
Summary
A bioequivalence study compares the bioavailability between a test and a reference drug product in terms of the rate and extent of drug absorption. Area under the plasma concentration-time curve (AUC) and maximum plasma concentration (Cmax) are the pharmacokinetic parameters that serve as characteristics for the assessment of the extent and rate of absorption, respectively. The experimental design of a bioequivalence study is usually a crossover and rarely a parallel or a paired comparative. The statistical assessment of bioequivalence is based on the 90% confidence interval for the ratio of the test mean to the reference mean for AUC and Cmax The aims of this paper are to: (i) investigate alternative designs to a crossover design for conducting bioequivalence studies; (ii) propose the statistical analysis of different designs for bioequivalence studies on the same products; and (iii) discuss their usefulness for the approval of new generic drug products. For this purpose, three case studies are illustrated and analysed. The first case study concerns the investigation of the merits of a crossover design relative to a parallel group design for highly variable drugs using as an example a bioequivalence study of tamoxifen products. The second case study concerns the pooled statistical analysis of two bioequivalent studies of the same levodopa products. The analyses of the individual studies failed to meet the regulatory criteria for bioequivalence. The one study design was a paired comparative and the other one a crossover. Under some assumptions the crossover design may be considered as a paired comparative and the data from the two studies may be analysed together as a paired comparative design. The third case study concerns the statistical pooled analysis of two bioequivalent studies of the same clodronate products. The one study was a three-period crossover pilot study and it was used to identify the variability of the active substance. Then, this variability was used to determine the number of subjects for the main pivotal study which was a two-period crossover. The pilot study design was converted into a two-period crossover design and the data from the two studies were analysed together as a two-period crossover design. The original data of the studies were modified accordingly.
Similar content being viewed by others

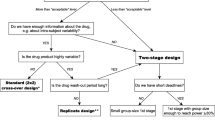
References
Chow S.C., Liu J.P. (1992): Design and analysis of bioavailability and bioequivalence studies.
Schuirmann D.J. (1990): Statistical evaluation of bioequivalence studies. In: Sharma K.N., Sharma K.K., Sen P. Eds. Generic Drugs, Bioequivalence and Pharmacokinetics, UCMS, Delhi, pp 119–125.
CPMP: Note for guidance on the investigation of bioavailability and bioequivalence. In: The Rules Governing Medicinal Products for Human Use in the Member States of the European Community, Volume III, Addendum No 2, Guidelines on the Quality, Safety & Efficacy of Medicinal Products for Human Use pp 149–168.
Metzler C.M. (1974): Bioavailability — a problem in equivalence. Biometrics, 30, 309–317.
Jones B., Kenward M.G. (1995): Design and Analysis of Crossover Trials. London: Chapman and Hall.
Dilleti E., Hauschke D., Steinijans V.W. (1992): Sample size determination for bioequivalence assessment by means of confidence intervals. Int. J. Clin. Pharmacol. Ther. Toxicol., 29, 51–58.
Jones B., Jarvis P., Lewis J.A., Ebbutt A.F. (1996): Trials to assess equivalence: the importance of rigorous methods. BMJ, 313, 36–39.
Mead R. (1988): Design of Experiments. Cambridge: Cambridge University Press.
Midha K.K, Blume H. (1993): Report of Consensus Meeting: Biointernational ′92, Conference on Bioavailability, Bioequivalence and Pharmacokinetic studies. Eur. J. Pharm. Sci., 1, 165–171.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Zintzaras, E., Bouka, P. Bioequivalence studies: biometrical concepts of alternative designs and pooled analysis. Eur. J. Drug Metab. Pharmacokinet. 24, 225–232 (1999). https://doi.org/10.1007/BF03190024
Received:
Issue Date:
DOI: https://doi.org/10.1007/BF03190024