
Abstract
Cystic fibrosis (CF) results from the absence or dysfunction of a single protein, the CF transmembrane conductance regulator (CFTR). CFTR plays a critical role in the regulation of ion transport in a number of exocrine epithelia. Improvement or restoration of CFTR function, where it is deficient, should improve the CF phenotype. There are >1000 reported disease-causing mutations of the CFTR gene. Recent investigations have afforded a better understanding of the mechanism of dysfunction of many of these mutant CFTRs, and have allowed them to be classified according to their mechanism of dysfunction. These data, as well as an enhanced understanding of the role of CFTR in regulating epithelial ion transport, have led to the development of therapeutic strategies based on pharmacologic enhancement or repair of mutant CFTR dysfunction. The strategy, termed ‘protein repair therapy’, is aimed at improving the regulation of epithelial ion transport by mutant CFTRs in a mutation-specific fashion. The grouping of CFTR gene mutations, according to mechanism of dysfunction, yields some guidance as to which pharmacologic repair agents may be useful for specific CFTR mutations. Recent data has suggested that combinations of pharmacologic repair agents may be necessary to obtain clinically meaningful CFTR repair. Nevertheless, such strategies to improve mutant CFTR function hold great promise for the development of novel therapies aimed at correcting the underlying pathophysiology of CF.


Similar content being viewed by others
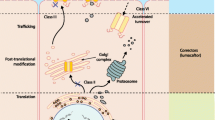
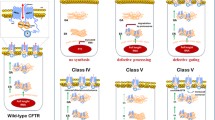

Notes
The use of trade names is for product identification purposes only and does not imply endorsement.
References
Schwiebert EM, Egan ME, Hwang TH, et al. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell 1995; 81: 1063–73
Reisin IL, Prat AG, Abraham EH, et al. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J Biol Chem 1994; 269: 20584–91
Braunstein GM, Roman RM, Clancy JP, et al. Cystic fibrosis transmembrane conductance regulator facilitates ATP release by stimulating a separate ATP release channel for autocrine control of cell volume regulation. J Biol Chem 2001; 276: 6621–30
Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. Am J Physiol 1998; 275 (1 Pt 1): C323–6
Choi JY, Muallem D, Kiselyov K, et al. Aberrant CFTR-dependent HCO3− transport in mutations associated with cystic fibrosis. Nature 2001; 410: 94–7
Konstas AA, Koch JP, Tucker SJ, et al. Cystic fibrosis transmembrane conductance regulator-dependent up-regulation of Kir1.1 (ROMK) renal K+ channels by the epithelial sodium channel. J Biol Chem 2002; 277: 25377–84
Lu M, Leng Q, Egan ME, et al. CFTR is required for PKA-regulated ATP sensitivity of Kir1.1 potassium channels in mouse kidney. J Clin Invest 2006; 116: 797–807
Briel M, Greger R, Kunzelmann K. Cl− transport by cystic fibrosis transmembrane conductance regulator (CFTR) contributes to the inhibition of epithelial Na+ channels (ENaCs) in Xenopus oocytes co-expressing CFTR and ENaC. J Physiol 1998; 508 (Pt 3): 825–36
Ismailov II, Awayda MS, Jovov B, et al. Regulation of epithelial sodium channels by the cystic fibrosis transmembrane conductance regulator. J Biol Chem 1996; 271: 4725–32
Ji HL, Chalfant ML, Jovov B, et al. The cytosolic termini of the beta- and gamma-ENaC subunits are involved in the functional interactions between cystic fibrosis transmembrane conductance regulator and epithelial sodium channel. J Biol Chem 2000; 275: 27947–56
Jiang Q, Li J, Dubroff R, et al. Epithelial sodium channels regulate cystic fibrosis transmembrane conductance regulator chloride channels in Xenopus oocytes. J Biol Chem 2000; 275: 13266–74
Suaud L, Li J, Jiang Q, et al. Genistein restores functional interactions between delta F508-CFTR and ENaC in Xenopus oocytes. J Biol Chem 2002; 277: 8928–33
