.2003.07.022
Wachinger M, Saermark T, Erfle V (1992) Influence of amphipathic peptides on the HIV-1 production in persistently infected T lymphoma cells. FEBS Lett 309:235–241. https://doi.org/10.1016/0014-5793(92)80780-k
Hood JL, Jallouk AP, Campbell N et al (2013) Cytolytic nanoparticles attenuate HIV-1 infectivity. Antivir Ther 18:95–103. https://doi.org/10.3851/imp2346
Sample CJ, Hudak KE, Barefoot BE et al (2013) A mastoparan-derived peptide has broad-spectrum antiviral activity against enveloped viruses. Peptides 48:96–105. https://doi.org/10.1016/j.peptides.2013.07.014
El-Bitar AM, Sarhan MM, Aoki C et al (2015) Virocidal activity of Egyptian scorpion venoms against hepatitis C virus Hepatitis viruses. Virol J 12:1–9. https://doi.org/10.1186/s12985-015-0276-6
Yan R, Zhao Z, He Y et al (2011) A new natural α-helical peptide from the venom of the scorpion Heterometrus petersii kills HCV. Peptides 32:11–19. https://doi.org/10.1016/j.peptides.2010.10.008
Hong W, Li T, Song Y et al (2014) Inhibitory activity and mechanism of two scorpion venom peptides against herpes simplex virus type 1. Antiviral Res 102:1–10. https://doi.org/10.1016/j.antiviral.2013.11.013
Li Q, Zhao Z, Zhou D et al (2011) Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides 32:1518–1525. https://doi.org/10.1016/j.peptides.2011.05.015
Zhao Z, Hong W, Zeng Z et al (2012) Mucroporin-M1 inhibits hepatitis B virus replication by activating the mitogen-activated protein kinase (MAPK) pathway and down-regulating HNF4α in vitro and in vivo. J Biol Chem 287:30181–30190. https://doi.org/10.1074/jbc.m112.370312
Chen Y, Cao L, Zhong M et al (2012) Anti-HIV-1 activity of a new scorpion venom peptide derivative Kn2-7. PLoS One 7:1–9. https://doi.org/10.1371/journal.pone.0034947
Zeng Z, Zhang R, Hong W et al (2018) Histidine-rich modification of a scorpion-derived peptide improves bioavailability and inhibitory activity against HSV-1. Theranostics 8:199–211. https://doi.org/10.7150/thno.21425
Chernysh S, Kim SI, Bekker G et al (2002) Antiviral and antitumor peptides from insects. Proc Natl Acad Sci 99:12628–12632. https://doi.org/10.1073/pnas.192301899
Kuczer M, Midak-Siewirska A, Zahorska R et al (2011) Further studies on the antiviral activity of alloferon and its analogues. J Pept Sci 17:715–719. https://doi.org/10.1002/psc.1388
Kuczer M, Majewska A, Zahorska R (2013) New alloferon analogues: synthesis and antiviral properties. Chem Biol Drug Des 81:302–309. https://doi.org/10.1111/cbdd.12020
Marcocci ME, Amatore D, Villa S et al (2018) The amphibian antimicrobial peptide temporin b inhibits in vitro herpes simplex virus 1 infection. Antimicrob Agents Chemother 62:1–13. https://doi.org/10.1128/aac.02367-17
Shartouny JR, Jacob J (2018) Mining the tree of life: host defense peptides as antiviral therapeutics. Semin Cell Dev Biol 88:147–155. https://doi.org/10.1016/j.semcdb.2018.03.001
Egal M, Conrad M, MacDonald DL et al (1999) Antiviral effects of synthetic membrane-active peptides on herpes simplex virus, type 1. Int J Antimicrob Agents 13:57–60
Dean RE, O’Brien LM, Thwaite JE et al (2010) A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides 31:1966–1972. https://doi.org/10.1016/j.peptides.2010.07.028
Belaid A, Aouni M, Khelifa R et al (2002) In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J Med Virol 66:229–234. https://doi.org/10.1002/jmv.2134