Reddy MM, Light MJ, Quinton PM. Activation of the epithelial Na+ channel (ENaC) requires CFTR Cl− channel function. Nature 1999; 402: 301–4
Egan M, Flotte T, Afione S, et al. Defective regulation of outwardly rectifying Cl− channels by protein kinase A corrected by insertion of CFTR. Nature 1992; 358: 581–4
Egan ME, Schwiebert EM, Guggino WB. Differential expression of ORCC and CFTR induced by low temperature in CF airway epithelial cells. Am J Physiol 1995; 268 (1 Pt 1): C243–51
Tarran R, Loewen ME, Paradiso AM, et al. Regulation of murine airway surface liquid volume by CFTR and Ca2+ activated Cl− conductances. J Gen Physiol 2002; 120: 407–18
Paradiso AM, Ribeiro CM, Boucher RC. Polarized signaling via purinoceptors in normal and cystic fibrosis airway epithelia. J Gen Physiol 2001; 117: 53–67
Matsui H, Grubb BR, Tarran R, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998; 95: 1005–15
Worlitzsch D, Tarran R, Ulrich M, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 2002; 109: 317–25
Tarran R, Grubb BR, Parsons D, et al. The CF salt controversy: in vivo observations and therapeutic approaches. Mol Cell 2001; 8: 149–58
Hopf A, Schreiber R, Mall M, et al. Cystic fibrosis transmembrane conductance regulator inhibits epithelial Na+ channels carrying Liddle’s syndrome mutations. J Biol Chem 1999; 274: 13894–9
Kunzelmann K, Kiser GL, Schreiber R, et al. Inhibition of epithelial Na+ currents by intracellular domains of the cystic fibrosis transmembrane conductance regulator. FEBS Lett 1997; 400: 341–4
Yan W, Samaha FF, Ramkumar M, et al. Cystic fibrosis transmembrane conductance regulator differentially regulates human and mouse epithelial sodium channels in Xenopus oocytes. J Biol Chem 2004; 279: 23183–92
Nagel G, Szellas T, Riordan JR, et al. Non-specific activation of the epithelial sodium channel by the CFTR chloride channel. EMBO Rep 2001; 2: 249–54
Nagel G, Barbry P, Chabot H, et al. CFTR fails to inhibit the epithelial sodium channel ENaC expressed in Xenopus laevis oocytes. J Physiol 2005; 564: 671–82
Bachhuber T, Konig J, Voelcker T, et al. Cl− interference with the epithelial N+ channel ENaC. J Biol Chem 2005; 280: 31587–94
Tarran R, Button B, Picher M, et al. Normal and cystic fibrosis airway surface liquid homeostasis: the effects of phasic shear stress and viral infections. J Biol Chem 2005; 280: 35751–9
Kunzelmann K, Schreiber R, Boucherot A. Mechanisms of the inhibition of epithelial Na+ channels by CFTR and purinergic stimulation. Kidney Int 2001; 60: 455–61
Kunzelmann K, Bachhuber T, Regeer R, et al. Purinergic inhibition of the epithelial Na+ transport via hydrolysis of PIP2. FASEB J 2005; 19: 142–3
Huang P, Gilmore E, Kultgen P, et al. Local regulation of cystic fibrosis transmembrane regulator and epithelial sodium channel in airway epithelium. Proc Am Thorac Soc 2004; 1: 33–7
Devor DC, Pilewski JM. UTP inhibits Na+ absorption in wild-type and delta F508 CFTR-expressing human bronchial epithelia. Am J Physiol 1999; 276 (4 Pt 1): C827–37
Mall M, Grubb BR, Harkema JR, et al. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 2004; 10: 487–93
Baker E, Jeunemaitre X, Portal AJ, et al. Abnormalities of nasal potential difference measurement in Liddle’s syndrome. J Clin Invest 1998; 102: 10–4
Cystic fibrosis mutation database [online]. Available from URL: http://www.genet.sickkids.on.ca/cftr/app [Accessed 2006 Sep 13]
Rubenstein RC, Zeitlin PL. Use of protein repair therapy in the treatment of cystic fibrosis. Curr Opin Pediatr 1998; 10: 250–5
Zeitlin PL. Novel pharmacologic therapies for cystic fibrosis. J Clin Invest 1999; 103: 447–52
Suaud L, Yan W, Rubenstein RC. Abnormal regulatory interactions of I148T-CFTR and the epithelial Na+ channel in Xenopus oocytes. Am J Physiol Cell Pysiol. In press
Monaghan KG, Highsmith WE, Amos J, et al. Genotype-phenotype correlation and frequency of the 3199del6 cystic fibrosis mutation among I148T carriers: results from a collaborative study. Genet Med 2004; 6: 421–5
Rohlfs EM, Zhou Z, Sugarman EA, et al. The I148T CFTR allele occurs on multiple haplotypes: a complex allele is associated with cystic fibrosis. Genet Med 2002; 4: 319–23
Claustres M, Altieri JP, Guittard C, et al. Are p.I148T, p.R74W and p.D1270N cystic fibrosis causing mutations? BMC Med Genet 2004; 5: 19
Hamosh A, Rosenstein BJ, Cutting GR. CFTR nonsense mutations G542X and W1282X associated with severe reduction of CFTR mRNA in nasal epithelial cells. Hum Mol Genet 1992; 1: 542–4
Hamosh A, Trapnell BC, Zeitlin PL, et al. Severe deficiency of cystic fibrosis transmembrane conductance regulator messenger RNA carrying nonsense mutations R553X and W1316X in respiratory epithelial cells of patients with cystic fibrosis. J Clin Invest 1991; 88: 1880–5
Cystic Fibrosis Foundation Patient Registry: annual data report to the Center Directors. Bethesda (MD): Cystic Fibrosis Foundation, 2004
Shoshani T, Augarten A, Gazit E, et al. Association of a nonsense mutation (W1282X), the most common mutation in the Ashkenazi Jewish cystic fibrosis patients in Israel, with presentation of severe disease. Am J Hum Genet 1992; 50: 222–8
Kalman YM, Kerem E, Darvasi A, et al. Difference in frequencies of the cystic fibrosis alleles, delta F508 and W1282X, between carriers and patients. Eur J Hum Genet 1994; 2: 77–82
Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med 1996; 2: 467–9
Bedwell DM, Kaenjak A, Benos DJ, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 1997; 3: 1280–4
Du M, Jones JR, Lanier J, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr−/−mouse carrying a human CFTR-G542X transgene. J Mol Med 2002; 80: 595–604
Clancy JP, Bebok Z, Ruiz F, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med 2001; 163: 1683–92
Knowles MR, Paradiso AM, Boucher RC. In vivo nasal potential difference: techniques and protocols for assessing efficacy of gene transfer in cystic fibrosis. Hum Gene Ther 1995; 6: 445–55
Wilschanski M, Famini C, Blau H, et al. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med 2000; 161: 860–5
Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 2003; 349: 1433–41
PTC Therapeutics [online]. Available from URL: http://www.ptcbio.com [Accessed 2006 Sep 20]
Pasyk EA, Foskett JK. Mutant (delta F508) cystic fibrosis transmembrane conductance regulator Cl− channel is functional when retained in endoplasmic reticulum of mammalian cells. J Biol Chem 1995; 270: 12347–50
Cheng SH, Gregory RJ, Marshall J, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990; 63: 827–34
Denning GM, Anderson MP, Amara JF, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992; 358: 761–4
Brown CR, Hong-Brown LQ, Biwersi J, et al. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1996; 1: 117–25
Sato S, Ward CL, Krouse ME, et al. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 1996; 271: 635–8
Bebok Z, Venglarik CJ, Panczel Z, et al. Activation of delta F508 CFTR in an epithelial monolayer. Am J Physiol 1998; 275 (2 Pt 1): C599–607
Zhang XM, Wang XT, Yue H, et al. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem 2003; 278: 51232–42
Cheng SH, Fang SL, Zabner J, et al. Functional activation of the cystic fibrosis trafficking mutant delta F508-CFTR by overexpression. Am J Physiol 1995; 268 (4 Pt 1): L615–24
Moyer BD, Loffing-Cueni D, Loffing J, et al. Butyrate increases apical membrane CFTR but reduces chloride secretion in MDCK cells. Am J Physiol 1999; 277 (2 Pt 2): F271–6
Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest 1997; 100: 2457–65
Jiang C, Fang SL, Xiao YF, et al. Partial restoration of cAMP-stimulated CFTR chloride channel activity in delta F508 cells by deoxyspergualin. Am J Physiol 1998; 275 (1 Pt 1): C171–8