Lorin C, Saidi H, Belaid A et al (2005) The antimicrobial peptide Dermaseptin S4 inhibits HIV-1 infectivity in vitro. Virology 334:264–275. https://doi.org/10.1016/j.virol.2005.02.002
Bergaoui I, Zairi A, Tangy F et al (2013) In vitro antiviral activity of dermaseptin S4 and derivatives from amphibian skin against herpes simplex virus type 2. J Med Virol 85:272–281. https://doi.org/10.1002/jmv.23450
Mechlia MB, Belaid A, Castel G et al (2018) Dermaseptins as potential antirabies compounds. Vaccine 2018:1–7. https://doi.org/10.1016/j.vaccine.2018.01.066
Monteiro JMC, Oliveira MD, Dias RS et al (2018) The antimicrobial peptide HS-1 inhibits dengue virus infection. Virology 514:79–87. https://doi.org/10.1016/j.virol.2017.11.009
Holthausen DJ, Lee SH, Kumar VT et al (2017) An amphibian host defense peptide is virucidal for human H1 hemagglutinin-bearing influenza viruses. Immunity 46:587–595. https://doi.org/10.1016/j.immuni.2017.03.018
Yasin B, Pang M, Turner JS et al (2000) Evaluation of the inactivation of infectious herpes simplex virus by host-defense peptides. Eur J Clin Microbiol Infect Dis 19:187–194. https://doi.org/10.1007/s100960050457
Carriel-Gomes MC, Kratz JM, Barracco MA et al (2007) In vitro antiviral activity of antimicrobial peptides against herpes simplex virus 1, adenovirus, and rotavirus. Mem Inst Oswaldo Cruz 102:469–472. https://doi.org/10.1590/s0074-02762007005000028
Lu Z, Van Wagoner RM, Harper MK et al (2011) Mirabamides E–H, HIV-inhibitory depsipeptides from the sponge Stelletta clavosa. J Nat Prod 74:185–193. https://doi.org/10.1021/np100613p
Migliolo L, Silva ON, Silva PA et al (2012) Structural and functional characterization of a multifunctional alanine-rich peptide analogue from pleuronectes americanus. PLoS One 7:e47047. https://doi.org/10.1371/journal.pone.0047047
Vilas Boas LCP, de Lima LMP, Migliolo L et al (2017) Linear antimicrobial peptides with activity against herpes simplex virus 1 and Aichi virus. Biopolymers 108:1–20. https://doi.org/10.1002/bip.22871
Holly MK, Diaz K, Smith JG (2017) Defensins in Viral Infection and Pathogenesis. Annu Rev Virol 4:369–391. https://doi.org/10.1146/annurev-virology-101416-041734
Wang W, Owen SM, Rudolph DL et al (2004) Activity of alpha- and theta-defensins against primary isolates of HIV-1. J Immunol 173:515–520. https://doi.org/10.4049/jimmunol.173.1.515
Wu Z, Cocchi F, Gentles D et al (2005) Human neutrophil α-defensin 4 inhibits HIV-1 infection in vitro. FEBS Lett 579:162–166. https://doi.org/10.1016/j.febslet.2004.11.062
Salvatore M, García-Sastre A, Ruchala P et al (2007) α-defensin inhibits influenza virus replication by cell-mediated mechanism(s). J Infect Dis 196:835–843. https://doi.org/10.1086/521027
Meyer-Hoffert U, Schwarz T, Schröder J-M, Gläser R (2008) Expression of human beta-defensin-2 and -3 in verrucae vulgares and condylomata acuminata. J Eur Acad Dermatol Venereol 22:1050–1054. https://doi.org/10.1111/j.1468-3083.2008.02675.x
Howell MD, Streib JE, Leung DYM (2007) Antiviral activity of human beta-defensin 3 against vaccinia virus. J Allergy Clin Immunol 119:1022–1025. https://doi.org/10.1016/j.jaci.2007.01.044
Quiñones-Mateu ME, Lederman MM, Feng Z et al (2003) Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS 17:F39–F48. https://doi.org/10.1097/01.aids.0000096878.73209.4f
Sørensen OE, Follin P, Johnsen AH et al (2001) Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97:3951–3959. https://doi.org/10.1182/blood.v97.12.3951
Barlow PG, Svoboda P, Mackellar A et al (2011) Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS One 6:1–9. https://doi.org/10.1371/journal.pone.0025333
Tripathi S, Wang G, White M et al (2015) Antiviral activity of the human cathelicidin, LL-37, and derived peptides on seasonal and pandemic influenza A viruses. PLoS One 10:1–17. https://doi.org/10.1371/journal.pone.0124706
Matsumura T, Sugiyama N, Murayama A et al (2016) Antimicrobial peptide LL-37 attenuates infection of hepatitis C virus. Hepatol Res 46:924–932. https://doi.org/10.1111/hepr.12627
Alagarasu K, Patil PS, Shil P et al (2017) In-vitro effect of human cathelicidin antimicrobial peptide LL-37 on dengue virus type 2. Peptides 92:23–30. https://doi.org/10.1016/j.peptides.2017.04.002
Ahmed A, Siman-Tov G, Keck F et al (2019) Human cathelicidin peptide LL-37 as a therapeutic antiviral targeting Venezuelan equine encephalitis virus infections. Antiviral Res 164:61–69. https://doi.org/10.1016/j.antiviral.2019.02.002
He M, Zhang H, Li Y et al (2018) Cathelicidin-derived antimicrobial peptides inhibit Zika virus through direct inactivation and interferon pathway. Front Immunol 9:1–12. https://doi.org/10.3389/fimmu.2018.00722
Sousa FH, Casanova V, Findlay F et al (2017) Cathelicidins display conserved direct antiviral activity towards rhinovirus. Peptides 95:76–83. https://doi.org/10.1016/j.peptides.2017.07.013
Gordon YJ, Huang LC, Romanowski EG et al (2005) Human cathelicidin (LL-37), a multifunctional peptide, is expressed by ocular surface epithelia and has potent antibacterial and antiviral activity. Curr Eye Res 30:385–394. https://doi.org/10.1080/02713680590934111
Van der Strate BWA, Beljaars L, Molema G et al (2001) Antiviral activities of lactoferrin. Antiviral Res 52:225–239. https://doi.org/10.1016/s0166-3542(01)00195-4
Li S, Zhou H, Huang G, Liu N (2009) Inhibition of HBV infection by bovine lactoferrin and iron-, zinc-saturated lactoferrin. Med Microbiol Immunol 198:19–25. https://doi.org/10.1007/s00430-008-0100-7
Chen JM, Fan YC, Lin JW et al (2017) Bovine lactoferrin inhibits dengue virus infectivity by interacting with heparan sulfate, low-density lipoprotein receptor, and DC-SIGN. Int J Mol Sci 18:1–13. https://doi.org/10.3390/ijms18091957
Carvalho CAM, Casseb SMM, Gonçalves RB et al (2017) Bovine lactoferrin activity against Chikungunya and Zika viruses. J Gen Virol 98:1749–1754. https://doi.org/10.1099/jgv.0.000849
Andersen JH, Osbakk SA, Vorland LH et al (2001) Lactoferrin and cyclic lactoferricin inhibit the entry of human cytomegalovirus into human fibroblasts. Antiviral Res 51:141–149. https://doi.org/10.1016/s0166-3542(01)00146-2
Mistry N, Drobni P, Näslund J et al (2007) The anti-papillomavirus activity of human and bovine lactoferricin. Antiviral Res 75:258–265. https://doi.org/10.1016/j.antiviral.2007.03.012
Marr AK, Jenssen H, Moniri MR et al (2009) Bovine lactoferrin and lactoferricin interfere with intracellular trafficking of Herpes simplex virus-1. Biochimie 91:160–164. https://doi.org/10.1016/j.biochi.2008.05.016
Shestakov A, Jenssen H, Nordström I, Eriksson K (2012) Lactoferricin but not lactoferrin inhibit herpes simplex virus type 2 infection in mice. Antiviral Res 93:340–345. https://doi.org/10.1016/j.antiviral.2012.01.003