Kelley TJ, Al Nakkash L, Cotton CU, et al. Activation of endogenous delta F508 cystic fibrosis transmembrane conductance regulator by phosphodiesterase inhibition. J Clin Invest 1996; 98: 513–20
Maitra R, Shaw CM, Stanton BA, et al. Increased functional cell surface expression of CFTR and delta F508-CFTR by the anthracycline doxorubicin. Am J Physiol Cell Physiol 2001; 280: C1031–7
Jacobson KA, Guay-Broder C, van Galen PJ, et al. Stimulation by alkylxanthines of chloride efflux in CFPAC-1 cells does not involve A1 adenosine receptors. Biochemistry 1995; 34: 9088–94
Zaman K, McPherson M, Vaughan J, et al. S-nitrosoglutathione increases cystic fibrosis transmembrane regulator maturation. Biochem Biophys Res Commun 2001; 284: 65–70
Andersson C, Gaston B, Roomans GM. S-nitrosoglutathione induces functional delta F508-CFTR in airway epithelial cells. Biochem Biophys Res Commun 2002; 297: 552–7
Dormer RL, Derand R, McNeilly CM, et al. Correction of delta F508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci 2001; 114: 4073–81
Egan ME, Glockner-Pagel J, Ambrose C, et al. Calcium-pump inhibitors induce functional surface expression of delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat Med 2002; 8: 485–92
Egan ME, Pearson M, Weiner SA, et al. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 2004; 304: 600–2
Kelley TJ, Thomas K, Milgram LJ, et al. In vivo activation of the cystic fibrosis transmembrane conductance regulator mutant delta F508 in murine nasal epithelium. Proc Natl Acad Sci U S A 1997; 94: 2604–8
Fischer H, Fukuda N, Barbry P, et al. Partial restoration of defective chloride conductance in delta F508 CF mice by trimethylamine oxide. Am J Physiol Lung Cell Mol Physiol 2001; 281: L52–7
Zeitlin PL, Diener-West M, Rubenstein RC, et al. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol Ther 2002; 6: 119–26
Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res 1991; 29: 147–50
Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of delta F508-CFTR. Am J Physiol Cell Physiol 2000; 278: C259–67
Rubenstein RC, Lyons BM. Sodium 4-phenylbutyrate downregulates HSC70 expression by facilitating mRNA degradation. Am J Physiol Lung Cell Mol Physiol 2001; 281: L43–51
Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes delta F508 CFTR trafficking. Am J Physiol Lung Cell Mol Physiol 2001; 281: L58–68
Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (buphenyl) in delta F508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med 1998; 157: 484–90
Song Y, Sonawane ND, Salinas D, et al. Evidence against rescue of defective delta F508-CFTR cellular processing by curcumin in cell culture and mouse models. J Biol Chem 2004 Sep 24; 279(39): 40629–33
Grubb BR, Gabriel SE, Mengos A, et al. SERCA pump inhibitors do not correct biosynthetic arrest of delta F508CFTR in cystic fibrosis. Am J Respir Cell Mol Biol 2005 Mar; 34(3): 355–63
Norez C, Antigny F, Becq F, et al. Maintaining low Ca level in the endoplasmic reticulum restores abnormal endogenous delta F508-CFTR trafficking in airway epithelial cells. Traffic 2006; 7: 562–73
Lipecka J, Norez C, Bensalem N, et al. Rescue of delta F508-CFTR (cystic fibrosis transmembrane conductance regulator) by curcumin: involvement of the keratin 18 network. J Pharmacol Exp Ther 2006; 317: 500–5
Berger AL, Randak CO, Ostedgaard LS, et al. Curcumin stimulates cystic fibrosis transmembrane conductance regulator Cl− channel activity. J Biol Chem 2005; 280: 5221–6
Yang H, Shelat AA, Guy RK, et al. Nanomolar affinity small molecule correctors of defective delta F508-CFTR chloride channel gating. J Biol Chem 2003; 278: 35079–85
Van Goor F, Straley KS, Cao D, et al. Rescue of delta F508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 2006 Jun; 290(6): L1117–30
Gonzalez JE, Oades K, Leychkis Y, et al. Cell-based assays and instrumentation for screening ion-channel targets. Drug Discov Today 1999; 4: 431–9
Logan J, Hiestand D, Daram P, et al. Cystic fibrosis transmembrane conductance regulator mutations that disrupt nucleotide binding. J Clin Invest 1994; 94: 228–36
Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993; 73: 1251–4
Hwang TC, Wang F, Yang IC, et al. Genistein potentiates wild-type and delta F508-CFTR channel activity. Am J Physiol 1997; 273 (3 Pt 1): C988–98
Illek B, Zhang L, Lewis NC, et al. Defective function of the cystic fibrosis-causing missense mutation G551D is recovered by genistein. Am J Physiol 1999; 277 (4 Pt 1): C833–9
Urban D, Irwin W, Kirk M, et al. The effect of isolated soy protein on plasma biomarkers in elderly men with elevated serum prostate specific antigen. J Urol 2001; 165: 294–300
Galietta LV, Jayaraman S, Verkman AS. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol 2001; 281: C1734–42
Caci E, Folli C, Zegarra-Moran O, et al. CFTR activation in human bronchial epithelial cells by novel benzoflavone and benzimidazolone compounds. Am J Physiol Lung Cell Mol Physiol 2003; 285: L180–8
Ma T, Vetrivel L, Yang H, et al. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J Biol Chem 2002; 277: 37235–41
Galietta LJ, Springsteel MF, Eda M, et al. Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem 2001; 276: 19723–8
Sheppard DN, Rich DP, Ostedgaard LS, et al. Mutations in CFTR associated with mild-disease-form Cl− channels with altered pore properties. Nature 1993; 362: 160–4
Highsmith WE, Burch LH, Zhou Z, et al. A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. N Engl J Med 1994; 331: 974–80
Highsmith WE, Burch LH, Zhou Z, et al. Identification of a splice site mutation (2789 + 5 G > A) associated with small amounts of normal CFTR mRNA and mild cystic fibrosis. Hum Mutat 1997; 9: 332–8
Silvis MR, Picciano JA, Bertrand C, et al. A mutation in the cystic fibrosis transmembrane conductance regulator generates a novel internalization sequence and enhances endocytic rates. J Biol Chem 2003; 278: 11554–60
Lukacs GL, Chang XB, Bear C, et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane: determination of functional half-lives on transfected cells. J Biol Chem 1993; 268: 21592–8
Swiatecka-Urban A, Brown A, Moreau-Marquis S, et al. The short apical membrane half-life of rescued delta F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of delta F508-CFTR in polarized human airway epithelial cells. J Biol Chem 2005; 280: 36762–72
Dalemans W, Barbry P, Champigny G, et al. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 1991; 354: 526–8
Li C, Ramjeesingh M, Reyes E, et al. The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat Genet 1993; 3: 311–6
Randak C, Welsh MJ. An intrinsic adenylate kinase activity regulates gating of the ABC transporter CFTR. Cell 2003; 115: 837–50
Lim M, McKenzie K, Floyd AD, et al. Modulation of delta F508 CFTR trafficking and function with 4-PBA and flavonoids. Am J Respir Cell Mol Biol 2004 Sep; 31(3): 351–7
Mall M, Hipper A, Greger R, et al. Wild type but not delta F508 CFTR inhibits Na+ conductance when coexpressed in Xenopus oocytes. FEBS Lett 1996; 381: 47–52
Suaud L, Carattino M, Kleyman TR, et al. Genistein improves regulatory interactions between G551D-cystic fibrosis transmembrane conductance regulator and the epithelial sodium channel in Xenopus oocytes. J Biol Chem 2002; 277: 50341–7
Acknowledgments
The author’s research is supported by grants from the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (grant numbers R01–DK58046 and R01–DK54354), the Cystic Fibrosis Foundation, and an Established Investigator Award from the American Heart Association.
The author has no conflict of interest that is directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rubenstein, R.C. Targeted Therapy for Cystic Fibrosis. Mol Diag Ther 10, 293–301 (2006). https://doi.org/10.1007/BF03256204
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF03256204