Wang WY, Wong JH, Ip DTM et al (2016) Bovine lactoferrampin, human lactoferricin, and lactoferrin 1-11 inhibit nuclear translocation of HIV integrase. Appl Biochem Biotechnol 179:1202–1212. https://doi.org/10.1007/s12010-016-2059-y
Borst EM, Ständker L, Wagner K et al (2013) A peptide inhibitor of cytomegalovirus infection from human hemofiltrate. Antimicrob Agents Chemother 57:4751–4760. https://doi.org/10.1128/aac.00854-13
Sala A, Ardizzoni A, Ciociola T et al (2018) Antiviral activity of synthetic peptides derived from physiological proteins. Intervirology 61:166–173. https://doi.org/10.1159/000494354
Porto WF, Silva ON, Franco OL (2012) Prediction and rational design of antimicrobial peptides. In: Faraggi E (ed) Protein structure. InTech, London, pp 377–396
Beltrán Lissabet JF, Belén LH, Farias JG (2019) AntiVPP 1.0: a portable tool for prediction of antiviral peptides. Comput Biol Med 107:127–130. https://doi.org/10.1016/j.compbiomed.2019.02.011
Jose GG, Larsen IV, Gauger J et al (2013) A cationic peptide, TAT-Cd0, inhibits herpes simplex virus type 1 ocular infection in vivo. Investig Ophthalmol Vis Sci 54:1070–1079. https://doi.org/10.1167/iovs.12-10250
Cui X, Wu Y, Fan D et al (2018) Peptides P4 and P7 derived from E protein inhibit entry of dengue virus serotype 2 via interacting with β3 integrin. Antiviral Res 155:20–27. https://doi.org/10.1016/j.antiviral.2018.04.018
Chew MF, Poh KS, Poh CL (2017) Peptides as therapeutic agents for dengue virus. Int J Med Sci 14:1342–1359. https://doi.org/10.7150/ijms.21875
Hrobowski YM, Garry RF, Michael SF (2005) Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol J 2:1–10. https://doi.org/10.1186/1743-422x-2-49
Lok SM, Costin JM, Hrobowski YM et al (2012) Release of dengue virus genome induced by a peptide inhibitor. PLoS One 7:5–12. https://doi.org/10.1371/journal.pone.0050995
Costin JM, Jenwitheesuk E, Lok S-M et al (2010) Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl Trop Dis 4:e721. https://doi.org/10.1371/journal.pntd.0000721
Roizman B, Taddeo B (2007) The strategy of herpes simplex virus replication and takeover of the host cell. In: Arvin A, Campadelli-Fiume G, Mocarski E et al (eds) Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge
Akkarawongsa R, Pocaro NE, Case G et al (2009) Multiple peptides homologous to herpes simplex virus type 1 glycoprotein B inhibit viral infection. Antimicrob Agents Chemother 53:987–996. https://doi.org/10.1128/aac.00793-08
Cetina-Corona A, López-Sánchez U, Salinas-Trujano J et al (2016) Peptides derived from glycoproteins H and B of herpes simplex virus type 1 and herpes simplex virus type 2 are capable of blocking herpetic infection in vitro. Intervirology 59:235–242. https://doi.org/10.1159/000464134
ONUSIDA (2018) Global AIDS monitoring 2018: indicators for monitoring the 2016 United Nations Political Declaration on HIV and AIDS. Geneva
Chong H, Xue J, Xiong S et al (2017) A lipopeptide HIV-1/2 fusion inhibitor with highly potent in vitro, ex vivo, and in vivo antiviral activity. J Virol 91:1–13. https://doi.org/10.1128/jvi.00288-17
Lemey P, Pybus OG, Wang B et al (2003) Tracing the origin and history of the HIV-2 epidemic. Proc Natl Acad Sci 100:6588–6592. https://doi.org/10.1073/pnas.0936469100
Menéndez-Arias L, Álvarez M (2014) Antiretroviral therapy and drug resistance in human immunodeficiency virus type 2 infection. Antiviral Res 102:70–86. https://doi.org/10.1016/j.antiviral.2013.12.001
Markosyan RM (2005) Time-resolved imaging of HIV-1 env-mediated lipid and content mixing between a single virion and cell membrane. Mol Biol Cell 16:5502–5513. https://doi.org/10.1091/mbc.e05-06-0496
He Y, Xiao Y, Song H et al (2008) Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 283:11126–11134. https://doi.org/10.1074/jbc.m800200200
Fung HB, Guo Y (2004) Enfuvirtide: a fusion inhibitor for the treatment of HIV infection. Clin Ther 26:352–378
Naider F, Anglister J (2009) Peptides in the treatment of AIDS. Curr Opin Struct Biol 19:473–482. https://doi.org/10.1016/j.sbi.2009.07.003
Borrego P, Calado R, Marcelino JM et al (2013) An ancestral HIV-2/simian immunodeficiency virus peptide with potent HIV-1 and HIV-2 fusion inhibitor activity. AIDS 27:1081–1090. https://doi.org/10.1097/qad.0b013e32835edc1d
Vincent N, Tardy J-C, Livrozet J-M et al (2005) Depletion in antibodies targeted to the HR2 region of HIV-1 glycoprotein gp41 in sera of HIV-1-seropositive patients treated with T20. J Acquir Immune Defic Syndr 38:254–262
Su Y, Chong H, Qiu Z et al (2015) Mechanism of HIV-1 resistance to short-peptide fusion inhibitors targeting the Gp41 pocket. J Virol 89:5801–5811. https://doi.org/10.1128/jvi.00373-15
Brauer F, Schmidt K, Zahn RC et al (2013) A rationally engineered anti-HIV peptide fusion inhibitor with greatly reduced immunogenicity. Antimicrob Agents Chemother 57:679–688. https://doi.org/10.1128/aac.01152-12
Bártolo I, Diniz AR, Borrego P et al (2018) Evaluation of the fusion inhibitor P3 peptide as a potential microbicide to prevent HIV transmission in women. PLoS One 13:1–23. https://doi.org/10.1371/journal.pone.0195744
Xiong S, Borrego P, Ding X et al (2017) A helical short-peptide fusion inhibitor with highly potent activity against human immunodeficiency virus type 1 (HIV-1), HIV-2, and Simian immunodeficiency. Virus 91:1–15. https://doi.org/10.1128/jvi.01839-16
Hsieh I, Hartshorn KL (2016) The role of antimicrobial peptides in influenza virus infection and their potential as antiviral and immunomodulatory therapy. Pharmaceuticals 9:1–15. https://doi.org/10.3390/ph9030053
Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team, Dawood FS, Jain S et al (2009) Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 360:2605–2615. https://doi.org/10.1056/nejmoa0903810
Koszalka P, Tilmanis D, Hurt AC (2017) Influenza antivirals currently in late-phase clinical trial. Influenza Other Respi Viruses 11:240–246. https://doi.org/10.1111/irv.12446
Vanderlinden E, Naesens L (2014) Emerging antiviral strategies to interfere with influenza virus entry. Med Res Rev 34:301–339. https://doi.org/10.1002/med.21289
Lin D, Luo Y, Yang G et al (2017) Potent influenza A virus entry inhibitors targeting a conserved region of hemagglutinin. Biochem Pharmacol 144:35–51. https://doi.org/10.1016/j.bcp.2017.07.023
López-Martínez R, Ramírez-Salinas GL, Correa-Basurto J, Barrón BL (2013) Inhibition of influenza A virus infection in vitro by peptides designed in silico. PLoS One 8:e76876. https://doi.org/10.1371/journal.pone.0076876
Koday MT, Nelson J, Chevalier A et al (2016) A computationally designed hemagglutinin stem-binding protein provides in vivo protection from influenza independent of a host immune response. PLoS Pathogens 12:e1005409. https://doi.org/10.1371/journal.ppat.1005409
Friedman N, Alter H, Hindiyeh M et al (2018) Human coronavirus infections in Israel: epidemiology, clinical symptoms and summer seasonality of HCoV-HKU1. Viruses 10:515. https://doi.org/10.3390/v10100515
Chinese SARS Molecular Epidemiology Consortium TCSME (2004) Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303:1666–1669. https://doi.org/10.1126/science.1092002
Kim KH, Tandi TE, Choi JW et al (2017) Middle East respiratory syndrome coronavirus (MERS-CoV) outbreak in South Korea, 2015: epidemiology, characteristics and public health implications. J Hosp Infect 95:207–213. https://doi.org/10.1016/j.jhin.2016.10.008
Zumla A, Hui DS, Perlman S (2015) Middle East respiratory syndrome. Lancet 386:995–1007. https://doi.org/10.1016/s0140-6736(15)60454-8
Liu C, Feng Y, Gao F et al (2006) Characterization of HCoV-229E fusion core: implications for structure basis of coronavirus membrane fusion. Biochem Biophys Res Commun 345:1108–1115. https://doi.org/10.1016/j.bbrc.2006.04.141
Gao J, Lu G, Qi J et al (2013) Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of middle east respiratory syndrome coronavirus. J Virol 87:13134–13140. https://doi.org/10.1128/jvi.02433-13
Lu L, Liu Q, Zhu Y et al (2014) Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat Commun 5:1–12. https://doi.org/10.1038/ncomms4067
Sun Y, Zhang H, Shi J et al (2017) Identification of a novel inhibitor against Middle East respiratory syndrome coronavirus. Viruses 9:255. https://doi.org/10.3390/v9090255
Xia S, Xu W, Wang Q et al (2018) Peptide-based membrane fusion inhibitors targeting HCoV-229E spike protein HR1 and HR2 domains. Int J Mol Sci 19:1–15. https://doi.org/10.3390/ijms19020487
Fields GB (2001) Introduction to Peptide Synthesis. In: Current Protocols in Protein Science. John Wiley & Sons, Inc., Hoboken, NJ, USA, pp 18.1.1–18.1.9
Pattabiraman VR, Bode JW (2011) Rethinking amide bond synthesis. Nature 480:471–479. https://doi.org/10.1038/nature10702
Di L (2014) Strategic approaches to optimizing peptide ADME properties. AAPS J 17:134–143. https://doi.org/10.1208/s12248-014-9687-3
Papo N, Oren Z, Pag U et al (2002) The consequence of sequence alteration of an amphipathic a—helical antimicrobial peptide and its diastereomers. J Biol Chem 277:33913–33921. https://doi.org/10.1074/jbc.m204928200
Lembo D, Donalisio M, Civra A et al (2018) Nanomedicine formulations for the delivery of antiviral drugs: a promising solution for the treatment of viral infections. Expert Opin Drug Deliv 15:93–114. https://doi.org/10.1080/17425247.2017.1360863
Ron-Doitch S, Sawodny B, Kühbacher A et al (2016) Reduced cytotoxicity and enhanced bioactivity of cationic antimicrobial peptides liposomes in cell cultures and 3D epidermis model against HSV. J Control Release 229:163–171. https://doi.org/10.1016/j.jconrel.2016.03.025
Acknowledgements
This work was supported by fellowships from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (LCPVB and RLAB), Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil (NCN), Fundação de Amparo a Pesquisa do Distrito Federal (FAPDF), and Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT). Authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vilas Boas, L.C.P., Campos, M.L., Berlanda, R.L.A. et al. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 76, 3525–3542 (2019). https://doi.org/10.1007/s00018-019-03138-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03138-w